Discovery of potent, proteolytically stable, and cell permeable human sirtuin peptidomimetic inhibitors containing Nε-thioacetyl-lysine†
Brett M.
Hirsch
a,
Caroline A.
Gallo
a,
Zhanwen
Du
b,
Zhenghe
Wang
b and
Weiping
Zheng
*a
aDepartment of Chemistry, University of Akron, Akron, OH 44325, USA. E-mail: wzheng@uakron.edu; Tel: +1 330 972 2193
bDepartment of Genetics and Case Comprehensive Cancer Center, Case Western Reserve University, Cleveland, OH 44106, USA
First published on 18th August 2010
Abstract
Inhibitors of sirtuin-catalyzed NAD+-dependent protein lysine deacetylation reaction possess great value for facilitating deciphering the biology of sirtuins and as potential therapeutic agents for metabolic and age-related diseases and cancer. Built upon our previously discovered potent Nε-thioacetyl-lysine (ThAcK)-containing 18-amino acid peptidic human sirtuin inhibitor 1, we performed a structure–activity-relationship (SAR) study with the goal of transforming this potent peptidic inhibitor to potent yet proteolytically stable, cell permeable, and low molecular weight ThAcK-containing peptidomimetic inhibitors. Specifically, in this SAR study, we have identified two such peptidomimetic inhibitors (8 and 9) that exhibited potent in vitro and in vivo human sirtuin inhibition and whose molecular weights are in the 500 Da range.
Introduction
Silent information regulator (Sir2) enzymes (or sirtuins) are a family of protein deacetylases that can catalyze the NAD+-dependant protein lysine Nε-deacetylation, with the formation of lysine Nε-deacetylated protein species and small molecule products, i.e. nicotinamide and 2′-O-acetyl-ADP-ribose. Following its release into the bulk solution, this latter product was found to undergo a non-enzymatic isomerization to 3′-O-acetyl-ADP-ribose and eventually reach a roughly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar mixture of the two ADP-ribose derivatives (Fig. 1, X = O).1 These enzymes are known to be present in all the three kingdoms of life with the yeast Sir2 being the founding family member. In humans, seven Sir2 homologs (i.e. SIRT1-7) have been identified. While the first protein substrates identified were the histone proteins, it is now known that sirtuins also accept many non-histone proteins as their physiological substrates, including various transcription factors and metabolic enzymes.1c,2 The sirtuin-catalyzed protein lysine Nε-deacetylation has been shown to play important roles in multiple biological processes such as gene transcription, apoptosis, DNA repair, metabolism, aging, neurodegeneration, and HIV-1 replication.2e,3 Therefore, there has been a tremendous interest in identifying inhibitors for this enzymatic deacetylation reaction.1c,4 These inhibitors could be potential novel therapeutics for metabolic and age-related diseases and cancer. They could also be used as chemical biological tools toward a fuller exploration of sirtuin biology and pharmacology.
:1 molar mixture of the two ADP-ribose derivatives (Fig. 1, X = O).1 These enzymes are known to be present in all the three kingdoms of life with the yeast Sir2 being the founding family member. In humans, seven Sir2 homologs (i.e. SIRT1-7) have been identified. While the first protein substrates identified were the histone proteins, it is now known that sirtuins also accept many non-histone proteins as their physiological substrates, including various transcription factors and metabolic enzymes.1c,2 The sirtuin-catalyzed protein lysine Nε-deacetylation has been shown to play important roles in multiple biological processes such as gene transcription, apoptosis, DNA repair, metabolism, aging, neurodegeneration, and HIV-1 replication.2e,3 Therefore, there has been a tremendous interest in identifying inhibitors for this enzymatic deacetylation reaction.1c,4 These inhibitors could be potential novel therapeutics for metabolic and age-related diseases and cancer. They could also be used as chemical biological tools toward a fuller exploration of sirtuin biology and pharmacology.
