 Open Access Article
Open Access ArticleSystem-level strategies for studying the metabolism of Mycobacterium tuberculosis
Dany J. V.
Beste
and
Johnjoe
McFadden
*
Faculty of Health and Medical Sciences, University of Surrey, Guildford GU2 7XH, UK. E-mail: d.beste@surrey.ac.uk; j.mcfadden@surrey.ac.uk; Fax: +44 (0)1483-300374; Tel: +44 (0)1483-696494
First published on 11th October 2010
Abstract
Despite decades of research many aspects of the biology of Mycobacterium tuberculosis remain unclear and this is reflected in the antiquated tools available to treat and prevent tuberculosis and consequently this disease remains a serious public health problem. Important discoveries linking M. tuberculosis's metabolism and pathogenesis have renewed interest in this area of research. Previous experimental studies were limited to the analysis of individual genes or enzymes whereas recent advances in computational systems biology and high throughput experimental technologies now allow metabolism to be studied on a genome scale. Here we discuss the progress being made in applying system level approaches to studying the metabolism of this important pathogen. The information from these studies will fundamentally change our approach to tuberculosis research and lead to new targets for therapeutic drugs and vaccines.
 Dany J. V. Beste | Dany J. V. Beste started her career as a state registered Biomedical Scientist after obtaining a BSc in Biochemistry from the University of Surrey. She spent several years working at a number of different diagnostic and reference laboratories in the UK including the Streptococcus and Diphtheria Reference Laboratory and the Hospital of Tropical Medicine where she learnt lots of useful practical skills which would prove invaluable in her subsequent research career. Dany first became interested in studying tuberculosis whilst working as a VSO microbiology lecturer at the Malawi College of Health Sciences. After obtaining a masters degree at the London School of Hygiene and Tropical Medicine, Dany achieved her goal by studying for a PhD in mycobacterial genetics and systems biology and is currently a Senior Research Fellow in Mycobacterial Systems Biology at the University of Surrey. Dany has developed and characterised a chemostat model for the growth of Mycobacterium tuberculosis which is amenable to systems biology studies and had a key role in the development of the first genome scale model of M. tuberculosis. The goal of her current Wellcome Trust research project is to use a systems biology approach to decipher what M. tuberculosis metabolises in vivo. |
 Johnjoe McFadden | Johnjoe McFadden received his PhD from Imperial College and although his first post-doc was in the area of human molecular genetics he moved on to applying molecular genetic tools to the study of microbes. He applied RFLP analysis to mycobacterial molecular epidemiology and was able to demonstrate, for instance, that mycobacteria isolated from human Crohn's disease were identical to Mycobacterium paratuberculosis isolated from cattle with Johne's disease. After moving to the University of Surrey where he is now a Professor in Molecular Genetics he applied similar approaches to examine the molecular epidemiology of meningococcal meningitis in the UK and developed several molecular diagnostic tools. As molecular diagnostics and epidemiology became established in clinical laboratories, his research shifted to examining mechanisms of virulence in mycobacteria and the meningococcus. In recent years his work has concentrated on functional genomic and systems biology approaches to understanding bacterial virulence. Researchers in his laboratory have performed metabolic analysis of the TB bacillus and constructed the first genome-scale model of metabolism in Mycobacterium tuberculosis. Transposon mutagenesis was used to identify genes involved in control of growth rate, which is a key component of persistence in this pathogen. He has also recently developed collaboration with engineers to investigate the application of nanotechnology in medicine. In addition to his laboratory-based research, Johnjoe McFadden has been active in popularising science in writing articles for national (mostly the Guardian) and international press. |
Introduction
Tuberculosis (TB) is a disease which plagued ancient Egyptians and still remains a major threat to human health thousands of years later. The control of tuberculosis has been significantly hindered by the limited resources available for the prevention and treatment of tuberculosis. A truly effective vaccine is lacking as the 90 year old Mycobacterium bovis bacillus Calmette–Guerin live attenuated vaccine is not universally protective and does not produce immunity against re-infection or reactivation. Lengthy (6–9 months) and complex (three or more different drugs) treatment is required using currently available anti-TB drugs. The economic and logistic burden of administering these drug regimens in industrially undeveloped countries where TB is most prevalent is enormous and combined with poor patient compliance are important factors in the emergence of drug resistant TB isolates which are causing ongoing epidemics. These factors underscore the urgent need for the development of novel and effective therapeutics and vaccines and new approaches will be required to achieve these goals.Mycobacterium tuberculosis is an unusual bacterial pathogen which has the remarkable ability to cause both acute life threatening disease and also clinically latent infections which can persist for the lifetime of the human host. Unlike many pathogens M. tuberculosis does not rely on the production of specific toxins to cause disease but rather the secret of this bacterium's great success seems to be the ability to adapt and survive within the changing and adverse environment provided by the human host during the course of an infection. It is becoming apparent that key to this adaptation is the metabolic reprogramming of M. tuberculosis during both the acute and chronic phase of TB disease and therefore a more complete understanding of mycobacterial metabolism remains a major goal of TB research.
