Sirtuin mechanism and inhibition: explored with Nε-acetyl-lysine analogs†
Brett M.
Hirsch
and
Weiping
Zheng
*
Department of Chemistry, University of Akron, 190 E. Buchtel Commons, Akron, OH 44325, USA. E-mail: wzheng@uakron.edu
First published on 15th September 2010
Abstract
Silent information regulator 2 (Sir2) enzymes or sirtuins are a family of intracellular protein deacetylases that can catalyze the β-nicotinamide adenine dinucleotide (β-NAD+)-dependent deacetylation of Nε-acetyl-lysine on protein substrates, with the formation of lysine Nε-deacetylated protein species and small molecule products, i.e.nicotinamide and 2′-O-acetyl-ADP-ribose (2′-O-AADPR). These enzymes are evolutionarily conserved among all the three kingdoms of life, with the yeast Sir2 being the founding family member. In humans, seven sirtuins, i.e. SIRT1-7, have been identified. The past a few years have witnessed a tremendous interest in investigating the unique mechanism for the sirtuin-catalyzed deacetylation reaction. We have also seen a lot of research employing different strategies to identify different types of the inhibitors for this enzymatic deacetylation reaction. These inhibitors hold great potential toward a fuller exploration of sirtuin biology and pharmacology as well as toward developing novel therapeutics for metabolic and age-related diseases and cancer. Here we would like to review the significant contributions that the judicious use of a variety of Nε-acetyl-lysine analogs has been able to make toward our enhanced mechanistic understanding and capability of pharmacological exploitation of the sirtuin-catalyzed deacetylation reaction.
1. Introduction
Nε-acetylation and deacetylation on specific lysine side chains have been recognized as an important post-translational means to exert structural and functional regulation of proteins.1–16 The first proteins identified that were subject to this regulatory switch were histone proteins. This same regulatory switch was subsequently found to be able to modulate the structure/function of many non-histone proteins. Since the mid-90's, two growing superfamilies of enzymes, i.e.protein acetyltransferases and protein deacetylases, have been shown to be responsible for catalyzing the lysine Nε-acetylation and deacetylation reactions, respectively (Fig. 1).3–13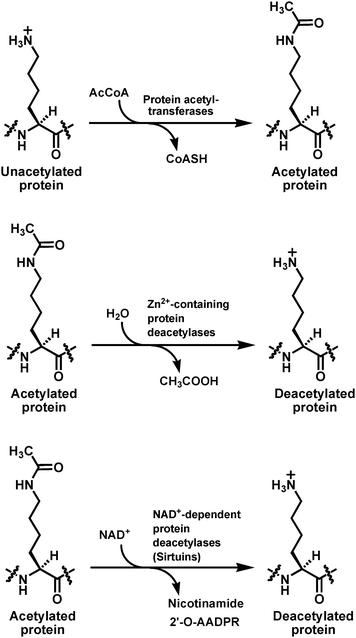 | ||
| Fig. 1 The lysine Nε-acetylation and deacetylation reactions catalyzed respectively by protein acetyltransferases and the two families of the protein deacetylases. AcCoA, acetyl-coenzyme A; CoASH, coenzyme A; NAD+, β-nicotinamide adenine dinucleotide; 2′-O-AADPR, 2′-O-acetyl-ADP-ribose. | ||
Multiple transcriptional coactivators such as p300, cAMP-response element binding protein (CREB) binding protein (CBP), and p300/CBP-associated factor (PCAF) were shown to possess an intrinsic protein acetyltransferase activity. The yeast transcriptional repressors reduced potassium dependency 3 (Rpd3), histone deacetylase 1 (Hda1), and silent information regulator 2 (Sir2) were shown to possesses a protein deacetylase activity. While acetyl-coenzyme A (AcCoA) is used as the universal cofactor to donate the acetyl group during lysine Nε-acetylation catalyzed by all the protein acetyltransferases, the enzymatic lysine Nε-deacetylation is accomplished by the deacetylase enzymes that use either Zn2+ or β-nicotinamide adenine dinucleotide (β-NAD+ or just NAD+ as used below throughout this article) as the catalytic cofactor. While the Zn2+-containing metallo-deacetylases catalyze the Zn2+-assisted hydrolysis of the amide bond of the acetyl-lysine side chain to afford the deacetylated product and acetic acid, the NAD+-dependent deacetylases achieve the deacetylation of the acetyl-lysine side chain by catalyzing the ultimate transfer of the acetyl group onto the 2′-OH of the nicotinamide ribose of NAD+, which is coupled to the cleavage of nicotinamide from NAD+, thus affording three enzymatic products, i.e.nicotinamide, the deacetylated product, and 2′-O-acetyl-ADP-ribose (2′-O-AADPR) (Fig. 1). Rpd3 and Hda1 are the two founding members of the Zn2+-containing protein deacetylase enzyme family that also includes the eleven human homologs (HDAC1-11, HDAC stands for histone deacetylase). Sir2 is the founding member of the NAD+-dependent protein deacetylase enzyme family that is also known as the sirtuin family.
Based on sequence homology and phylogenetic analysis, protein deacetylases can also be categorized into four classes, i.e. classes I, II, III, and IV.12,17,18 Class I (including human HDAC1-3 and HDAC8), II (including human HDAC4-7, HDAC9, and HDAC10), and IV (including human HDAC11) constitute the Zn2+-containing family. Of note, human class II can also be further categorized into class IIa (HDAC4, HDAC5, HDAC7, and HDAC9) and class IIb (HDAC6 and HDAC10). Class III constitutes the NAD+-dependent sirtuin family.
Sirtuin family enzymes have been identified in organisms from all the three kingdoms of life,9–12,17–29 and include the human enzymes: SIRT1-7; the bacterial enzymes: CobB from multiple bacterial species, Sir2Tm from Thermatoga maritima, and Rv1151c from Mycobacterium tuberculosis; the archaeal enzymes: Sir2Af1 and Sir2Af2 from Archaeoglobus fulgidus; the parasitic enzymes: PfSir2 from Plasmodium falciparum, EhSir2a from Entamoeba histolytica, SIR2rp1-3 from Trypanosoma and Leishmania parasites, and the Sir2 homologs from Cryptosporidium parasites; the yeast enzymes Sir2 and Hst1-4; and the Sir2 homologs found in other fungal species such as Candida albicans and Candida glabrata. Endogenous acetylated protein substrates have been identified for most prokaryotic and eukaryotic sirtuin enzymes.22–43
Eukaryotic sirtuins have been found in different intracellular compartments including nucleus (e.g. human SIRT6 and SIRT7), mitochondrion (e.g. human SIRT3, SIRT4, and SIRT5), and cytoplasm (e.g. human SIRT2 and yeast Hst2). Moreover, for the seven human sirtuins, they are all ubiquitously expressed in different organs/tissues.44
The sirtuin-catalyzed lysine Nε-deacetylation reaction has been shown to play important roles in multiple biological processes such as gene transcription, apoptosis, DNA repair, metabolism, aging, neurodegeneration, and HIV-1 replication;8,12,45–50 the past a few years have thus witnessed a tremendous interest in employing different strategies to identify different types of the inhibitors for the sirtuin-catalyzed deacetylation reaction, as evidenced by the appearance of a number of review articles on the topic.30,51–64 These inhibitors hold great potential toward a fuller exploration of sirtuin biology and pharmacology as well as toward developing novel therapeutics for metabolic and age-related diseases and cancer. We have also seen a lot of research investigating the unique mechanism for the sirtuin-catalyzed deacetylation reaction, also as evidenced by the appearance of a number of review articles on the topic.9–11,13,30,65–70 This mechanistic investigation has played an important role in facilitating the rational design of sirtuin inhibitors. While quite a few excellent reviews have already been published since 2008 to cover an in-depth discussion of the different types of the sirtuin inhibitors and/or the catalytic mechanism of the sirtuin-catalyzed deacetylation reaction,30,52–63,65–68 we felt it would be a worthwhile endeavor to survey the significant contributions that the judicious use of a variety of analogs of Nε-acetyl-lysine, the very substrate for the sirtuin-catalyzed acetyl group removal (Fig. 1), has been able to make toward the mechanistic investigation and the inhibitor development for the sirtuin-catalyzed deacetylation reaction.
2. Sirtuin mechanistic probes containing Nε-acetyl-lysine analogs
Fig. 2 depicts the proposed chemical mechanism for the sirtuin-catalyzed lysine Nε-deacetylation reaction.30,65–68 This chemical mechanistic scheme is consistent with the demonstration with human SIRT2 and yeast Hst2 that the sirtuin-catalyzed deacetylation reaction obeys an ordered sequential kinetic mechanism, in that the chemical step occurs only after the ordered binding of the acetylated substrate (first) and NAD+ (second), and nicotinamide is the first product to be released.71 It was also demonstrated that, following the initial release of nicotinamide, the deacetylated product and 2′-O-AADPR can be randomly released from the sirtuin active site. Following the release of 2′-O-AADPR into the bulk solution, it undergoes a non-enzymatic isomerization to 3′-O-AADPRvia intramolecular transesterification to eventually reach a roughly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar mixture of 2′- and 3′-O-AADPR, as supported by the chromatographic/spectroscopic identification and quantification of these two compounds in a sirtuin deacetylation assay mixture.72,73 2′- and 3′-O-AADPR were also both shown to be present in solution as the α and β-anomers (∼1:1 molar ratio) due to the fast epimerization at the C1′ position.