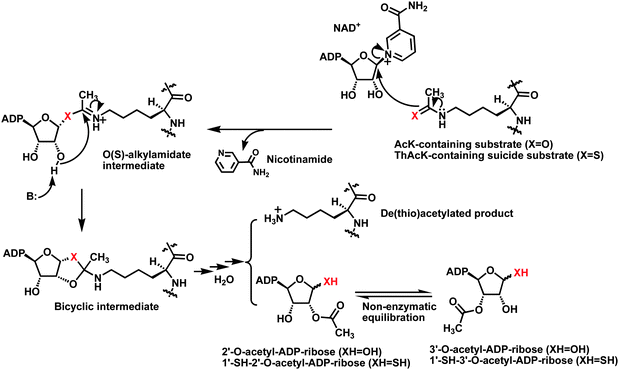 | ||
| Fig. 1 The proposed chemical mechanism for the sirtuin-catalyzed lysine Nε-de(thio)acetylation reaction. When a ThAcK-containing suicide substrate (X = S) is used, the corresponding S-alkylamidate intermediate is stalled along the reaction coordinate. ADP, adenosine diphosphate; B: refers to a general base. | ||
Our laboratory previously demonstrated that simply replacing Nε-thioacetyl-lysine (ThAcK) for Nε-acetyl-lysine (AcK) in a peptide substrate could achieve very potent inhibition of various human sirtuins.5 The ThAcK-containing peptides were also shown by us and others in biochemical and structural studies to behave as mechanism-based inhibitors (or suicide substrates) of sirtuins.5a,6–8 Specifically, a ThAcK-containing peptide can be recognized by a sirtuin as a substrate, but its ThAcK residue is converted by a sirtuin to a stalled S-alkylamidate intermediate (Fig. 1, X = S), a mimic of the O-alkylamidate intermediate formed from AcK during the normal sirtuin catalysis (Fig. 1, X = O). As a result, the sirtuin enzymatic activity is inhibited. A sluggish sirtuin-catalyzed formation of the dethioacetylated product and 1′-SH-2′-O-acetyl-ADP-ribose was also observed.
Since linear peptides are susceptible to protease/peptidase-catalyzed peptide bond cleavage and are not cell permeable in general,9 we embarked on a structure–activity-relationship (SAR) study with the goal of transforming the potent ThAcK-containing peptide-based inhibitor to potent yet proteolytically stable and cell permeable ThAcK-containing peptidomimetic inhibitors. Since it was shown that the molecular weight can also impact a compound's membrane permeability and a lower molecular weight could promote membrane permeability better than a higher molecular weight,10 it was also our interest to develop the ThAcK-containing peptidomimetic inhibitors with low molecular weights. For our SAR study reported here, we used peptide 1 (Fig. 2) as the starting point since it was shown previously by us to be a very potent inhibitor for human SIRT1.5
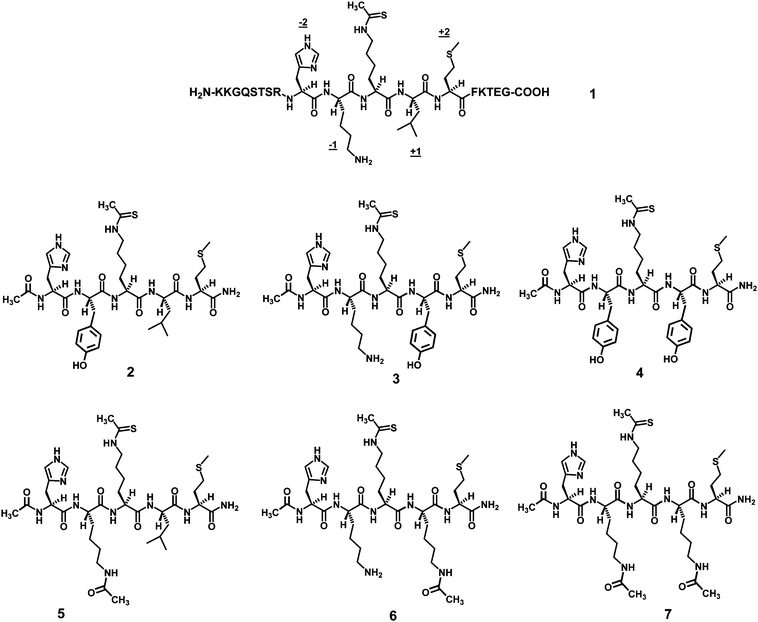 | ||
| Fig. 2 Structures of the ThAcK-containing peptidic human sirtuin inhibitors involved in the current study. The four positions immediately flanking the ThAcK residue in peptide 1 are also indicated, i.e. the −2, −1, +1, and +2 positions. | ||
Results and discussion
Since it was indicated from previous structural and biochemical studies that (i) AcK and the two immediately flanking amino acid residues on each side of AcK were the amino acid residues of a peptide substrate that made predominant binding interactions with a sirtuin,8,11 and (ii) a ThAcK-containing peptide can behave as a mechanism-based sirtuin inhibitor in that the first obligatory step for its inhibitory action is its binding to the sirtuin active site as a substrate,5a,6–8 the first step we took to modify 1 was to truncate this 18-amino acid peptide to a 5-amino acid peptide with the central ThAcK residue. In addition to this modification, we also modified the side chains at the two positions closest to the central ThAcK residue (i.e. the −1 and the +1 position, Fig. 2) as follows: the native side chains at these two positions (i.e. those of lysine and leucine residues) were replaced by those of tyrosine and AcK. With the expected increase in the lipophilicity at these two positions of a penta-peptide following such replacement, a SAR examination of this structural modification would be able to allow us to assess the feasibility of developing ThAcK-containing molecules with an enhanced chance of cell permeability. Tyrosine and AcK were chosen as the replacing residues because their side chains were more hydrophobic than the native side chains, yet they are still capable of mediating hydrogen bonding interactions due to the presence of the hydrogen bonding donors and acceptors. Furthermore, in our opinion tyrosine is the best choice for this purpose among all the standard amino acids with uncharged polar side chains. AcK was chosen further because it is a simple neutral Nε-acetylated analog of lysine. The penta-peptides thus designed are shown in Fig. 2 (i.e. peptides 2–7).All the penta-peptides were synthesized using the 9-fluorenylmethoxycarbonyl (Fmoc) chemistry-based solid phase peptide synthesis (SPPS)12 and purified by the preparative high pressure liquid chromatography (HPLC). The resulting peptides were screened for their SIRT1 inhibitory potency by a HPLC-based SIRT1 inhibition assay,5 and the data are recorded in Table 1.