Whilst recent increases in research funding has progressed our understanding of the basic biology of M. tuberculosis this has not yet impacted on the global TB trends which remain at staggering levels. A possible reason why it has been difficult to translate basic research into effective strategies for combating tuberculosis is that TB research has until recently, focused on studying individual parameters in isolation which can consequently result in an overestimation of the importance of these factors. This effect may be particularly profound for a persistent pathogen such as M. tuberculosis which lacks classical virulence factors. The systems biology framework which investigates the dynamic interactions of many components provides an alternative and complementary strategy to the more traditional reductionist approaches to TB research. The central component of all systems biology approaches is a mathematical model which instantiates knowledge about a particular system that can be used to provide a scaffold for the integration and interpretation of “omic” scale datasets but is also capable of generating novel hypotheses and predictions that can be experimentally tested in an iterative cycle of hypothesis generation, prediction and model refinement (Fig. 1).
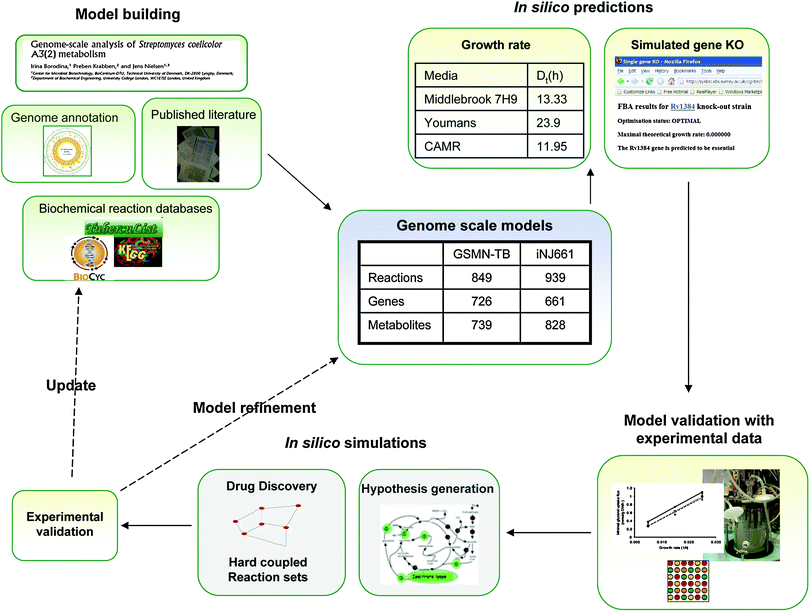 | ||
| Fig. 1 The iterative process of modeling the metabolism of the pathogen Mycobacterium tuberculosis. A cycle of model building and experimental validation, refinement of the model for the identification of drug targets and hypothesis generation is shown. The drawing includes the properties of the two available genome scale models.28,29 | ||
Metabolic model building
The ultimate goal of system biology approaches to studying TB is to construct a complete model of infection incorporating both the pathogen and host, but this is currently infeasible as the information about the different components to be included in the model is lacking. Studies with other organisms have demonstrated that metabolism is, by far, the best understood cellular network and is thereby an excellent starting point for a systems-based approach.1–3However, metabolism is complex. Even the simplest organisms synthesize many hundreds of metabolites connected by a similar number of enzyme-catalyzed reactions. Each reaction is described by a set of kinetic parameters (e.g. Km, Vmax) which, in combination with substrate/product concentrations, determine its rate. Although Km values are constants (for a particular substrate/product combination) and may be determined experimentally, intracellular concentrations of substrate, products and enzyme (influencing Vmax) vary over wide ranges and are not easily measured. Even a single enzyme reaction is therefore a highly dynamic system; and systems of just a few reactions steps are usually mathematically described by a set of ordinary differential equation with a large number of parameters and variables whose values are extremely challenging to measure experimentally. Kinetic models have therefore only been applied to the dynamics of small well-defined systems, such as glycolysis in Escherichia coli4 that are very far from being genome-scale.
However, it is relatively straightforward to generate a metabolic network that describes the biochemical reactions that an organism is predicted to be capable of performing, in terms of stoichiometric formulas. The genome annotation predicts all those genes encoding enzymes that are readily linked to databases describing the predicted biochemical conversions performed by these enzymes. These stoichiometric reactions can then be incorporated into pathways that may be integrated into a network to construct a genome-scale metabolic network that effectively describes the chain of reactions responsible for inputting nutrients and transforming them into the biomass of the cell, waste products and energy.