:1 molar mixture of 2′- and 3′-O-AADPR, as supported by the chromatographic/spectroscopic identification and quantification of these two compounds in a sirtuin deacetylation assay mixture.72,73 2′- and 3′-O-AADPR were also both shown to be present in solution as the α and β-anomers (∼1:1 molar ratio) due to the fast epimerization at the C1′ position.
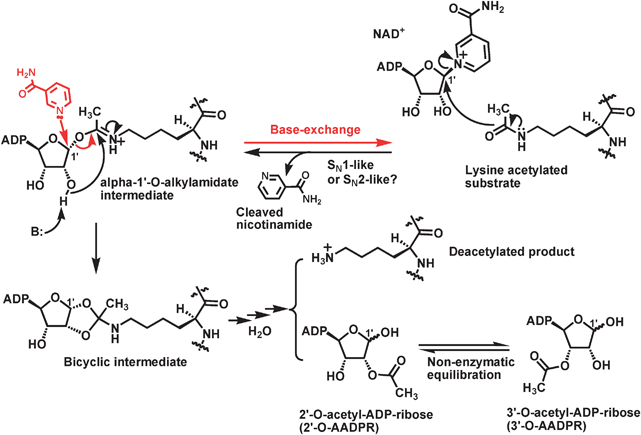 | ||
| Fig. 2 The proposed chemical mechanism for the sirtuin-catalyzed deacetylation reaction. ADP, adenosine diphosphate; B: refers to a general base. | ||
The key features of the proposed chemical mechanism shown in Fig. 2 are (i) the formation of the α-1′-O-alkylamidate intermediate, (ii) the formation the bicyclic intermediate, and (iii) the collapse of this bicyclic intermediate in the presence of water. In this regard, several fundamental mechanistic questions can be asked: (i) are these intermediates really formed? (ii) How are these intermediates formed? (iii) How is the bicyclic intermediate resolved in the presence of water ultimately into the deacetylated product and 2′-O-AADPR?
2.1 The formation of the α-1′-O-alkylamidate intermediate
Biochemical and structural studies during the past several years have generated data supporting the formation of the α-1′-O-alkylamidate intermediate along the sirtuin catalytic coordinate. For example, besides the deacetylation reaction, sirtuin is also known to be able to catalyze the base-exchange reaction, i.e. the regeneration of NAD+ and the acetylated substrate following the condensation of the free nicotinamide molecule with a sirtuin catalytic intermediate.74,75 This base-exchange reaction was observed only in the presence of the acetylated substrate.74 The depicted nucleophilic reaction in Fig. 2 between nicotinamide and the proposed α-1′-O-alkylamidate intermediate is consistent with the experimental observation and with the fact that the anomeric C1′ of this intermediate can be an electrophilic position susceptible to the attack of a nucleophile from the β-face. This susceptibility to the nucleophilic attack at the C1′ position of this proposed intermediate also offers a satisfying explanation for the formation of β-1′-O-methyl-ADP-ribose in a sirtuin deacetylation assay mixture containing methanol and the Hst2[H135A] mutant.76 This mutant of yeast Hst2 lost the catalytically essential H135 side chain that was proposed to serve as a general base to activate viaproton abstraction the 2′-OH group for its nucleophilic attack intramolecularly from the α-face onto another electrophilic position (i.e. the iminium carbon) of the α-1′-O-alkylamidate intermediate to form the bicyclic intermediate (Fig. 2) (also see section 2.2). As a result, the conversion of the α-1′-O-alkylamidate intermediate to the bicyclic intermediate was expected to be diminished significantly with this mutant. Indeed, the deacetylation rate with Hst2[H135A] was found to be ∼100-fold slower than that with the wild-type Hst2.75 However, the generation of the α-1′-O-alkylamidate intermediate with Hst2[H135A] was suggested to be only minimally impeded as compared to that with the wild-type Hst2 since the base-exchange reaction with the mutant was only ∼3-fold slower than that with the wild-type.75 Therefore, the enzyme bound α-1′-O-alkylamidate intermediate could be partitioned onto the formation of β-1′-O-methyl-ADP-ribose following its interception by methanol from the assay mixture via intermolecular nucleophilic attack from the β-face onto the anomeric C1′ position.Even though α-1′-O-alkylamidate has two electrophilic centers, i.e. the anomeric C1′ carbon and the iminium carbon, the observed regioselective intermolecular reaction between this enzyme bound intermediate and other nucleophiles (e.g.nicotinamide and methanol) at the anomeric C1′ position could be explained by the geometric constraints at the sirtuin active site. Methanol and nicotinamide were suggested to bind to the same pocket within the active site of the Plasmodium falciparum sirtuin PfSir2 and to interact with the same electrophilic center (i.e. the anomeric C1′ carbon) based on the observed direct competition between these two nucleophiles for the reaction with α-1′-O-alkylamidate.77
The formation of the α-1′-O-alkylamidate intermediate also received support from the structural studies with the different sirtuin homologs from different species. For example, in the X-ray crystal structure of the ternary complex of an acetylated peptide derived from the human p53 protein, NAD+, and the bacterial sirtuin Sir2Tm, it was observed that the acetyl-lysine side chain amide oxygen was positioned at the α-face of the nicotinamide ribose of the bound NAD+ and it was within the van der Waals distance (3.2 Å) from the C1′ position.78 This relative positioning of the bound acetylated substrate and NAD+ within a sirtuin active site hinted that the formation of the α-1′-O-alkylamidate intermediate is possible following the direct interaction of the two bound reactants.
Despite the above-described informative biochemical and structural data supporting the formation of the α-1′-O-alkylamidate intermediate along the sirtuin catalytic coordinate, the existence of this proposed intermediate was more directly ascertained by studies with the use of the peptides containing two acetyl-lysine analogs, i.e. the histone H3 peptide containing [18O]acetyl-lysine and the human p53 peptide containing thioacetyl-lysine (Fig. 3).
![The chemical structures of [18O]-acetyl-lysine and thioacetyl-lysine. [18O]-acetyl-lysine was incorporated into position X in the following histone H3 peptide sequence for the study in ref. 76: H2N-KSTGGXAPRKQCONH2; Thioacetyl-lysine was incorporated into position X in the following p53 peptide sequence for the study in ref. 79: H2N-KKGQSTSRHKXLMFKTEG-COOH. The chemical structure of 1′-18OH-2′-O-acetyl-ADP-ribose and the stick model of the α-1′-S-alkylamidate intermediate trapped in Sir2Tm active site are also shown.](/image/article/2011/MB/c0mb00033g/c0mb00033g-f3.gif) | ||
| Fig. 3 The chemical structures of [18O]-acetyl-lysine and thioacetyl-lysine. [18O]-acetyl-lysine was incorporated into position X in the following histone H3 peptide sequence for the study in ref. 76: H2N-KSTGGXAPRKQCONH2; Thioacetyl-lysine was incorporated into position X in the following p53 peptide sequence for the study in ref. 79: H2N-KKGQSTSRHKXLMFKTEG-COOH. The chemical structure of 1′-18OH-2′-O-acetyl-ADP-ribose and the stick model of the α-1′-S-alkylamidate intermediate trapped in Sir2Tm active site are also shown. | ||
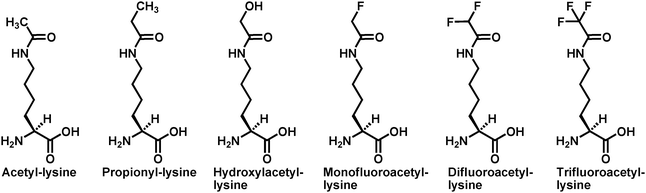 | ||
| Fig. 4 The chemical structures of acetyl-lysine and its analogs used in ref. 85 to investigate if there is a nucleophilic participation of acetyl-lysine for the sirtuin-catalyzed nicotinamide cleavage reaction. Acetyl-lysine and its analogs were all incorporated into position X in the following histone H3 peptide sequence: H2N-KSTGGXAPRKQ-COOH. | ||
When the [18O]-acetylated H3 peptide was used as the acetylated substrate in a sirtuin deacetylation assay with yeast Hst2, it was found that the 18O label was transferred onto the C1′ position of NAD+, thus yielding the 2′-O-AADPR analog with 1′-OH labeled with 18O (i.e.1′-18OH-2′-O-acetyl-ADP-ribose, Fig. 3).76 This experimental observation strongly argued that there was an obligatory covalent bond formation between the side chain amide oxygen of acetyl-lysine and the C1′ of NAD+ for the sirtuin-catalyzed deacetylation reaction. When the crystal of the bacterial sirtuin Sir2Tm with the bound p53 peptide containing thioacetyl-lysine was soaked with a cryoprotective solution containing NAD+, the catalytically active Sir2Tm in the crystal state was able to catalyze the covalent bond formation between the side chain thioacetyl sulfur of thioacetyl-lysine and the C1′ of NAD+, with the accompanying nicotinamide cleavage from NAD+. However, the formed α-1′-S-alkylamidate intermediate was long-lived enough to be trapped within the crystal without further catalytic conversions.79 The successful solution of this 2.5 Å Sir2Tm crystal structure (Fig. 3) provided the conclusive evidence for the existence of the α-1′-S-alkylamidate intermediate that is stalled following the Sir2Tm-catalyzed nicotinamide cleavage, thus providing the first structural evidence for the formation of the α-1′-O-alkylamidate intermediate along the sirtuin catalytic coordinate.