As shown in Table 1, this quick SIRT1 inhibition screening revealed that all the ThAcK-containing penta-peptides still maintained the potent SIRT1 inhibition exhibited by the ThAcK-containing 18-amino acid lead peptide inhibitor 1, as evidenced by the modest (within ∼3-fold) change in the IC50 values from 1 to the penta-peptides. It should be noted that the AcK residue at the −1 and the +1 position in the last three penta-peptides in Fig. 2 was not deacetylated by SIRT1 under our sirtuin inhibition assay condition (data not shown). Since it is now known that a ThAcK-containing peptide can be a mechanism-based sirtuin inhibitor in that it is first treated as a substrate at a sirtuin active site, our result is consistent with the current literature view on the normal peptide substrate recognition within a sirtuin active site: AcK and the two immediately flanking amino acid residues on each side of AcK were the amino acid residues that made predominant binding interactions within a sirtuin active site.8,11 Our current finding also reinforced our previous discovery of a modest (∼6-fold) decline in SIRT1 inhibitory potency for H2N-HK-ThAcK-LM-COOH as compared to peptide 15b and the discovery of some potent ThAcK-containing SIRT1 inhibitory penta-peptides by Kiviranta et al.13
The data in Table 1 also indicates that the −1 and the +1 position in a ThAcK-containing penta-peptide can both be replaced with the neutral, bulkier, and more hydrophobic side chains without a dramatic change in the SIRT1 inhibitory potency. Given the previous demonstration that the −1 position relative to AcK played an important role in peptide substrate recognition by bacterial Sir2Tm and yeast Hst2,11c our observed lack of a dramatic change in SIRT1 inhibitory potency following replacing the basic lysine at the −1 position with the neutral residues tyrosine and AcK therefore suggested that different sirtuins could have quite different modes of substrate side chain recognition at their active sites.
However, as another possible explanation for the lack of a dramatic change in SIRT1 inhibitory potency following the substitution of tyrosine and AcK for lysine at the −1 position, it could be argued that the basic histidine residue at the −2 position could compensate for the charge neutralization at the −1 position. Therefore, we were next interested in removing this histidine residue from the penta-peptide bearing a −1 modification, and for this purpose we chose peptide 7 as the template because it was found to be the most potent SIRT1 inhibitor among all the penta-peptides bearing a −1 modification (i.e. peptides 2, 4, 5, and 7). Furthermore, we also decided to remove the methionine residue at the +2 position of 7 to further decrease the molecular weight. The resulting neutral peptidomimetic compound 8 (Fig. 3) could still be a potent SIRT1 inhibitor (i) since the AcK substitution for leucine at the +1 position is apparently highly favored as evidenced by the very strong SIRT1 inhibitory potency of peptide 6 (even 2.4-fold more potent than peptide 1) and (ii) if a charge interaction involving the −1 and the −2 position is indeed not indispensible. Compound 8 was then synthesized using the Fmoc chemistry-based SPPS, purified by the preparative HPLC, and assayed for its SIRT1 inhibitory potency by the HPLC-based SIRT1 inhibition assay, and the data is recorded in Table 2.