On their own genome-scale metabolic networks are essentially descriptive, describing the set of metabolic reactions and their connectivity but cannot actually simulate any metabolic process. To do that they must incorporate metabolite concentrations and, for dynamic systems, all the kinetic parameters discussed above which is infeasible at a genome scale. However, if a critical assumption is made, that all metabolite concentrations are held constant, then the underlying dynamics are hugely simplified and the system may be solvable. Consider the simple set of stoichiometric conversions, A → 2B → C + D. If this is a dynamic system then knowing the concentration of A tells us nothing about the concentrations of B, C or D without knowledge of the detailed kinetic parameters. Yet if the concentrations of A, B, C and D are unchanging (the system is at steady-state) then the rate of flux from A → 2B must equal the rate of flux from 2B → C + D, so knowing the concentration of A (or the flux towards A) uniquely determines the concentration (or the fluxes) of all the downstream metabolites. Steady state systems are thereby described by far fewer parameters and variables than dynamic systems.
Of course a genome scale model is far more complex than the above system but the entire set of stoichiometric conversions for an organism is available from its genome annotation and standard biochemical literature. It is therefore possible to build a model consisting of all the stoichiometric reactions predicted by the annotation and link these pathways and networks connected by flux values between each reaction. Such a network can be incorporated into a mathematical model that, once parametrized by measurements such as substrate uptake or biomass production rates, can be solved by standard linear algebra tools (rather than the differential calculus required by kinetic models). However, because metabolic networks contain multiple branch points and parallel pathways there is not a unique solution but a vast space of possible solutions (the system is underdetermined). It is therefore necessary to apply constraint based approaches which reduce the solution space and thereby predict metabolic capabilities or internal fluxes.5–10 Flux balance analysis (FBA) uses the procedure of optimization to reduce the solution space (for a review of FBA we recommend11,12), whereas metabolic flux analysis (MFA) applies additional measurements as constraints. The application of both of these methods to M. tuberculosis will be discussed below.
There are of course limitations to these approaches such as the requirement for steady or quasi-steady state conditions. Also, since no consideration is made of either transcriptional, translational, metabolic regulation or enzyme kinetics the predictive capabilities of constraint based models are limited to situations when these factors are not significantly influencing reaction rates.3 Nevertheless, this approach has been successfully applied to predict the metabolic capabilities of many different cellular systems5–10 and has also been used in metabolic engineering.13,14
Metabolism of M. tuberculosis
Application of metabolic modeling approaches to M. tuberculosis is aided by the fact that metabolism is a reasonably well studied system even in mycobacteria. Moreover, metabolism has been shown to be involved in the virulence of M. tuberculosis, playing a key role in the development and maintenance of both acute and persistent TB infections.15–17 It is perhaps not surprising therefore that several modeling efforts in tuberculosis have focused on metabolism.Much of what is known about metabolism in M. tuberculosis has been gleaned from conventional biochemical and molecular studies over many decades. The pathogen appears typical of bacteria of the Actinomycetales order, with a predominantly aerobic metabolism that is able to catabolize a wide range of substrates to generate biomass and energy. The genome encodes all the enzymes of the Embden–Meyerhof–Parnas pathway (EMP) and pentose phosphate pathway (PPP) and has a complete, or nearly complete TCA cycle (see below). The pathogen also encodes a functional glyoxylate shunt as well as several enzymes connecting the TCA cycle and glycolysis that may be used for either anaplerosis or gluconeogenesis.
There are however several features of central metabolism in M. tuberculosis that appear to be unusual. Although the link between glycolysis and the TCA cycle is complete in M. tuberculosis, the closely related pathogen, M. bovis, lacks a functional pyruvate kinase and is therefore unable to deliver sugars from glycolysis to the TCA cycle and thus is unable to utilize carbohydrates as the sole carbon source.18 This function is therefore unnecessary in vivo as this pathogen causes very similar disease in humans to M. tuberculosis. The role of isocitrate lyase has been intensively studied since the demonstration that both of the isocitrate lyase genes encoded by this pathogen, icl1 and icl2 (although some strains only have icl1), play an essential role in virulence.15,16 This finding has been generally interpreted to be due to this enzymes role in the glyoxylate shunt and a metabolic shift in the principal carbon source from carbohydrates to fat in the host. However the role of the isocitrate lyases may be more complex than just fat catabolism as these enzymes also function as methyl citrate lyases in the methyl citrate cycle.19 In addition, it has also been demonstrated that the glyoxylate shunt functions concurrently with an oxidative TCA cycle which is completed by an anaerobic α-ketoglutarate ferredoxin oxidoreductase (KOR).20 The TCA cycle also seems to be atypical since α-ketoglutarate dehydrogenase (KDH) activity has not be detected. It has been proposed that M. tuberculosis can either complete the oxidative TCA using KOR, an enzyme usually associated with the reductive TCA cycle, or use an alternative pathway from α-ketoglutarate to succinate via succinic semialdehyde.20,21Fig. 2 illustrates the central metabolic pathways of M. tuberculosis, as understood in 2010.