The synthesis and incorporation of thioacetyl-lysine into a peptide sequence to investigate the functional consequences of replacing acetyl-lysine with thioacetyl-lysine in a sirtuin peptide substrate was first reported in 2006.80 In this study, the p53 peptides containing either acetyl-lysine or thioacetyl-lysine were evaluated side-by-side for their processing by human SIRT1. It was found that SIRT1 was able to accept the thioacetylated p53 peptide as its substrate since SIRT1 was able to catalyze the thioacetyl group removal (or dethioacetylation) from the thioacetylated peptide and the nicotinamide cleavage from NAD+ when the thioacetylated peptide was used in the assay. However, the SIRT1-catalyzed dethioacetylation was found to be ∼400-fold slower than the SIRT1-catalyzed deacetylation from the acetylated p53 peptide, yet the SIRT1-catalyzed nicotinamide formation was found to be only ∼7-fold slower when the thioacetylated p53 peptide was used instead of the acetylated p53 peptide in the assay. These findings thus suggested that the thioacetylated peptide is able to be processed as a substrate by SIRT1, but to form a catalytically less competent, longer-lived intermediate following the nicotinamide cleavage step along the reaction coordinate, as compared to the normal processing of the acetylated peptide substrate by SIRT1.
Following these studies with human SIRT1, we also found with the bacterial sirtuin Sir2Tm that the Sir2Tm-catalyzed dethioacetylation from the afore-mentioned thioacetylated p53 peptide was profoundly slower as compared to the Sir2Tm-catalyzed deacetylation from the corresponding acetylated p53 peptide, yet again a minimally impeded Sir2Tm-catalyzed nicotinamide cleavage was observed when the thioacetylated peptide was used instead of the acetylated peptide in the assay (Zheng, unpublished results).
The phenomenon of the profound and the minimal impact respectively on the enzymatic dethioacetyltion and the enzymatic nicotinamide cleavage when a thioacetylated peptide was used instead of its acetylated counterpart in a sirtuin assay was also observed with the yeast sirtuin Hst2.81 In this study, when the thioacetylated and the acetylated histone H3 peptides were evaluated for their processing by Hst2, it was found that the Hst2-catalyzed dethioacetylation was ∼80-fold slower than the Hst2-catalyzed deacetylation, however, the Hst2-catalyzed nicotinamide cleavage was only ∼1.5-fold slower when the thioacetylated peptide was used instead of the acetylated peptide in the assay. Furthermore, the overall steady-state rate (kcat) of the Hst2-catalyzed dethioacetylation was shown to be ∼2000-fold slower as compared to the rate of nicotinamide production under single-turnover conditions. In this same study, the identity of the stalled catalytic intermediate was directly revealed via mass spectroscopic detection to be the α-1′-S-alkylamidate intermediate, a mimic of the naturally occurring α-1′-O-alkylamidate intermediate (Fig. 2). Through computational modeling, the sluggish commitment of this intermediate to the deacetylation chemistry was proposed to be due to (i) the longer C–S glycosidic linkage than the C–O linkage, so that the 2′-OH group is positioned farther away from the electrophilic iminium carbon in the α-1′-S-alkylamidate intermediate; (ii) the altered attacking angle of 2′-OH onto the iminium carbon in α-1′-S-alkylamidate as compared to that in α-1′-O-alkylamidate; and (iii) the intrinsically weaker electrophilicy of the iminium carbon in α-1′-S-alkylamidate than that in α-1′-O-alkylamidate.
As mentioned above within this section, when Hst2[H135A] was used in a sirtuin deacetylation assay, due to the loss of the catalytically essential H135 side chain, the conversion of the α-1′-O-alkylamidate intermediate to the downstream bicyclic intermediate was significantly discouraged, as evidenced by the significantly lower deacetylation rate with the mutant than that with the wild-type, yet its generation from the substrates was suggested to be only minimally impeded since the base-exchange reaction with the mutant was only slightly slower than that with the wild-type.75 However, the further conversion of this enzyme bound α-1′-O-alkylamidate intermediate was shown to be still possible.75,76 Besides the above-mentioned interception of this intermediate with other nucleophiles such as methanol from an assay mixture, this intermediate could also be intercepted by water to generate ADP-ribose and to regenerate acetylated substrate. Still, this intermediate could also be intercepted by nicotinamide to regenerate the acetylated substrate and NAD+ since the base exchange reaction was shown not to be that compromised with this mutant enzyme as compared to that with the wild-type enzyme.75 Therefore, the α-1′-O-alkylamidate intermediate formed within the active site of Hst2[H135A] still had multiple alternative fates.
However, the α-1′-S-alkylamidate intermediate is a relatively stable species once it is formed within the sirtuin active site from the thioacetylated substrate and NAD+ because of its sluggish conversion to the downstream bicyclic intermediate and because its anomeric C1′ position is predicted not to be that susceptible to a nucleophilic substitution reaction due to the presence of a C–S glycosidic linkage instead of a C–O glycosidic linkage. Consistent with this prediction, a variety of glycosidases have been shown not to be capable of hydrolyzing the C–S glycosidic linkage as efficiently as the C–O glycosidic linkage.82–84 To support this prediction experimentally, when a SIRT1 deacetylation assay was performed with a thioacetylated p53 peptide and NAD+ in the presence of methanol, we were unable to detect with mass spectrometry the formation of β-1′-O-methyl-ADP-ribose and ADP-ribose (Zheng, unpublished results). Furthermore, as Sauve indicated in his recent review, nicotinamide base exchange was not supported when the thioacetylated peptide instead of the acetylated peptide was used in the sirtuin assay.65
The next related mechanistic issue is how a sirtuin enzyme can promote the formation of the α-1′-O-alkylamidate intermediate via the alkylation of the generally weak nucleophilic side chain amide oxygen of acetyl-lysine with the nicotinamide moiety of NAD+ as the leaving group of this enzymatic reaction. It could be imagined that a sirtuin enzyme could handle this challenge by presenting a strong electrophile in the form of a fully dissociated and enzyme stabilized oxacarbenium ion intermediate or a transition state with a strong oxacabenium ion character. If the sirtuin active site can stabilize a fully dissociated oxacarbenium ion as an intermediate, then the reaction is SN1-like and the nucleophilic participation from the acetyl-lysine side chain amide oxygen would be minimal. On the other hand, nucleophilic participation would be expected for a SN2-like mechanism. In order to determine whether or not there is such a nucleophilic participation of acetyl-lysine, a series of acetyl-lysine analogs substituted at the acetyl α-carbon with substituents with similar steric size but different electron withdrawing power (Fig. 4) were employed in a study with the yeast sirtuin Hst2.85
When the histone H3 peptides containing acetyl-lysine or its analogs at position 14 were subjected to the nictotinamide cleavage assay with Hst2, it was found that the nicotinamide formation rate is negatively related to the electron withdrawing power of the α-substituent, and thus the nicotinamide formation rate is positively related to the nucleophilicity of the side chain amide oxygen. When the log values of the nicotinamide formation rates were plotted against the inductive Taft constants (σ*) of the α-substituents, a linear free energy relationship with a negative slop (ρ* = −1.9) was obtained. In this same study, it was also found that the Hst2 active site was able to favorably bind most side chain modified acetyl-lysine analogs, based on the Kd measurement. Furthermore, for those analog-containing peptides with which the steady-state turnover rates (kcat's) were measurable with Hst2, each of them was found to be capable of supporting the nicotinamide formation with a first-order rate similar to the kcat value, suggesting the shift of the rate-limiting step from the final product release with the acetyl-lysine peptide76 to the nicotinamide cleavage with the acetyl-lysine analogs. These analog-containing peptides were also found to be converted by Hst2 to their corresponding 2′-O-AADPR analogs, with no observed enzymatic formation of ADP-ribose. A SN2-like mechanism was suggested based on the data from this study for the sirtuin-catalyzed nicotinamide cleavage. A SN2-like mechanism seems to be consistent with the observed positioning of the acetyl-lysine side chain amide oxygen at the α-face of the nicotinamide ribose of NAD+ and within the van der Waals distance (3.2 Å) from the C1′ position of NAD+ in the structure of a ternary complex of Sir2Tm with the two bound substrates.78 However, it was unclear from the study with Hst285 about the transition state structure for the suggested SN2-like mechanism.