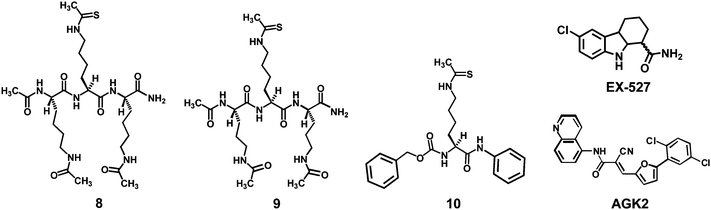 | ||
| Fig. 3 Structures of the ThAcK-containing peptidomimetic human sirtuin inhibitors 8 and 9, as well as the three reference compounds (10, EX-527, and AGK2) used in the current study. | ||
| Compound | 8 | 9 | 10 | EX-527 | AGK2 | |
|---|---|---|---|---|---|---|
| a See the ESI for the sirtuin inhibition assay details.† b The compound reported by Suzuki et al.14 c The compound reported by Napper et al.15 d The compound reported by Outeiro et al.19 e N.D., Not determined in the current study. | ||||||
| IC50 (μM) | SIRT1 | 2.1 ± 0.4 | 5.9 ± 1.6 | 140 ± 21 | 0.76 ± 0.12 | N.D.e |
| SIRT2 | 2.4 ± 0.01 | 2.9 ± 1.3 | N.D. | N.D. | 55 ± 42 | |
| SIRT3 | 4.5 ± 1.0 | 4.3 ± 0.1 | N.D. | N.D. | N.D. | |
Compound 8 also turned out to be a fairly potent low micromolar SIRT1 inhibitor under our assay condition. This result tends to support the notion that a positive charge at the −1 and the −2 position is dispensible for a favorable SIRT1 substrate binding interaction. In order to assess the SIRT1 inhibitory potency of compound 8 further, we synthesized and tested the reference compound 10 (Fig. 3) which was disclosed recently by Suzuki et al.,14 and is the only currently reported small molecule non-peptidic ThAcK-containing human sirtuin inhibitor. When 10 was evaluated under our SIRT1 inhibition assay condition, we found that it was a ∼67-fold weaker SIRT1 inhibitor than our compound 8 (Table 2). We also evaluated another reference compound under our SIRT1 inhibition assay condition, that is EX-527 (Fig. 3) which was reported by Napper et al.15 and represents the most potent and selective SIRT1 inhibitor in literature. We found that EX-527 indeed was a very potent sub-micromolar SIRT1 inhibitor (Table 2). However, we also found that our compound 8 is only ∼2.8-fold weaker as an SIRT1 inhibitor than EX-527. In our opinion, the much higher IC50 values we obtained for the reference compounds 10 and EX-527 than those reported14,15 (i.e. 140 μM vs. 2.7 μM; 0.76 μM vs. 0.098 μM) resulted from the use of the much higher [S]/Km ratios (i.e. the ratio of the substrate concentration over its Km value) in our HPLC-based assay, so that it takes more of an inhibitor to inhibit the binding of the substrate to the active site of an enzyme. Furthermore, 10 was pre-incubated with NAD+ in the presence of SIRT1 before the addition of the acetylated peptide substrate in the SIRT1 inhibition assay procedure that Suzuki et al. used.14
Compound 8 was next examined for its proteolytic stability. For this purpose, we used pronase as the proteolytic enzyme preparation since it is a mixture of a variety of different types of proteases/peptidases and as such it has very broad substrate specificity.16 When the pronase digestion reaction was monitored by HPLC, a fast time-dependent degradation was observed for the control peptide NH2-HK-AcK-LM-COOH with all the starting peptide being degraded within 5 min since the start of the digestion reaction. However, essentially no degradation occurred within 30 min for compound 8 (Fig. 4). This finding suggested that the peptidomimetic compound 8 is much more proteolytically stable than a linear peptide. The proteolytic stability of 8 could have resulted from the fact that all of its three amino acid residues (i.e. ThAcK and AcK) are non-standard residues.
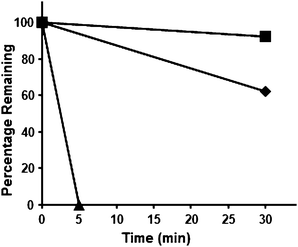 | ||
| Fig. 4 The proteolytic stability of 8 and 9, as assessed by the pronase digestion assay. ■, degradation profile of 8; ♦, degradation profile of 9; ▲, degradation profile of the control: NH2-HK-AcK-LM-COOH. | ||
As can be seen in Fig. 3, compound 8 has two AcK residues. We did not observe SIRT1-catalyzed deacetylation of this compound under our sirtuin inhibition assay condition. However, in order to more rigorously examine the stability of compound 8 toward SIRT1, we subjected compound 8 to a HPLC-based time course assay with SIRT1. As indicated from the resulting HPLC traces (see ESI†), compound 8 was essentially resistant to SIRT1. Of note, a longer reaction time (4 h) was employed in this time course assay than that used (5 min) in the sirtuin inhibition assay with SIRT1 (see ESI†).