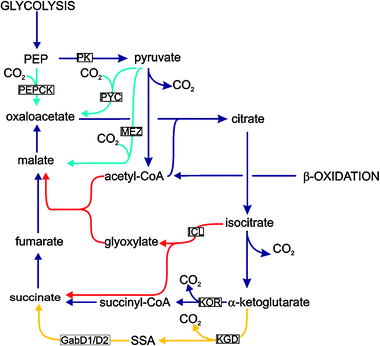 | ||
| Fig. 2 A metabolic map of central metabolism in M. tuberculosis showing the reactions surrounding the TCA cycle. The standard TCA cycle is shown in blue with the variant (SSA) pathway in yellow. Anaplerotic/gluconeogenic reactions are shown in turquoise with the glyoxylate shunt in red. Only enzymes mentioned in the text are indicated, including KOR (α-ketoglutarate ferredoxin oxidoreductase), KGD (α-ketoglutarate decarboxylase), GabD1/D2 (succinic semialdehyde dehydrogenase), ICL (isocitrate lyase) and MEZ (malic enzyme (malate dehydrogenase, decarboxylating)), PEPCK (phosphoenolpyruvate carboxykinase), PK (pyruvate kinase) and PYC (pyruvate carboxylase). | ||
Experimental systems
Systems biology is an iterative procedure of experimental data acquisition, model building, hypothesis generation and experimental verification (Fig. 1). One of the constraints upon this approach surrounds the experimental basis of this work. Models should be developed and validated with accurate and reproducible data, which can be obtained from standard high throughput methods such as transcriptomics, proteomics and metabolomics. However, it is important to note that the mathematical underpinning of flux-based predictions derived from modeling approaches such as FBA or MFA assume that the system is in metabolic steady-state, which can usually only be met by growing the organism in steady state (typically in a chemostat or batch cultivated cells in the exponential growth phase where the growth rate is maximal) and flux-based predictions are only strictly valid for these systems. Determination of substrate utilization and biomass productions rates are also essential for most flux-based predictions so it is usually necessary to grow the test organism in defined media with simple and readily assayable carbon and nitrogen sources.Our group has developed a system for growing mycobacteria in a carbon limited chemically defined minimal medium and demonstrated that biomass composition of the pathogen is a function of the growth rate.22 This adds a further level of complexity to M. tuberculosis models as, to model the organisms growing at different growth rates, alternative biomass formulae must be incorporated.
However, it should be emphasized that many systems-based studies of metabolism do not depend on the steady-state assumption. Indeed, systems-based investigations into the intracellular metabolism of pathogens can be performed using model free approaches.23–25
Modeling the metabolism of M. tuberculosis
The first M. tuberculosis constraint based model was constructed by Raman et al. (2005) and consisted of all the reactions in mycolic acid synthesis.26 This sub-model of metabolism was composed of 219 reactions which involved 197 metabolites, catalysed by 28 enzymes. FBA was used to simulate mycolic acid metabolism and to identify potential drug targets in these pathways. The study illustrates the use of optimization in FBA. As already discussed, FBA solves the excess solution space problem by finding the flux solution of the network that reaches the optimal value of the ‘objective function’. Deciding on an appropriate objective function is a perquisite for the successful application of FBA. Objective functions include maximization or minimization of ATP production; maximization of redox potential; maximization of the rate of synthesis of a particular product, or minimization of nutrient uptake. But the most commonly used objective function in FBA is maximization of growth rate, which of course makes the assumption that microbial cells do indeed maximize their growth rate. Although this assumption could of course be criticized on many grounds, the use of a objective function that maximizes growth rate or biomass formation has been shown to result in a good predictive accuracy in several experimental systems, including nutrient limited chemostat culture of E. coli.27 Its use is more problematic for slow growing pathogens, such as M. tuberculosis, since it is not at clear that these organisms do indeed maximize their growth rate. Raman et al. (2005) used two objective functions that optimized the production of mycolic acids. The first, termed C1, optimized production of only the most abundant mycolate, whereas the objective function C2 maintained the known ratios of different mycolates. To test the predictive accuracy of these objective functions in silico deletions were performed and compared to transposon site hybridization mutagenesis data. The highest predictive accuracy was obtained with the objective function C1. FBA identified 16 essential genes in this study and this primary list was then filtered to remove any genes encoding proteins which were complemented by homologues and also those with close homologues in the human proteome. This feasibility analysis identified seven potential drugs targets for anti-TB drug design.Although targeting a small sub-system such as mycolic acid synthesis can yield valuable information on specific pathways, it has limited value in elucidating the metabolic capability of M. tuberculosis. This latter objective is best approached by constructing a genome-scale networks of metabolism.28,29 Two independent genome scale models of M. tuberculosis have been published to date (GSMN-TB and iNj661) using different reconstruction and validation approaches. The first published genome scale network was built by our group using Streptomyces coelicolor as a starting model.28 Orthology relationships were mapped between the related species using the KEGG databases and this preliminary model was further supplemented with data from the BioCyc database. This automatic process, however, accounted for only 57% of the final model. The remaining model was reconstructed by labour intensive manual curation based upon primary research publications, textbooks and review articles and also by picking the brains of experts in the field. The final model utilized two biomass formulations which were derived from published data of cell composition obtained from a variety of sources including our own chemostat-derived data obtained from fast and slow-growing BCG. BIOMASS 1 reflects the actual macromolecular composition of in vitro-grown M. tuberculosis, whereas BIOMASSe consisted of only those cellular components, such as DNA, RNA, protein, co-factors and cell wall skeleton that were considered to be essential for in vitro growth. The advantage of having these two biomass formulations is that the model could be used to predict gene essentiality both in vitro (with the minimal BIOMASSe as the objective function) and in vivo (with the more complete BIOMASS 1 as the objective function).