To address the issue of the transition state structure for the sirtuin-catalyzed nicotinamide cleavage reaction, an ab initioQM/MM molecular dynamics simulation of the Sir2Tm-catalyzed nicotinamide cleavage was performed very recently.86 This simulation study suggested that the enzymatic nicotinamide cleavage proceeds via a highly dissociative yet concerted nucleophilic displacement mechanism in that there is a modest nucleophilic participation of the acetyl-lysine for the nicotinamide cleavage on the one hand, and there is a highly dissociative transition state with a strong oxacarbenium ion character on the other. The mechanistic suggestion from this simulation study is consistent with a SN2-like mechanism and also with that proposed earlier in the literature.11,69,72 Despite this simulation study, an experimentally determined transition state structure for the sirtuin-catalyzed nicotinamide cleavage reaction is still lacking. Therefore, further studies are still needed to address this important sirtuin mechanistic issue.
2.2 The formation of the bicyclic intermediate
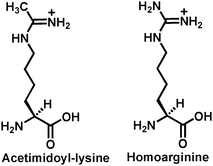
As compared to the voluminous biochemical/structural studies addressing the existence and the formation of the α-1′-O-alkylamidate intermediate in the proposed chemical mechanism for the sirtuin-catalyzed deacetylation (Fig. 2), more direct biochemical/structural evidence is still lacking for the existence and the formation of the bicyclic intermediate shown in Fig. 2. Nevertheless, the following biochemical evidence indirectly supporting the existence and the formation of this intermediate has been available. (i) The 14C or the 3H label was found to be transferred from [14C]acetyl-lysine or [3H]-acetyl-lysine in a peptide substrate onto the 2′-acetoxy carbonyl carbon or its methyl group of the sirtuin-catalyzed deacetylation product 2′-O-AADPR with the formation of 2′-O-[14C]acetyl-ADP-ribose or 2′-O-[3H]acetyl-ADP-ribose (Fig. 5);87,88 (ii) The origin of the acetylcarbonyl oxygen of 2′-O-AADPR is water as evidenced by the transfer of the 18O label in H218O onto this position;72,76 (iii) The 18O label of [18O]acetyl-lysine in a peptide substrate was found to be transferred onto the 1′-OH of 2′-O-AADPR with the formation of 1′-18OH-2′-O-acetyl-ADP-ribose (Fig. 3);76 (iv) As described above within section 2.1, as compared to the wild-type yeast Hst2, the Hst2[H135A] mutant was found to have significantly diminished deacetylase activity since it lost the catalytically essential H135 side chain that was proposed to serve as a general base to activate viaproton abstraction the 2′-OH group of α-1′-O-alkylamidate, presumably for its intramolecular nucleophilic addition reaction with α-1′-O-alkylamidate leading to the formation of the proposed bicyclic intermediate;75 (v) when the sirtuin peptide substrates containing homocitrulline instead of acetyl-lysine were examined under the sirtuin deacetylation assay condition, no decarbamoylated peptide (same as the deacetylated peptide) and 2′-O-carbamoyl-ADP-ribose (the analog of 2′-O-AADPR) were formed, instead ADP-ribose was found to be produced and the original homocitrulline peptide substrate was regenerated. A robust enzymatic nicotinamide cleavage was also observed. These results strongly suggested that an α-1′-O-alkylamidate intermediate analog was formed from the homocitrulline peptide substrate and NAD+, however, instead of being converted to the bicyclic intermediate leading to the formation of the decarbamoylated peptide and 2′-O-carbamoyl-ADP-ribose, it was intercepted by water at the anomeric C1′ position (Fig. 6).89,90 The inability of this α-1′-O-alkylamidate analog to support the formation of the 5-membered dioxo ring in the bicyclic intermediate was proposed to result from the lowered electrophilicity of the iminium carbon due to the electron-donating mesomeric effect from the neighboring NH2 group, so that the nucleophilic addition of the activated 2′-OH onto this iminium carbon is less favored.![The chemical structures of [14C]-acetyl-lysine, 2′-O-[14C]acetyl-ADP-ribose, [3H]-acetyl-lysine, and 2′-O-[3H]acetyl-ADP-ribose. [14C]-acetyl-lysine and [3H]-acetyl-lysine can be incorporated into a peptide.](/image/article/2011/MB/c0mb00033g/c0mb00033g-f5.gif) | ||
| Fig. 5 The chemical structures of [14C]-acetyl-lysine, 2′-O-[14C]acetyl-ADP-ribose, [3H]-acetyl-lysine, and 2′-O-[3H]acetyl-ADP-ribose. [14C]-acetyl-lysine and [3H]-acetyl-lysine can be incorporated into a peptide. | ||
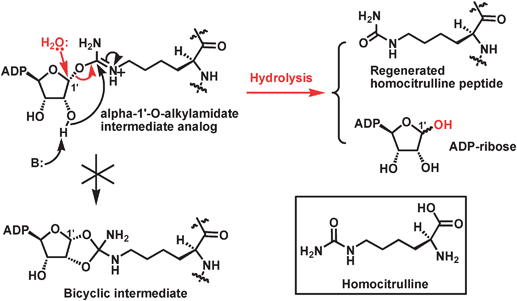 | ||
| Fig. 6 The proposed mechanism for the formation of ADP-ribose and the regeneration of homocitrulline peptide substrate. It is also indicated that the α-1′-O-alkylamidate intermediate analog is unable to be converted to the corresponding bicyclic intermediate. The chemical structure of homocitrulline is shown in the inset. | ||
The intramolecular nucleophilic attack of the 2′-OH onto the iminium carbon of the α-1′-O-alkylamidate intermediate with the formation of the 5-membered dioxo ring in the proposed bicyclic intermediate (Fig. 2) is also expected to be thermodynamically and geometrically favored, due to (i) the thermodynamically favored 5-membered ring transition state structure for the reaction and (ii) the geometrically favored relative positioning of the nucleophile and the electrophile, i.e. the 2′-OH and the anomeric position of α-1′-O-alkylamidate both assume the α-configuration. Moreover, the deprotonation of the 2′-OH by a general base is expected to further facilitate this intramolecular reaction due to the enhanced nucleophilicity of 2′-OH.
2.3 The collapse of the bicyclic intermediate
Three distinct pathways have been proposed to explain how the proposed bicyclic intermediate could be resolved in the presence of water to afford the deacetylated product and 2′-O-AADPR, as shown in Fig. 7.89,91 Pathway A follows a SN1-like mechanism in which a delocalized carbocation is formed from the cleavage of the indicated C–N bond before it is captured by the nucleophile (i.e.water). This C–N bond cleavage generates the deacetylated product and was proposed to be facilitated by a general acid, e.g. the protonated side chain of the H135 residue of Hst2 that acted as a general base earlier in the deacetylation reaction coordinate to acquire a proton from the 2′-OH of the α-1′-O-alkylamidate intermediate leading to the formation of the proposed bicyclic intermediate (also see above). The resulting hemi-orthoester intermediate then collapses in a way that the C–O1′ bond is cleaved, leading to the formation of 2′-O-AADPR. Pathway B follows a SN2-like mechanism in which the nucleophile (i.e.water) attacks the proposed bicyclic intermediate with the concerted cleavage of the indicated C–N bond also aided by a general acid, leading to the generation of the deacetylated product and the same hemi-orthoester intermediate as that in pathway A. The collapse of this hemi-orthoester intermediate then produces 2′-O-AADPR. Pathway C also follows a SN2-like mechanism in which water attacks as a nucleophile directly onto the proposed bicyclic intermediate, however with the concerted cleavage of the C–O1′ bond, leading to the formation of a distinct tetrahedral intermediate. The collapse of this intermediate then generates both the deacetylated product and 2′-O-AADPR in a single step.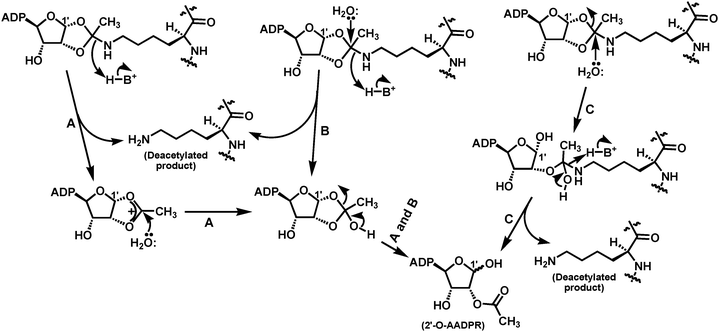 | ||
| Fig. 7 The proposed pathways for the collapse of the bicyclic intermediate in the presence of water to afford the deacetylated product and 2′-O-AADPR. | ||
Very few biochemical/structural data has been available so far in support of either of these three mechanistic proposals or the possible presence of other mechanism(s) accounting for the collapse of the proposed bicyclic intermediate. However, the following studies have nevertheless furnished some mechanistic clues for this last stage along the sirtuin-catalyzed deacetylation reaction coordinate.