It is now known that all the currently known protein deacetylase enzymes are either sirtuins or the Zn2+-containing classical family of protein deacetylases.2b,2c Of note, 11 classical enzymes (i.e. HDAC1-11 wherein HDAC stands for histone deacetylase) have been identified in humans. Furthermore, as we demonstrated previously, ThAcK could also be dethioacetylated by HDAC8 when placed within an appropriate structural context.5,17 Therefore, compound 8 was further subjected to the deacetylase time course assay with two different preparations of the human HDACs, i.e. the recombinant HDAC8 and the HeLa nuclear extract enriched in HDAC1 and HDAC2. The enzymatic reaction progress was also monitored by HPLC and graphed in Fig. S1.† It is apparent from these two graphs that compound 8 was essentially resistant to HDAC8 and nearly resistant to the HDACs (primarily HDAC1 and HDAC2) present in the HeLa nuclear extract, despite the observed ∼19% decline in the amount of the starting 8 in two hours in the assay with the HeLa nuclear extract.
Following the above in vitro studies, compound 8 was further evaluated in a cell-based assay to examine its capability of achieving SIRT1 inhibition inside the cell. For this purpose, we used the HCT116 human colon cancer cells that express the wild-type p53 protein and assessed the extent to enhance the level of the acetylation of this SIRT1 endogenous substrate protein at the K382 position18 following the treatment with compound 8. As shown in Fig. 5, 8 was able to enhance the p53 K382 acetylation level in a dose-dependent manner. Furthermore, based on visual inspection, it could be estimated that 100% p53 acetylation level increase was already achieved with 3 μM of 8. However, peptide 1 was shown not to be able to affect the p53 acetylation level even at 500 μM under our assay condition, which is consistent with it being a cell impermeable peptide that is unable to inhibit SIRT1 inside the cell. Consistent with our in vitro finding that 8 was more potent than 10 as a SIRT1 inhibitor (vide supra), this degree of p53 K382 acetylation level increase was also more profound than that observed previously by Suzuki et al.14 with 10 and the HCT116 human colon cancer cells. Based on visual inspection of the western blot image shown in the paper by Suzuki et al.,14 it could be estimated that 10 was unable to achieve a 100% p53 acetylation level increase at a concentration of 10 μM. In our opinion, the greater SIRT1 inhibition achieved with 8versus10 resulted from a rich presence in 8 of the functional moieties immediately flanking the ThAcK residue that would be able to mediate different types of binding interactions (e.g. hydrogen bonding and van der Waals contacts) with SIRT1 at its active site.
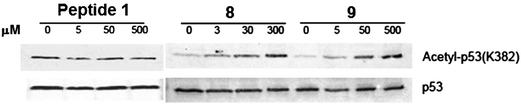 | ||
| Fig. 5 Western blot analysis of the p53 protein acetylation level change in HCT116 human colon cancer cells following the treatment with 1, 8, or 9. | ||
We were also interested in examining compound 8 for its inhibitory potency against the other two human sirtuins, i.e. SIRT2 and SIRT3. As can be seen in Table 2, 8 was also found to be a potent inhibitor against SIRT2 and SIRT3. We further found that 8 was a ∼23-fold more potent SIRT2 inhibitor than another reference compound under our assay condition, i.e. AGK2 reported by Outeiro et al. as a potent and selective SIRT2 inhibitor.19 Again, the much higher IC50 value we obtained for AGK2 than that reported19 (i.e. 55 μM vs. 3.5 μM) could also result from the use of the much higher [S]/Km ratios in our HPLC-based SIRT2 inhibition assay.
It should be pointed out that, even though the IC50 values rather than the Ki values were used in our current study to compare the sirtuin inhibitory potency of our peptidomimetic inhibitors to those of the reference compounds (i.e.10, EX-527, and AGK2), in our opinion, the former is a more appropriate parameter for this particular comparison because of the different inhibition kinetics. Specifically, while the ThAcK-containing sirtuin inhibitors are competitive inhibitors versus the acetyl-lysine substrate,6 EX-527 is a mixed-type inhibitor versus the acetyl-lysine substrate,15 and the sirtuin inhibition kinetics for AGK2 is currently unknown. Furthermore, different inhibitors were assayed in our current study against a given sirtuin enzyme under a single specified condition, the obtained IC50 values should thus enable the direct comparison of the inhibitory potency of the different inhibitors against the given sirtuin enzyme.