The final functional genome scale metabolic network of M. tuberculosis (GSMN-TB) consisted of 739 metabolites participating in 849 reactions and involves 726 genes (Fig. 1). The model is freely available as both an excel file or in sbml format, and is accessible via a user-friendly web tool for constraint-based simulations (http://sysbio.sbs.surrey.ac.uk/tb/). FBA-based predictions of in vitro gene essentiality using BIOMASSe as the objective function had a good correlation with experimental data obtained by global transposon mutagenesis,30 with an overall predictive accuracy of 78%.28
Quantitative validation of the model was performed using data from continuous culture chemostat experiments.28 The model predicted a lower rate of glycerol consumption than the experimentally determined values. A plausible explanation for the discrepancy was that, in addition to consumption of glycerol, the tubercle bacillus also utilized oleic acid released from hydrolysis of the Tween 80 dispersal agent present in the media. Opening an additional oleic acid transport flux corrected this discrepancy and unpublished data from our laboratory have confirmed that Tween 80 is indeed consumed in these experiments. The success of the approach also demonstrated that growth rate optimization was an appropriate objective function for M. tuberculosis at least in these in vitro conditions.
The second genome scale reconstruction of M. tuberculosis was carried out starting with the genome annotation and then using several databases combined with manual curation and yielded 939 reactions, catalysed by 543 enzymes encoded by 661 genes29 (Fig. 1). Although silicon iNj661 weighs in with 90 more reactions and less genes than the GSMN-TB the percentage of orphan reactions is approximately 20% in both networks suggesting that iNj661 has an expanded coverage. However, whilst iNj661 was very good at predicting the growth rate of M. tuberculosis in different media this model had a relatively poor predictive accuracy for experimental essentiality data (55%) as compared with GSMN-TB and is also limited by having only one biomass formulation.
Building upon the work described by Raman et al. (2005) hard-coupled reaction (HCR) sets were calculated for the iNj66I model in order to predict novel drug targets on a genome scale.29 Hard coupling reaction (HCR) sets are generated by a method which does not require an objective function and therefore overcomes any bias introduced by using a specific objective function to constrain the solution space. HCR's are groups of reactions which, due to mass conservation and connectivity constraints, must operate in unison. This strategy identified known and also potential new drug targets which require further analysis.
Application of genome-scale models to provide system-level insights into metabolism
Genome-scale models can also provide novel insights and predictions that are not readily apparent from a consideration of individual reactions and pathways. Model predictions can be structural, depending only on the network connectivity between metabolites (e.g. FBA-based gene/substrate essentiality predictions), or flux-based. The latter (e.g. predictions of internal fluxes, growth or substrate utilization rates) are usually generated by methods such as FBA and MFA and depend on the steady-state assumption and are thereby strictly applicable only to experimental systems in which this condition is satisfied (see above). Most gene essentiality predictions are structure-based and can yield some non-intuitive surprises. For instance, the gene encoding isocitrate lyase is (correctly) predicted to be essential for growth of fatty acids, since the product of their beta oxidation, acetate, can only be incorporated into biomass via the carbon-conserving glyoxylate shunt. However, the gene is not predicted to be essential for growth on complex fats, such as phospholipids and triglycerides, as sole carbon source. The explanation is apparent through examination of the flux solution. The shunt is no longer required because oxidation of these fats yields glycerol in addition to acetate, and glycerol can be incorporated to generate biomass without operation of the shunt. That the ICL gene is essential for growth in vivo suggests that either triglycerides and phospholipids are not in vivo substrates for M. tuberculosis, or, for unknown reasons, only the acetate product of their oxidation is available for incorporation into biomass.As an example of a useful flux-based prediction, flux variability analysis (FVA) was performed to assess the in silico metabolic response of M. tuberculosis to slow growth (Fig. 3) on Roisin's media with glycerol as sole carbon source. FVA is a variant of FBA which, instead of finding a single optimal solution, computes the range of fluxes in each reaction that are compatible with optimization of the objective function. The FVA predicted that whereas the relative fluxes through most reactions would be similar between slow and fast growth rate, a significant relative increase in flux through