By performing the sirtuin deacetylation assay in H218O, it was found that the 18O label was transferred onto the acetylcarbonyl oxygen of 2′-O-AADPR.72,76 This observation indicated that it is during the collapse of the bicyclic intermediate that water was involved since the formation of the α-1′-O-alkylamidate and the bicyclic intermediates (Fig. 2) does not involve water. The above finding with the use of H218O also suggested that water attacks at the tetrahedral carbon of the dioxo ring in the bicyclic intermediate, rather than at the C1′ position which is another plausible position susceptible to a nucleophilic attack. Another clue to this regioselective water attack comes from the study with the [18O]-acetylated H3 peptide (Fig. 3).76 Specifically, since the 18O label of the [18O]-acetylated H3 peptide was shown to be transferred onto the C1′ position of 2′-O-AADPR in a deacetylation assay with the yeast Hst2 enzyme, it could be concluded that the C1′–18O bond stays throughout the reaction coordinate once it is formed in the α-1′-O-alkylamidate intermediate from NAD+ and the [18O]-acetylated H3 peptide (also see above).
As depicted in Fig. 7 for both proposed pathways A and B, it seems that the deacetylated product is formed before 2′-O-AADPR, and thus it could also depart sirtuin active site before 2′-O-AADPR, which is somewhat inconsistent with the random product release sirtuin kinetic scheme demonstrated with human SIRT2 and yeast Hst2,71 unless the deacetylated product could be retained somehow by the sirtuin active site so that it is released after the departure of 2′-O-AADPR. This analysis could imply that the collapse of the bicyclic intermediate may follow another pathway such as the proposed pathway C that is also depicted in Fig. 7. This pathway could potentially lead to the simultaneous formation of the deacetylated product and 2′-O-AADPR.
It is obvious that further studies are still needed to pinpoint the pathway for the collapse of the bicyclic intermediate leading to the formation of the deacetylated product and 2′-O-AADPR.
3. Sirtuin substrate recognition probed with Nε-acetyl-lysine analogs
As described in section 2.1, multiple sirtuins (e.g.SIRT1, Sir2Tm, and Hst2) have been shown to be able to use the thioacetylated peptides as substrates to support a robust enzymatic nicotinamide cleavage. Furthermore, besides the deacetylase activity, a robust depropionylase activity catalyzing the Nε-depropionylation of propionyl-lysine (Fig. 4), a robust debutyrylase activity catalyzing the Nε-debutyrylation of butyryl-lysine (Fig. 8), and/or a robust dephenylacetylase activity catalyzing the Nε-dephenylacetylation of phenylacetyl-lysine (Fig. 8) have also been reported for several sirtuins, including the human SIRT1,90,92,93SIRT2,90,94SIRT3,90,94 the yeast Hst2,85,90 and the bacterial sirtuins Sir2Tm and CobB.94 In the in vitro activity assays employing the propionyl-lysine peptide,85,90protein,94 and small molecule,93 it was found that the sirtuin-catalyzed depropionylation reaction exhibited a steady-state rate at least ∼28% of that for the corresponding deacetylation reaction. Among all the sirtuins tested, Hst2 was found to exhibit the most proficient depropionylase activity with a kcat of 0.17 s−1, approaching that for its deacetylase activity (kcat = 0.2 s−1). However, Hst2 was also shown to be the least proficient debutyrylase among all the sirtuins tested in the in vitro activity assays with the butyryl-lysine peptide90 and small molecule.93 Specifically, while human SIRT1, SIRT2, and SIRT3 were shown to be able to catalyze debutyrylation with a steady-state rate ≥∼20% of that of the corresponding deacetylation, Hst2-catalyzed debutyrylation reaction rate was shown to be only ∼2% of that of the corresponding deacetylation reaction. In another in vitro activity assay with a phenylacetyl-lysine small molecule,93 human SIRT1 was also shown to be capable of catalyzing the dephenylacetylation reaction with a rate 56% of that of the corresponding deacetylation reaction. Furthermore, the SIRT1 depropionylase activity was also demonstrated in a recent cellular study with 293T cells.92 In this study, SIRT1 was shown to significantly depropionylate the p53 and p300 proteins inside the cell, based on a Western blotting analysis.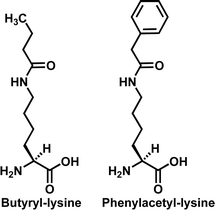 | ||
| Fig. 8 The chemical structures of butyryl-lysine and phenylacetyl-lysine. | ||
In another recent study, ethyl malonyl-lysine was shown to be capable of supporting the SIRT1-catalyzed nicotinamide cleavage from NAD+, with the formation of a stable lysine-ADP-ribose conjugate (Fig. 9).95 The SIRT1-catalyzed formation of this stable covalent conjugate was proposed to account for the mechanism-based SIRT1 inhibition by the ethyl malonyl-lysine compound shown in Fig. 9 (also see section 5).
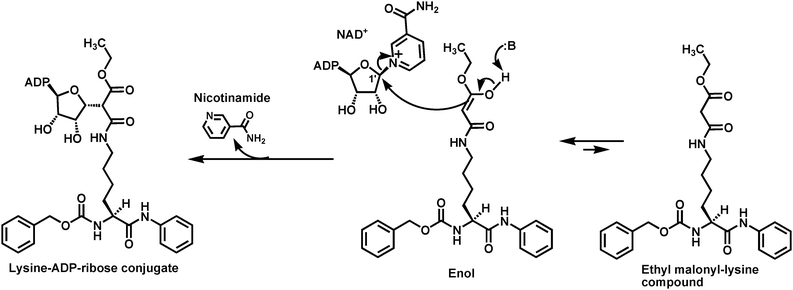 | ||
| Fig. 9 Illustrated formation of the stable lysine-ADP-ribose conjugate from the SIRT1-catalyzed nicotinamide cleavage supported by a small molecule containing ethyl malonyl-lysine. | ||
The above findings therefore suggested that the hydrophobic acetyl-lysine binding tunnel of the sirtuin active site is able to accommodate an acyl group that is bulkier and possibly also more hydrophobic than the acetyl group of acetyl-lysine.
In order to provide a quantitative measure of the molecular determinants of ligand binding at the acetyl-lysine binding tunnel, acetyl-lysine, thioacetyl-lysine, butyryl-lysine, homocitrulline, and six additional analogs with different Nε-acyl groups shown in Fig. 4 and 10 were incorporated into a histone H3 peptide sequence at position 14, and the resulting peptides were measured for their binding affinity (Kd) for Hst2. When the log Kd values were plotted against the hydrophobicity parameter π for each acyl group including the acetyl, a linear plot with a slope of −0.33 was obtained. This plot suggested that the acetyl-lysine binding tunnel tends to bind tighter to the acetyl-lysine analogs with more hydrophobic acyl groups.90 Since thioacetyl (π = 0.35), propionyl (π = 0.06), and butyryl (π = 0.33) are more hydrophobic than acetyl (π = −0.55), the peptides containing these three acyl groups were found to bind tighter with lower Kd values than the acetyl-lysine peptide.