Compound 8 was also found to be essentially resistant to SIRT2 and SIRT3 as indicated by the HPLC analysis traces from the time course assays with these two sirtuins and 8 (see ESI†). Again, a longer reaction time and a higher enzyme concentration and/or a higher substrate concentration were employed in the time course assays than those used in the sirtuin inhibition assay with SIRT2 and SIRT3 (see ESI†).
While roughly equal inhibitory potency was observed for SIRT1 and SIRT2, 8 was a ∼2-fold weaker inhibitor against SIRT3 than SIRT1. While this degree of selectivity between SIRT1 and SIRT3 is not spectacular, the overall inhibitory profile of 8 among these three human sirtuins could be beneficial given a very recent report suggesting that a SIRT1 and SIRT2 dual inhibitor would be more able to confer an anticancer benefit than an inhibitor highly selective for SIRT1, e.g. EX-527.20 Nevertheless, in order to develop sirtuin inhibitors as chemical biological tools for helping dissect the functions of individual sirtuins, a high degree of inhibitory selectivity among different sirtuins would be desirable. In this regard, our compound 8 could be less desirable. However, we believe that compound 8 could serve as a new platform toward developing selective sirtuin inhibitors, and the modular nature of 8 (i.e. consisting of two non-standard amino acid residues (i.e. AcK) immediately flanking ThAcK on its two sides) would make its further SAR study a feasible task.
To explore this feasibility in the current study, we changed the two AcK residues in 8 to Nδ-acetyl-ornithine, a very close structural analog of AcK. The resulting peptidomimetic compound 9 (Fig. 3) was also synthesized, purified, and tested for its sirtuin inhibitory potency as was compound 8. As shown in Table 2, compound 9 was found to maintain the SIRT2 and SIRT3 inhibitory potency of compound 8, however, it was a ∼2.8-fold weaker SIRT1 inhibitor than 8. These results suggested that even a very subtle structural change in the neighboring moieties of ThAcK in 8 can alter the inhibitory profile among different sirtuins. This result also lends support to the idea that different sirtuins could have quite different modes of substrate side chain recognition at their active sites.
In the subsequent time course assays with pronase, SIRT1, SIRT2, SIRT3, HDAC8, and HeLa nuclear extract, compound 9 was also found to be much more proteolytically stable than the linear peptide control even though it is less stable than compound 8 (Fig. 4). Again, the enhanced proteolytic stability of 9versus the linear peptide could also result from the fact that all the three amino acid residues (i.e. ThAcK and Nδ-acetyl-ornithine) in 9 are non-standard residues. Compound 9 was also found to be essentially resistant to SIRT1, SIRT2, and SIRT3 as indicated by the HPLC analysis traces from the sirtuin time course assays with 9 (see ESI†). Furthermore, compound 9 was found to be essentially resistant to HDAC8 and the HDACs (primarily HDAC1 and HDAC2) present in the HeLa nuclear extract (Fig. S1†). This result is not that surprising given the previously reported inability of the classical protein deacetylases to catalyze the acetyl removal from Nδ-acetyl-ornithine.21 Compound 9 was also found to be a cell permeable human sirtuin inhibitor as evidenced by its strong SIRT1 inhibition inside the HCT116 human colon cancer cells with an estimated (based on visual inspection) 100% p53 acetylation level increase achieved at 5 μM (Fig. 5), which was also more profound than that observed previously by Suzuki et al.14 with 10 and the HCT116 human colon cancer cells.