the isocitrate lyase reaction was expected at slow growth rate (Fig. 3), leading to the hypothesis that isocitrate lyase was involved in mycobacterial survival at slow growth rates. This was surprising since the glyoxylate shunt was not thought to be involved in catabolism of a 3-carbon compounds (such as glycerol) at any growth rate. However, experimental measurement of isocitrate lyase enzyme activity at both growth rates in the chemostat did indeed demonstrate increased isocitrate lyase activity at slow growth rate, which is consistent with the hypothesis that the glyoxylate shunt is involved in maintenance of slow growth. We are currently investigating this hypothesis further using 13C-MFA (Fig. 4) but the finding may also have relevance to the demonstration that isocitrate lyase is essential for survival of M. tuberculosis in vivo.15,16,31,32
 | ||
| Fig. 3 Flux variance analysis predicted an increased flux through the reaction catalysed by isocitrate lyase when M. tuberculosis is growing slowly in carbon limited conditions. Using the GSMN-TB we compared the predicted flux ratios for two different growth rates. A doubling time of 23 hours (dilution rate 0.03) was compared with a doubling time of 69 hours (dilution rate 0.01). Flux ratios for central metabolism were calculated by flux variability analysis (FVA) as the ratios of midpoints of flux ranges obtained for slow and fast growth rates. There was a large predicted (seven-fold) increase in flux through the isocitrate lyase reaction. This hypothesis was confirmed by experimentally demonstrating a significant increased specific isocitrate lyase activity in slow growing cells. | ||
 | ||
| Fig. 4 An experimental work flow for 13C Metabolic Flux Analysis (MFA). Media containing a mixture of unlabelled and labeled substrate is fed into steady state chemostat cultures. The label becomes distributed throughout the cells and after several volume changes the culture will reach isotopic steady state after which very little change to the labeling pattern will occur. Samples are taken for physiological measurements such as glycerol uptake rates and for measurement of the 13C enrichment of proteogenic amino acids from cell hydrolysates and sometimes also intracellular metabolite pools. An isotopomer model detailing the fate of each carbon atom in central metabolism is constructed and the experiment is simulated in silico. An algorithmic fitting procedure is used to adjust the fluxes in the isotopomer model to find the values of the intracellular fluxes that are consistent with the pattern of amino acid labeling. | ||
Using models to interrogate genome annotation
Genome-scale networks are usually constructed initially from genome annotation and are thereby subject to errors in that annotation. However, the metabolic model scrutinizes the metabolic component of genome annotation at a system level for functionality and can thereby be used to find pathway holes or inconsistencies in the annotation. There are several ‘orphan reactions’ in GSMN-TB, that is reactions that are required for network functionality but for which there is no annotated M. tuberculosis gene predicted to perform that function. For example, sulfolipid synthesis in M. tuberculosis generates the metabolite adenosine 3′,5′-bisphosphate (PAP in the model) which will accumulate and thereby become toxic (unbalanced in the model) if it is not catabolized. The model is therefore infeasible unless the reaction catalyzed by the enzyme 3′,5′-bisphosphate nucleotidase (which converts the metabolite to AMP and inorganic phosphate) is included in the network, as an orphan reaction. Examining model feasibility thereby generates clues to incomplete or incorrect genome annotation and may even provide novel drug targets that are not apparent in the genome annotation.In silico models also allow genome annotation to be scrutinized by systems-based experimental data. For example, the route for glycerol utilization is generally assumed to proceed via glycerol kinase (encoded by glpK) followed by dehydrogenation; however, the genome annotation of M. tuberculosis includes several alcohol dehydrogenases that could be involved in an alternative uptake pathway whereby glycerol is first oxidized by glycerol dehydrogenase before being phosphorylated (this pathway is annotated in the KEGG M. tuberculosis pathway map). However, incorporation of this pathway into the initial GSMN-TB model led to the prediction that the gene glpK is dispensable for growth on media with glycerol as sole carbon source. Global mutagenesis data demonstrated that glpK was in fact essential for growth on glycerol as sole carbon source, which was confirmed by construction of a glpK knock-out mutant.33 This information was incorporated into a refined GSMN-TB model in which the annotated alcohol dehydrogenases does not provide an alternative glycerol uptake pathway.
In this way, genome-scale models can be used to interrogate and correct genome annotations and the resulting consensus are used to update both model and genome annotation.