 | ||
| Fig. 10 The additional acetyl-lysine analogs used in ref. 90. | ||
A series of additional acetyl-lysine analogs (Fig. 11) were also employed to further address the substrate specificity of the sirtuin-catalyzed lysine Nε-deacetylation reaction. These analogs were incorporated into a p53 peptide sequence at position 382, and the resulting peptides were examined for their ability to support the nicotinamide cleavage and the deacetylation with the human SIRT1. While robust SIRT1-catalyzed nicotinamide cleavage and deacetylation were observed with the acetyl-lysine peptide, none of the acetyl-lysine analog peptides was able to support the nicotinamide cleavage or the deacetylation under the experimental condition of the study.96 No nicotinamide cleavage or deacetylation was also observed when acetyl-poly-ornithine was assayed with Hst2.89 It can be thus concluded from these observations that the sirtuin (e.g.SIRT1 or Hst2)-catalyzed nicotinamide cleavage and deacetylation have a very stringent requirement for the distance between the α-carbon and the side chain acetamido group, with that found in acetyl-lysine being optimal. It was also found in the study that SIRT1-catalyzed nicotinamide cleavage and deacetylation were stereospecifically supported by L-acetyl-lysineversus its D-isomer,96 consistent with the previously observed lack of Hst2-catalyzed deacetylation of acetyl-poly-D-lysine.88
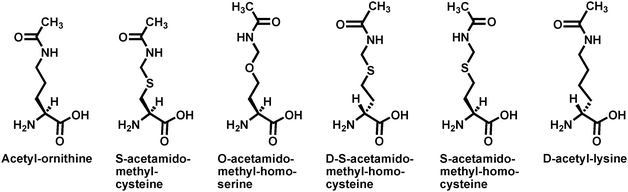 | ||
| Fig. 11 The acetyl-lysine analogs used in ref. 96. Acetyl-lysine and these analogs were all incorporated into position X in the following p53 peptide sequence: H2N-HKXLM-COOH. | ||
4. Sirtuin inhibitors containing Nε-thioacetyl-lysine
As described above, multiple sirtuins (e.g.SIRT1, Sir2Tm, and Hst2) have been shown to be capable of accepting the thioacetylated peptides as substrates to support the nicotinamide cleavage with the formation of a stalled species, i.e. α-1′-S-alkylamidate. Since this stalled intermediate is basically a covalent conjugate between the two substrates (i.e.NAD+ and the thioacetylated peptide), it likely behaves as a tight-binding bisubstrate analog sirtuin inhibitor. In this regard, the thioacetylated peptide substrate could be a bona fide mechanism-based sirtuin inhibitor or suicide substrate. The first indication that a thioacetylated peptide could be a potent sirtuin inhibitor was made when the human p53 peptide containing thioacetyl-lysine was found to be a potent SIRT1 inhibitor with an IC50 ∼2 μM.80 Subsequently, quite a few thioacetyl-lysine peptides have also been shown to be potent inhibitors against different sirtuins including Hst2, SIRT1, SIRT2, and SIRT3. Different degrees of inhibitory selectivity among different sirtuins (e.g. among SIRT1, SIRT2, and SIRT3) have also been demonstrated for some thioacetyl-lysine peptides. A list of the representative potent and/or selective thioacetyl-lysine peptide sirtuin inhibitors is shown in Fig. 12.80,81,98,99 Through in-depth kinetic studies with Hst2, the thioacetyl-lysine histone H3 peptide was found to be a competitive inhibitorversusacetyl-lysine peptide substrate with an inhibition constant (Kis) of 17 nM.81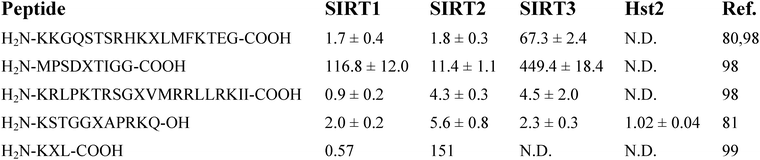 | ||
| Fig. 12 Representative potent and/or selective thioacetyl-lysine peptide sirtuin inhibitors. The numbers are the IC50(μM) values determined under the particular sirtuin inhibition assay conditions in different referenced studies. X in each peptide sequence is thioacetyl-lysine. N.D., not determined. | ||
Despite the identification of multiple peptide-based potent and selective thioacetyl-lysine-containing sirtuin inhibitors, due to the fact that linear peptides are susceptible to peptide bond cleavage catalyzed by peptidases/proteases and are not cell permeable in general,97 developing proteolytically stable and cell permeable thioacetyl-lysine-containing peptidomimetic or non-peptide sirtuin inhibitors undoubtedly represents a logical step toward developing thioacetyl-lysine-based chemical biological tools for deciphering the biology of the sirtuin enzymes and thioacetyl-lysine-based potential therapeutics for metabolic and age-related diseases and cancer.
There has so far been one report of a non-peptide thioacetyl-lysine-containing sirtuin inhibitor (Fig. 13).100 Under the authors’ assay condition, this compound was shown to be a potent SIRT1 inhibitor (IC50 = 2.7 μM) with 8.5-fold and >37-fold inhibitory selectivity versusSIRT2 and SIRT3, respectively. Furthermore, this compound was shown to be capable of inhibiting SIRT1 inside the human colon cancer cell line HCT116 in a concentration-dependent manner, as evidenced by the concentration-dependent elevation of the acetylation level of p53 in the HCT116 cells following the compound treatment.
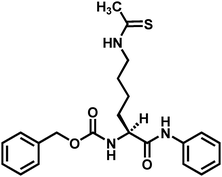 | ||
| Fig. 13 The chemical structure of the currently reported non-peptide thioacetyl-lysine-containing sirtuin inhibitor. | ||
Very recently, we have identified two thioacetyl-lysine-based proteolytically stable and cell permeable peptidomimetic inhibitors against the human sirtuins (Fig. 14).101 We found that these two compounds were both stronger SIRT1 inhibitors than the compound shown in Fig. 13.
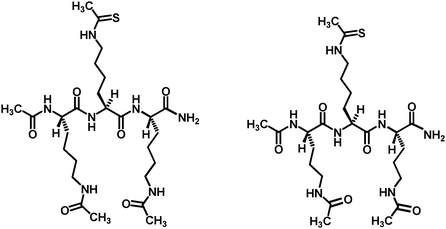 | ||
| Fig. 14 The chemical structures of the thioacetyl-lysine-containing peptidomimetic sirtuin inhibitors. | ||
5. Sirtuin inhibitors containing other Nε-acetyl-lysine analogs
Acetyl-lysine analogs other than thioacetyl-lysine have also been shown to be capable of conferring decent inhibitory action against sirtuin-catalyzed deacetylation reaction. A histone H3 peptide containing trifluoroacetyl-lysine (Fig. 4) (i.e.H2N-KSTGGXAPRKQ-COOH, wherein X is trifluoroacetyl-lysine) was found to be a competitive Hst2 inhibitorversus the acetyl-lysine peptide with a Kis of 4.8 μM,81 suggesting that trifluoroacetyl-lysine is able to compete directly with acetyl-lysine for binding to its binding tunnel at the Hst2 active site. However, over 5 orders of magnitude slower nicotinamide cleavage rate was observed in an Hst2 assay with this trifluoroacetyl-lysine peptide than that supported by the acetyl-lysine peptide, and this was suggested to be due to the strong electron-withdrawing nature of fluorine and the consequent low nucleophilicity of the trifluoroacetamide oxygen.85 However, it was also found that this trifluoroacetyl-lysine peptide (Kd = 3.3 μM) binds tighter to Hst2 than the acetyl-lysine peptide (Kd = 21 μM), and this was suggested to be due to the greater hydrophobicity (π = 0.02) of trifluoroacetyl than acetyl (π = −0.55).85,90 Therefore, a trifluoroacetyl-lysine peptide could be regarded as a dead-end sirtuin inhibitor.As described above, the SIRT1 active site was also recently shown to be capable of accommodating ethyl malonyl-lysine to support the nicotinamide cleavage from NAD+ with the formation of a stable lysine-ADP-ribose conjugate (Fig. 9).95 This conjugate was proposed to be derived from the nucleophilic reaction of the depicted enol form of the ethyl malonyl-lysine-based small molecule at the C1′ position of NAD+, and has been directly detected by mass spectrometry. Since this conjugate is also a stalled species within SIRT1 active site and is basically a covalent conjugate simultaneously occupying the binding sites for NAD+ and acetyl-lysine, it also likely behaves as a tight-binding bisubstrate analog sirtuin inhibitor. Therefore, the ethyl malonyl-lysine-based small molecule could also be a bona fide mechanism-based sirtuin inhibitor or suicide substrate. The ethyl malonyl-lysine-based small molecule was found to be a competitive SIRT1 inhibitorversus the acetyl-lysine substrate with a Kis of 9.5 μM,95 further suggesting that ethyl malonyl-lysine is able to compete directly with acetyl-lysine for binding to its binding tunnel at the SIRT1 active site.
It should be pointed out that the success of the mechanism-based SIRT1 inhibition by the ethyl malonyl-lysine compound lies in the hypothesized preferred α-C-alkylation to O-alkylation of the acetamide moiety of ethyl malonyl-lysine. The O-alkylation would potentially lead to the deacylation of this acetyl-lysine analog. Even though this hypothesis was not directly addressed experimentally in the original study,95 it is consistent with authors’ modeling studies showing that (i) the enol form depicted in Fig. 9, in which the enol OH forms a hydrogen bond with the side chain imidazole of a histidine residue, represented the most stable tautomer in the active site of the model sirtuin Hst2 and (ii) significantly larger size of the highest occupied molecular orbital (HOMO) was found at the α-carbon than the oxygen of the acetamide moiety. This hypothesis is also consistent with the literature precedents of preferred C- to O-alkylation of malonamic acid derivatives, as described in the study. Finally, the observed decent SIRT1 inhibition by the ethyl malonyl-lysine-based small molecule should argue for the predominant α-C-alkylation with minimal, if any, O-alkylation with this particular malonamic acid analog within the SIRT1 active site.
Under authors’ assay condition, the ethyl malonyl-lysine-based small molecule was also shown to be a selective SIRT1 inhibitor with 17-fold and >77-fold inhibitory selectivity versusSIRT2 and SIRT3, respectively.95 The ethyl malonyl-lysine-based small molecule was also shown to be capable of inhibiting SIRT1 inside the human colon cancer cell line HCT116 in a concentration-dependent manner, as evidenced by the concentration-dependent elevation of the acetylation level of p53 in the HCT116 cells following the compound treatment.95
Very recently, Nε-selenoacetyl-lysine was also shown to be able to confer a strong sirtuin inhibition.102 The selenoacetyl-lysine compounds (the first two structures shown in Fig. 15) were found in in vitro assays to be SIRT1 and SIRT2 inhibitors with similar or slightly better (5.5-fold) potency than their thioacetyl-lysine counterparts. In addition, the Nε-isothiovaleryl-lysine tripeptide (the third structure in Fig. 15) was also found in this in vitro study to be a SIRT1 and SIRT2 inhibitor with similar potency to its thioacetyl-lysine counterpart. Presumably, these compounds are able to inhibit the sirtuin deacetylase activityvia a similar mechanism to that by the thioacetyl-lysine sirtuin inhibitors.