Conclusions
In the current study, a rational SAR approach has been applied to our previously discovered potent ThAcK-containing 18-amino acid peptidic sirtuin inhibitor 1 with the identification of two still potent yet proteolytically stable and cell permeable ThAcK-containing low molecular weight peptidomimetic sirtuin inhibitors (8 and 9). These two compounds may serve as valuable pre-clinical anticancer candidate compounds on their own right, given the very recently demonstrated anticancer benefit of a SIRT1/2 dual inhibition20 and antagonistic effect of SIRT3 on p53-mediated cellular senescence via its p53 deacetylase activity.22 The exceptional inhibitory potency against human SIRT1/2/3 of these two compounds and the observed sirtuin inhibitory profile alteration following the subtle structural change from 8 to 9 also attest to the promise of finding potent and more selective cell permeable ThAcK-containing low molecular weight peptidomimetic sirtuin inhibitors in the future.Acknowledgements
The financial supports from the James L. and Martha J. Foght Endowment and the University of Akron Research Foundation (to W. Zheng) and the U.S. National Institutes of Health (R01-CA127590 to Z. Wang) are highly appreciated. We thank Prof. Tony Kouzarides (University of Cambridge, UK) for the GST-SIRT1 plasmid. We also thank Prof. Chrys Wesdemiotis and his research group at the University of Akron for the assistance with mass spectrometric analysis of the peptides synthesized in the current study. We wish to thank The Goodyear Corporation for donation of the NMR instrument (Varian Mercury 300) used in this work. We also wish to thank The National Science Foundation (CHE-8808587) for funds used to purchase the NMR instrument (Varian GEMINI 300) used in this work.Notes and references
- (a) A. A. Sauve, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1591 CrossRef CAS; (b) B. D. Sanders, B. Jackson and R. Marmorstein, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1604 CrossRef CAS; (c) B. C. Smith, W. C. Hallows and J. M. Denu, Chem. Biol., 2008, 15, 1002 CrossRef CAS; (d) A. A. Sauve, C. Wolberger, V. L. Schramm and J. D. Boeke, Annu. Rev. Biochem., 2006, 75, 435 CrossRef CAS; (e) A. A. Sauve and V. L. Schramm, Curr. Med. Chem., 2004, 11, 807 CrossRef CAS; (f) A. A. Sauve, I. Celic, J. Avalos, H. Deng, J. D. Boeke and V. L. Schramm, Biochemistry, 2001, 40, 15456 CrossRef CAS; (g) M. D. Jackson and J. M. Denu, J. Biol. Chem., 2002, 277, 18535 CrossRef CAS.
- (a) S. Greiss and A. Gartner, Mol. Cells, 2009, 28, 407 CrossRef CAS; (b) S. Thiagalingam, K. H. Cheng, H. J. Lee, N. Mineva, A. Thiagalingam and J. F. Ponte, Ann. N. Y. Acad. Sci., 2003, 983, 84 CrossRef CAS; (c) I. V. Gregoretti, Y. M. Lee and H. V. Goodson, J. Mol. Biol., 2004, 338, 17 CrossRef CAS; (d) R. A. Frye, Biochem. Biophys. Res. Commun., 2000, 273, 793 CrossRef CAS; (e) L. R. Saunders and E. Verdin, Oncogene, 2007, 26, 5489 CrossRef CAS.
- (a) J. Yu and J. Auwerx, Ann. N. Y. Acad. Sci., 2009, 1173, E10 CrossRef CAS; (b) M. C. Haigis and D. A. Sinclair, Annu. Rev. Pathol.: Mech. Dis., 2010, 5, 253 Search PubMed; (c) T. Finkel, C. X. Deng and R. Mostoslavsky, Nature, 2009, 460, 587 CrossRef CAS; (d) S. Imai and L. Guarente, Trends Pharmacol. Sci., 2010, 31, 212 CrossRef CAS; (e) T. Liu, P. Y. Liu and G. M. Marshall, Cancer Res., 2009, 69, 1702 CrossRef CAS.
- (a) O. Grubisha, B. C. Smith and J. M. Denu, FEBS J., 2005, 272, 4607 CrossRef CAS; (b) M. Porcu and A. Chiarugi, Trends Pharmacol. Sci., 2005, 26, 94 CrossRef CAS; (c) P. J. Elliott and M. Jirousek, Curr. Opin. Investig. Drugs, 2008, 9, 371 Search PubMed; (d) T. F. Outeiro, O. Marques and A. Kazantsev, Biochim. Biophys. Acta, 2008, 1782, 363 CAS; (e) J. C. Milne and J. M. Denu, Curr. Opin. Chem. Biol., 2008, 12, 11 CrossRef CAS; (f) R. C. Neugebauer, W. Sippl and M. Jung, Curr. Pharm. Des., 2008, 14, 562 CrossRef CAS; (g) Y. Cen, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1635 CrossRef CAS; (h) J. Schemies, U. Uciechowska, W. Sippl and M. Jung, Med. Res. Rev., 2009 Oct 12 Search PubMed [Epub ahead of print]; (i) F. J. Alcaín and J. M. Villalba, Expert Opin. Ther. Pat., 2009, 19, 283 Search PubMed; (j) J. Schemies, W. Sippl and M. Jung, Cancer Lett., 2009, 280, 222 CrossRef CAS; (k) P. A. Cole, Nat. Chem. Biol., 2008, 4, 590 CrossRef CAS; (l) B. G. Szczepankiewicz and P. Y. Ng, Curr. Top. Med. Chem., 2008, 8, 1533 CrossRef CAS; (m) S. Lavu, O. Boss, P. J. Elliott and P. D. Lambert, Nat. Rev. Drug Discovery, 2008, 7, 841 CrossRef CAS.