Use of genome-scale models to integrate and interpret functional genome data and predict metabolic states
The functional genomics revolution has provided the means to generate high-throughput transcriptomic, proteomic and metabolic datasets but one of the challenges of systems biology is to use these datasets to predict the metabolic state of a cell. Transcriptomics is often the most readily available dataset and therefore several researchers have developed methods that use genome-scale models to interrogate transcriptome data and predict aspects of the corresponding metabolic state. However, there are a number of problems with this approach as the mapping between transcriptome and metabolic flux will be affected by factors (e.g. post-transcriptional control) that are not measured in transcriptome experiments. Nevertheless, a number of structural or flux-based approaches have been developed that use models to interrogate transcriptome data. The simplest structural method is to use the model as a scaffold and then simply overlay the transcriptomic data of genes encoding metabolic enzymes onto the reactions catalyzed by those genes in a network. However, this approach does not utilize the systems properties of the networks and thereby generates pathway-centric interpretations. A more sophisticated method is the reporter metabolites approach which utilizes bi-partite graph analysis to associate genes with metabolites within a metabolic network and identify focal points of metabolism: network nodes that appear to be most affected by differentially expressed (enzyme-encoding) genes.34 We have recently developed a related method, Differential Producibility Analysis (DPA), which uses FBA to probe metabolite connectivity and identify similar focal points of metabolism. The method was used to extract metabolic signals from the transcriptional response of M. tuberculosis to the macrophage35 and predicted a general slowing down of central metabolism together with a remodeling of the bacterial surface as the pathogen adapts to its intracellular environment.Several flux-based methods have also been developed. The challenge with these approaches is that there is not a linear relationship between the level of mRNA encoding an enzyme and the metabolic flux through the reactions encoded by that enzyme. Nevertheless, approaches have recently been developed that attempt to predict metabolic state of organisms including M. tuberculosis from transcriptome data.36–38 In the M. tuberculosis study a large compendium of gene expression data from the tubercule bacillus growing in a variety of different conditions was used to constrain the MAP sub-model and also GSMN-TB.37 Fluxes were constrained using a function that was based on the level of transcript obtained for the gene encoding that enzyme in each experiment and this was used to limit the maximal flux through each enzymatic reaction. As the focus of the study was to predict impact on fatty acid metabolism, both total mycolic acid production and biomass production were used as objective functions. This ‘E-flux’ method applied to either the GSMN-TB or MAP correctly predicted that seven of the eight mycolic acid inhibitors tested in the experimental data negatively modulated mycolic acid metabolism regardless of which objective function was used. The study also predicted that several other novel compounds had an impact on mycolic acid metabolism. The validity of these predictions awaits further experimental testing.
Use of models to identify drug targets
Both the currently available genome-scale M. tuberculosis metabolic networks correctly identified the target of several anti-tuberculous drugs (e.g. isoniazid) as essential genes (e.g. inhA). Systems level approaches have also been applied in the drug discovery process39 and will undoubtedly facilitate the development of novel anti-TB therapeutics. Raman et al. (2008) have developed a very useful framework for drug discovery which incorporates the three published M. tuberculosis metabolic models as part of a comprehensive and integrated scheme for drug identification. TargetTB imposes sequential filters on an initial list of essential genes (a structural prediction) in order to generate several shortlists of putative drug targets.40 The first list was generated by combining data from a protein–protein interactome and FBA studies using both the available genome scale models (iNj661, GSMN-TB) and also the mycolic acid pathway (MAP) sub-model. This list was then filtered using sequence analysis to remove genes with homologues in humans; structural analysis to remove targets with similar binding sites to human proteins. The process identified 622 putative drug targets including several known and proposed candidate drug targets thereby validating the method. Further filtering generated a short list of broad spectrum antibacterial, TB specific targets and also candidate targets which could be important for treating persistent TB (216 targets).As an alternative to the above approaches which predict reactions as drug targets by evaluating their effects on metabolism, Kim et al. (2009)41 used a metabolite-centric approach to identify potential candidate drug targets in a number of pathogens including M. tuberculosis. Using constraint based flux analysis of the iNj661 model, essential metabolites were identified as metabolites which resulted in zero growth when removed from the model using growth rate as the objective function. Chokepoint analysis identifies enzymes that uniquely produce and/or consume a particular metabolite. Metabolites which were both essential and chokepoints were filtered in order to remove targets that could produce unwanted side effects. This analysis identified 413 essential metabolites, 554 chokepoints and a final list of 364 putative drug targets in M. tuberculosis. Chorismate synthesis was identified by this analysis as a broad range target. Chorismate is a key intermediate in the biosynthesis of a wide range of compounds, including aromatic amino acids, folate cofactors, menaquinones, ubiquinones, and siderophores. As a product of the shikimate pathway which is essential in higher plants, fungi, bacteria (including M. tuberculosis) and algae and is absent in mammals, chorismate production has been validated as a drug target by others.42,43 The shikimate pathway is essential for the growth of M. tuberculosis even with exogenous supplementation with amino acids44 and is therefore a very attractive anti-mycobacterial drug target. Mycolate synthesis was also identified as a prioritized drug target and its metabolism is already targeted by current anti-tubercular drugs.
In another study drug target combinations were identified using an iNj661 based reaction influence network in order to analyze the dependency of one protein on another by virtue of metabolite sharing.45 Highly influential proteins (those which are linked to a large number of other proteins through metabolite sharing) in the network were identified and their interaction with other influential proteins analyzed in order to ascertain influential pairs, triplets and quadruplets. This analysis was complemented with FBA which demonstrated that most of the identified combinations of proteins are also synthetic lethals in silico and may therefore be very good drug targets. Multi-target therapeutics can be more efficacious and less vulnerable to adaptive resistance because the biological system is less able to compensate for the action of two or more drugs simultaneously, and is of course the standard treatment for tuberculosis.