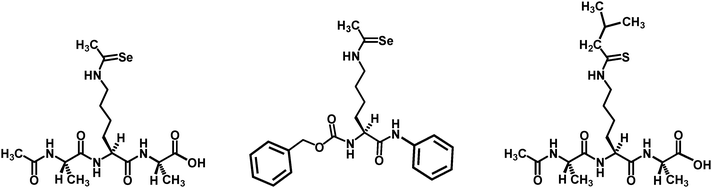 | ||
| Fig. 15 The chemical structures of the selenoacetyl-lysine and the isothiovaleryl-lysine sirtuin inhibitors. | ||
6. Summary
Due to the involvement of the sirtuin-catalyzed lysine deacetylation reaction in multiple (patho)physiological processes, this deacetylation reaction has been increasingly regarded as an important therapeutic target for developing potential therapeutics for metabolic and age-related diseases and cancer. In order to effectively design molecular entities targeting this enzymatic reaction as potential therapeutics or chemical biological tools for a further dissection of sirtuin biology, a detailed mechanistic understanding of this enzymatic reaction is undoubtedly an important research endeavor. Since the side chain acetyl group removal from acetyl-lysine is the very chemistry that this enzymatic reaction is about, it is satisfying to see that, during the past several years, a variety of acetyl-lysine analogs ranging from the simple isotopically labeled analogs to relatively sophisticated analogs (e.g.ethyl malonyl-lysine) have found exquisite uses in helping to elucidate the mechanism of sirtuin enzymes and/or in formulating novel sirtuin inhibitory strategies, especially the mechanism-based approach. It is expected that carefully designed novel acetyl-lysine analogs will continue to be employed to further enhance our mechanistic understanding and our capability of pharmacological exploitation of the reaction catalyzed by the fascinating class of enzymes called sirtuins.Acknowledgements
Our work described in this article was supported by the James L. and Martha J. Foght Endowment and the University of Akron.References
- T. Kouzarides, EMBO J., 2000, 19, 1176–1179 CrossRef CAS.
- S. L. Schreiber and B. E. Bernstein, Cell, 2002, 111, 771–778 CAS.
- S. Y. Roth, J. M. Denu and C. D. Allis, Annu. Rev. Biochem., 2001, 70, 81–120 CrossRef CAS.
- R. Marmorstein and S. Y. Roth, Curr. Opin. Genet. Dev., 2001, 11, 155–161 CrossRef CAS.
- X. J. Yang, Nucleic Acids Res., 2004, 32, 959–976 CrossRef CAS.
- C. E. Berndsen and J. M. Denu, Curr. Opin. Struct. Biol., 2008, 18, 682–689 CrossRef CAS.
- X. J. Yang and E. Seto, Nat. Rev. Mol. Cell Biol., 2008, 9, 206–218 CrossRef CAS.
- X. J. Yang and E. Seto, Oncogene, 2007, 26, 5310–5318 CrossRef CAS.
- G. Blander and L. Guarente, Annu. Rev. Biochem., 2004, 73, 417–435 CrossRef CAS.
- J. M. Denu, Curr. Opin. Chem. Biol., 2005, 9, 431–440 CrossRef CAS.
- A. A. Sauve, C. Wolberger, V. L. Schramm and J. D. Boeke, Annu. Rev. Biochem., 2006, 75, 435–465 CrossRef CAS.
- L. R. Saunders and E. Verdin, Oncogene, 2007, 26, 5489–5504 CrossRef CAS.
- S. C. Hodawadekar and R. Marmorstein, Oncogene, 2007, 26, 5528–5540 CrossRef CAS.
- M. A. Glozak, N. Sengupta, X. Zhang and E. Seto, Gene, 2005, 363, 15–23 CrossRef CAS.
- K. Batta, C. Das, S. Gadad, J. Shandilya and T. K. Kundu, Subcell. Biochem., 2007, 41, 193–212 Search PubMed.
- M. D. Shahbazian and M. Grunstein, Annu. Rev. Biochem., 2007, 76, 75–100 CrossRef.
- S. Thiagalingam, K. H. Cheng, H. J. Lee, N. Mineva, A. Thiagalingam and J. F. Ponte, Ann. N. Y. Acad. Sci., 2003, 983, 84–100 CrossRef CAS.
- I. V. Gregoretti, Y. M. Lee and H. V. Goodson, J. Mol. Biol., 2004, 338, 17–31 CrossRef CAS.
- S. Greiss and A. Gartner, Mol. Cells, 2009, 28, 407–415 CrossRef CAS.
- R. A. Frye, Biochem. Biophys. Res. Commun., 2000, 273, 793–798 CrossRef CAS.
- J. Gu, J. Y. Deng, R. Li, H. Wei, Z. Zhang, Y. Zhou, Y. Zhang and X. E. Zhang, Biochemistry (Moscow), 2009, 74, 743–748 CrossRef CAS.
- L. Figueiredo and A. Scherf, Chromosome Res., 2005, 13, 517–524 CrossRef CAS.
- S. Dam and A. Lohia, Cell. Microbiol., 2010, 12, 1002–1014 CrossRef CAS.
- J. A. García-Salcedo, P. Gijón, D. P. Nolan, P. Tebabi and E. Pays, EMBO J., 2003, 22, 5851–5862 CrossRef CAS.
- D. Sereno, B. Vergnes, F. Mathieu-Daude, A. Cordeiro da Silva and A. Ouaissi, Parasitol. Res., 2006, 100, 1–9 CrossRef CAS.
- J. Pérez-Martín, J. A. Uría and A. D. Johnson, EMBO J., 1999, 18, 2580–2592 CrossRef CAS.
- P. Trojer, E. M. Brandtner, G. Brosch, P. Loidl, J. Galehr, R. Linzmaier, H. Haas, K. Mair, M. Tribus and S. Graessle, Nucleic Acids Res., 2003, 31, 3971–3981 CrossRef CAS.
- R. Domergue, I. Castaño, A. De Las Peñas, M. Zupancic, V. Lockatell, J. R. Hebel, D. Johnson and B. P. Cormack, Science, 2005, 308, 866–870 CrossRef CAS.
- H. Yasukawa and K. Yagita, Parasitol. Res., 2010, 107, 707–712 CrossRef.
- B. C. Smith, W. C. Hallows and J. M. Denu, Chem. Biol., 2008, 15, 1002–1013 CrossRef CAS.
- C. Schlicker, M. Gertz, P. Papatheodorou, B. Kachholz, C. F. W. Becker and C. Steegborn, J. Mol. Biol., 2008, 382, 790–801 CrossRef CAS.
- T. Nakagawa, D. J. Lomb, M. C. Haigis and L. Guarente, Cell, 2009, 137, 560–570 CrossRef CAS.
- E. Michishita, R. A. McCord, E. Berber, M. Kioi, H. Padilla-Nash, M. Damian, P. Cheung, R. Kusumoto, T. L. A. Kawahara, J. C. Barrett, H. Y. Chang, V. A. Bohr, T. Ried, O. Gozani and K. F. Chua, Nature, 2008, 452, 492–496 CrossRef CAS.
- B. Yang, B. M. Zwaans, M. Eckersdorff and D. B. Lombard, Cell Cycle, 2009, 8, 2662–2663 CAS.
- E. Michishita, R. A. McCord, L. D. Boxer, M. F. Barber, T. Hong, O. Gozani and K. F. Chua, Cell Cycle, 2009, 8, 2664–2666 CAS.
- K. Yamagata and I. Kitabayashi, Biochem. Biophys. Res. Commun., 2009, 390, 1355–1360 CrossRef CAS.
- J. Wang and J. Chen, J. Biol. Chem., 2010, 285, 11458–11464 CrossRef CAS.
- R. Li, J. Gu, Y. Y. Chen, C. L. Xiao, L. W. Wang, Z. P. Zhang, L. J. Bi, H. P. Wei, X. D. Wang, J. Y. Deng and X. E. Zhang, Mol. Microbiol., 2010, 76, 1162–1174 CrossRef CAS.
- V. J. Starai, I. Celic, R. N. Cole, J. D. Boeke and J. C. Escalante-Semerena, Science, 2002, 298, 2390–2392 CrossRef CAS.
- K. Zhao, X. Chai and R. Marmorstein, J. Biol. Chem., 2003, 278, 26071–26077 CrossRef CAS.
- H. Cimen, M. J. Han, Y. Yang, Q. Tong, H. Koc and E. C. Koc, Biochemistry, 2010, 49, 304–311 CrossRef CAS.
- Y. Yang, H. Cimen, M. J. Han, T. Shi, J. H. Deng, H. Koc, O. M. Palacios, L. Montier, Y. Bai, Q. Tong and E. C. Koc, J. Biol. Chem., 2010, 285, 7417–7429 CrossRef CAS.
- N. Shulga, R. Wilson-Smith and J. G. Pastorino, J. Cell Sci., 2010, 123(6), 894–902 CrossRef CAS.
- E. Michishita, J. Y. Park, J. M. Burneskis, J. C. Barrett and I. Horikawa, Mol. Biol. Cell, 2005, 16, 4623–4635 CrossRef CAS.