- (a) D. G. Fatkins, A. D. Monnot and W. Zheng, Bioorg. Med. Chem. Lett., 2006, 16, 3651 CrossRef CAS; (b) D. G. Fatkins and W. Zheng, Int. J. Mol. Sci., 2008, 9, 1 Search PubMed.
- B. C. Smith and J. M. Denu, Biochemistry, 2007, 46, 14478 CrossRef CAS.
- W. F. Hawse, K. G. Hoff, D. G. Fatkins, A. Daines, O. V. Zubkova, V. L. Schramm, W. Zheng and C. Wolberger, Structure, 2008, 16, 1368 CrossRef CAS.
- L. Jin, W. Wei, Y. Jiang, H. Peng, J. Cai, C. Mao, H. Dai, W. Choy, J. E. Bemis, M. R. Jirousek, J. C. Milne, C. H. Westphal and R. B. Perni, J. Biol. Chem., 2009, 284, 24394 CrossRef CAS.
- M. Goodman and S. Ro, in Burger's Medicinal Chemistry and Drug Discovery, Vol. 1, Principles and Practice; M. E. Wolff, ed.; John Wiley & Sons, Inc.: USA 1995, 803–861, 5th edition Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3 CrossRef CAS.
- (a) J. L. Avalos, I. Celic, S. Muhammad, M. S. Cosgrove, J. D. Boeke and C. Wolberger, Mol. Cell, 2002, 10, 523 CrossRef CAS; (b) K. G. Hoff, J. L. Avalos, K. Sens and C. Wolberger, Structure, 2006, 14, 1231 CrossRef CAS; (c) M. S. Cosgrove, K. Bever, J. L. Avalos, S. Muhammad, X. Zhang and C. Wolberger, Biochemistry, 2006, 45, 7511 CrossRef CAS; (d) A. L. Garske and J. M. Denu, Biochemistry, 2006, 45, 94 CrossRef CAS.
- D. A. Wellings and E. Atherton, Methods Enzymol., 1997, 289, 44 CAS.
- P. H. Kiviranta, T. Suuronen, E. A. Wallén, J. Leppänen, J. Tervonen, S. Kyrylenko, A. Salminen, A. Poso and E. M. Jarho, J. Med. Chem., 2009, 52, 2153 CrossRef CAS.
- T. Suzuki, T. Asaba, E. Imai, H. Tsumoto, H. Nakagawa and N. Miyata, Bioorg. Med. Chem. Lett., 2009, 19, 5670 CrossRef CAS.
- A. D. Napper, J. Hixon, T. McDonagh, K. Keavey, J. F. Pons, J. Barker, W. T. Yau, P. Amouzegh, A. Flegg, E. Hamelin, R. J. Thomas, M. Kates, S. Jones, M. A. Navia, J. O. Saunders, P. S. DiStefano and R. Curtis, J. Med. Chem., 2005, 48, 8045 CrossRef CAS.
- Roche Applied Science, Pronase: Product Description, 2006. And references cited therein Search PubMed.
- D. G. Fatkins and W. Zheng, Anal. Biochem., 2008, 372, 82 CrossRef CAS.
- H. Vaziri, S. K. Dessain, E. Ng Eaton, S. I. Imai, R. A. Frye, T. K. Pandita, L. Guarente and R. A. Weinberg, Cell, 2001, 107, 149 CrossRef CAS.
- T. F. Outeiro, E. Kontopoulos, S. M. Altmann, I. Kufareva, K. E. Strathearn, A. M. Amore, C. B. Volk, M. M. Maxwell, J. C. Rochet, P. J. McLean, A. B. Young, R. Abagyan, M. B. Feany, B. T. Hyman and A. G. Kazantsev, Science, 2007, 317, 516 CrossRef CAS.
- B. Peck, C. Y. Chen, K. K. Ho, P. Di Fruscia, S. S. Myatt, R. C. Coombes, M. J. Fuchter, C. D. Hsiao and E. W. Lam, Mol. Cancer Ther., 2010, 9, 844 CrossRef CAS.
- D. Riester, D. Wegener, C. Hildmann and A. Schwienhorst, Biochem. Biophys. Res. Commun., 2004, 324, 1116 CrossRef CAS.
- S. Li, M. Banck, S. Mujtaba, M. M. Zhou, M. M. Sugrue and M. J. Walsh, PLoS One, 2010, 5, e10486 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Fig. S1, Copies of HPLC traces. See DOI: 10.1039/c0md00089b |
| This journal is © The Royal Society of Chemistry 2010 |