- J. Yu and J. Auwerx, Ann. N. Y. Acad. Sci., 2009, 1173, E10–19 CrossRef CAS.
- M. C. Haigis and D. A. Sinclair, Annu. Rev. Pathol.: Mech. Dis., 2010, 5, 253–295 Search PubMed.
- B. Schwer and E. Verdin, Cell Metab., 2008, 7, 104–112 CrossRef CAS.
- T. Finkel, C. X. Deng and R. Mostoslavsky, Nature, 2009, 460, 587–591 CrossRef CAS.
- S. Imai and L. Guarente, Trends Pharmacol. Sci., 2010, 31, 212–220 CrossRef CAS.
- T. Liu, P. Y. Liu and G. M. Marshall, Cancer Res., 2009, 69, 1702–1705 CrossRef CAS.
- M. Porcu and A. Chiarugi, Trends Pharmacol. Sci., 2005, 26, 94–103 CrossRef CAS.
- P. J. Elliott and M. Jirousek, Curr. Opin. Investig. Drugs, 2008, 9, 371–378 Search PubMed.
- T. F. Outeiro, O. Marques and A. Kazantsev, Biochim. Biophys. Acta, 2008, 1782, 363–369 CAS.
- J. C. Milne and J. M. Denu, Curr. Opin. Chem. Biol., 2008, 12, 11–17 CrossRef CAS.
- S. Imai and W. Kiess, Front. Biosci., 2009, 14, 2983–2995 CrossRef CAS.
- R. C. Neugebauer, W. Sippl and M. Jung, Curr. Pharm. Des., 2008, 14, 562–573 CrossRef CAS.
- Y. Cen, Biochim. Biophys. Acta, 2010, 1804, 1635–1644 CAS.
- J. Schemies, U. Uciechowska, W. Sippl and M. Jung, Med. Res. Rev., 2009 Search PubMed Oct 12. [Epub ahead of print].
- F. J. Alcaín and J. M. Villalba, Expert Opin. Ther. Pat., 2009, 19, 283–294 Search PubMed.
- J. Schemies, W. Sippl and M. Jung, Cancer Lett., 2009, 280, 222–232 CrossRef CAS.
- P. A. Cole, Nat. Chem. Biol., 2008, 4, 590–597 CrossRef CAS.
- B. G. Szczepankiewicz and P. Y. Ng, Curr. Top. Med. Chem., 2008, 8, 1533–1544 CrossRef CAS.
- S. Lavu, O. Boss, P. J. Elliott and P. D. Lambert, Nat. Rev. Drug Discovery, 2008, 7, 841–853 CrossRef CAS.
- O. Grubisha, B. C. Smith and J. M. Denu, FEBS J., 2005, 272, 4607–4616 CrossRef CAS.
- A. A. Sauve, Biochim. Biophys. Acta, 2010, 1804, 1591–1603 CAS.
- B. D. Sanders, B. Jackson and R. Marmorstein, Biochim. Biophys. Acta, 2010, 1804, 1604–1616 CAS.
- R. Marmorstein and R. C. Trievel, Biochim. Biophys. Acta, 2009, 1789, 58–68 CAS.
- B. C. Smith and J. M. Denu, Biochim. Biophys. Acta, 2009, 1789, 45–57 CAS.
- A. A. Sauve and V. L. Schramm, Curr. Med. Chem., 2004, 11, 807–826 CrossRef CAS.
- R. Marmorstein, Biochem. Soc. Trans., 2004, 32, 904–909 CrossRef CAS.
- M. T. Borra, M. R. Langer, J. T. Slama and J. M. Denu, Biochemistry, 2004, 43, 9877–9887 CrossRef CAS.
- A. A. Sauve, I. Celic, J. Avalos, H. Deng, J. D. Boeke and V. L. Schramm, Biochemistry, 2001, 40, 15456–15463 CrossRef CAS.
- M. D. Jackson and J. M. Denu, J. Biol. Chem., 2002, 277, 18535–18544 CrossRef CAS.
- J. Landry, J. T. Slama and R. Sternglanz, Biochem. Biophys. Res. Commun., 2000, 278, 685–690 CrossRef CAS.
- M. D. Jackson, M. T. Schmidt, N. J. Oppenheimer and J. M. Denu, J. Biol. Chem., 2003, 278, 50985–50998 CrossRef CAS.
- B. C. Smith and J. M. Denu, Biochemistry, 2006, 45, 272–282 CrossRef CAS.
- J. B. French, Y. Cen and A. A. Sauve, Biochemistry, 2008, 47, 10227–10239 CrossRef CAS.
- K. G. Hoff, J. L. Avalos, K. Sens and C. Wolberger, Structure, 2006, 14, 1231–1240 CrossRef CAS.
- W. F. Hawse, K. G. Hoff, D. G. Fatkins, A. Daines, O. V. Zubkova, V. L. Schramm, W. Zheng and C. Wolberger, Structure, 2008, 16, 1368–1377 CrossRef CAS.
- D. G. Fatkins, A. D. Monnot and W. Zheng, Bioorg. Med. Chem. Lett., 2006, 16, 3651–3656 CrossRef CAS.
- B. C. Smith and J. M. Denu, Biochemistry, 2007, 46, 14478–14486 CrossRef CAS.
- H. Yuasa, M. Izumi and H. Hashimoto, Curr. Top. Med. Chem., 2009, 9, 76–86 CrossRef CAS.
- N. J. Parry, D. E. Beever, E. Owen, I. Vandenberghe, J. Van Beeumen and M. K. Bhat, Biochem. J., 2001, 353, 117–127 CAS.
- H. Driguez, Top. Curr. Chem., 1997, 187, 85–116 CAS.
- B. C. Smith and J. M. Denu, J. Am. Chem. Soc., 2007, 129, 5802–5803 CrossRef CAS.
- P. Hu, S. Wang and Y. Zhang, J. Am. Chem. Soc., 2008, 130, 16721–16728 CrossRef CAS.
- M. T. Borra and J. M. Denu, Methods Enzymol., 2004, 376, 171–187 CAS.
- A. N. Khan and P. N. Lewis, J. Biol. Chem., 2005, 280, 36073–36078 CrossRef CAS.
- A. N. Khan and P. N. Lewis, J. Biol. Chem., 2006, 281, 11702–11711 CrossRef CAS.
- B. C. Smith and J. M. Denu, J. Biol. Chem., 2007, 282, 37256–37265 CrossRef CAS.
- J. L. Avalos, J. D. Boeke and C. Wolberger, Mol. Cell, 2004, 13, 639–648 CrossRef CAS.
- Z. Cheng, Y. Tang, Y. Chen, S. Kim, H. Liu, S. S. Li, W. Gu and Y. Zhao, Mol. Cell. Proteomics, 2009, 8, 45–52 CrossRef CAS.
- B. Heltweg, F. Dequiedt, B. L. Marshall, C. Brauch, M. Yoshida, N. Nishino, E. Verdin and M. Jung, J. Med. Chem., 2004, 47, 5235–5243 CrossRef CAS.
- J. Garrity, J. G. Gardner, W. Hawse, C. Wolberger and J. C. Escalante-Semerena, J. Biol. Chem., 2007, 282, 30239–30245 CrossRef CAS.
- T. Asaba, T. Suzuki, R. Ueda, H. Tsumoto, H. Nakagawa and N. Miyata, J. Am. Chem. Soc., 2009, 131, 6989–6996 CrossRef CAS.
- N. Jamonnak, B. M. Hirsch, Y. Pang and W. Zheng, Bioorg. Chem., 2010, 38, 17–25 CrossRef CAS.
- M. Goodman and S. Ro, in Burger's Medicinal Chemistry and Drug Discovery, Vol. 1, Principles and Practice, ed. M. E. Wolff, John Wiley & Sons, Inc., USA, 5th edn, 1995, pp. 803–861 Search PubMed.
- D. G. Fatkins and W. Zheng, Int. J. Mol. Sci., 2008, 9, 1–11 Search PubMed.
- P. H. Kiviranta, T. Suuronen, E. A. Wallén, J. Leppänen, J. Tervonen, S. Kyrylenko, A. Salminen, A. Poso and E. M. Jarho, J. Med. Chem., 2009, 52, 2153–2156 CrossRef CAS.
- T. Suzuki, T. Asaba, E. Imai, H. Tsumoto, H. Nakagawa and N. Miyata, Bioorg. Med. Chem. Lett., 2009, 19, 5670–5672 CrossRef CAS.
- B. M. Hirsch, C. A. Gallo, Z. Du, Z. Wang and W. Zheng, MedChemComm, 2010, 1, 233–238 RSC.
- T. Huhtiniemi, T. Suuronen, M. Lahtela-Kakkonen, T. Bruijn, S. Jääskeläinen, A. Poso, A. Salminen, J. Leppänen and E. Jarho, Bioorg. Med. Chem., 2010, 18, 5616–5625 CrossRef CAS.
Footnote |
| † This article is part of a themed issue of Molecular BioSystems on Post-translational modifications. |
| This journal is © The Royal Society of Chemistry 2011 |