Another promising application of systems biology approaches in the drug discovery process is in the simulation of therapeutic intervention. By developing a combined model consisting of enzyme inhibition kinetics, metabolic simulation by FBA of iNj661 and cell growth dynamics, Fang et al. (2009) successfully simulated drug inhibition of M. tuberculosis. Two metabolic inhibitors 3-nitropropionate and 5′-O-(N-salicylsulfamoyl) adenosine which target the enzyme isocitrate lyase and salicyl-AMP ligase, respectively, were chosen to test the model as these have been identified as important targets for therapeutic intervention and experimental data are also available for these inhibitors.46 The validation analysis showed that the model had good predictivity at determining the experimental dose–response curve of both these drugs. A limitation to this study was that there was some overlap between the experimental data used for construction and validation of this model. However, this study demonstrates the successful integration of three different phenotypic components, a key strength of systems biology approaches.47
Future challenges
The current models of M. tuberculosis metabolism are heavily influenced by the genome annotation, which, as we have already discussed, is likely to have many errors and is incomplete in many pathways. Inconsistencies between model predictions and experimental data are already apparent and indicate that there is much that remains to be learnt about the metabolism of this important pathogen.A key question is how M. tuberculosis actually catabolises its substrates and makes biomass both in vitro and in vivo. Although clues can be gleaned from the methods described above even FBA-based methods are hugely underdetermined so a great number of flux solutions are usually compatible with available data. The most powerful currently available method for directly measuring internal fluxes is 13C-MFA (Fig. 4). In this method, an organism is grown with a 13C-labelled substrate. The label is incorporated into internal metabolites and products of central metabolism, such as the proteogenic amino acids. The positional labeling patterns (which carbon atoms are labeled) of the amino acids and/or metabolites (as determined by either mass spectrometry or NMR) are then used as additional constraints in MFA to solve the internal fluxes and thereby reconstruct the paths through central metabolism that the carbon took inside the cells (Fig. 4). 13C-MFA has already proved to be an invaluable tool in metabolic engineering and has been used to successfully identify novel or unusual pathways in bacteria48,49 and has enormous potential for studying the metabolism of M. tuberculosis. In particular 13C-MFA could be used to investigate the metabolism of M. tuberculosis under conditions relevant to the in vivo situation such as low oxygen or using fatty acids as carbon sources. Such experiments could help answer questions surrounding the operation of the TCA cycle and may also identify entirely novel or unusual pathways in operation during these conditions which maybe prone to inhibition by targeted chemotherapy.
Currently it is not possible to apply many of the tools of systems biology directly to examine the metabolism of M. tuberculosis in vivo due to the requirement for steady state growth conditions for stoichiometry-based modeling approaches (FBA and MFA) and the problems associated with separating the metabolism of the bacterium from that of the host. There are however developments being made in this area and it is expected that studying host–pathogen interactions on a systems scale will eventually be possible.49 The development of kinetic models would allow the application of modeling approaches to study of dynamic systems, such as infection, but, as discussed above, measuring parameters for such models remains a major challenge. For now, it appears that the best approach may be to utilize a combination of both in vitro and in vivo studies, utilizing the in vitro studies to develop mapping rules that can be applied to the more limited data available in vivo and thereby discover the answer to vitally important questions, such as what and how M. tuberculosis eats in vivo.
The principle challenge for systems biology of M. tuberculosis is to construct more complete models that integrate genome, transcriptome, proteome, metabalome, physiology and structure and interactions with the host. Such multilevel models are beyond current capabilities but there are already models of transcriptional networks38 that could be integrated with metabolic models. Also, metabolic models of human cells have been constructed7,50 which should soon allow the building of combined host–pathogen cellular models that incorporate at least metabolite exchange between pathogen and host cell. While fully integrated models of tuberculosis are still a very distant goal, systems biology efforts can (and are beginning to) have an enormous impact on both our understanding of the metabolic potential of M. tuberculosis and the success of the drug discovery process which could lead to significant advances in the treatment of tuberculosis.
References
- A. M. Feist, M. J. Herrgard, I. Thiele, J. L. Reed and B. O. Palsson, Reconstruction of biochemical networks in microorganisms, Nat. Rev. Microbiol., 2009, 7, 129–143 Search PubMed.
- D. Bumann, System-level analysis of Salmonella metabolism during infection, Curr. Opin. Microbiol., 2009, 12, 559–567 CrossRef CAS.
- M. Durot, P. Bourguignon and V. Schachter, Genome-scale models of bacterial metabolism: reconstruction and applications, FEMS Microbiol. Rev., 2008, 33, 164–190.
- K. Bettenbrock, S. Fischer, A. Kremling, K. Jahreis, T. Sauter and E. D. Gilles, A quantitative approach to catabolite repression in Escherichia coli, J. Biol. Chem., 2006, 281, 2578–2584 CAS.
- Y. K. Oh, B. O. Palsson, S. M. Park, C. H. Schilling and R. Mahadevan, Genome-scale reconstruction of metabolic network in Bacillus subtilis based on high-throughput phenotyping and gene essentiality data, J. Biol. Chem., 2007, 282, 28791–28799 CrossRef CAS.
- M. AbuOun, P. F. Suthers, G. I. Jones, B. R. Carter, M. P. Saunders, C. D. Maranas, M. J. Woodward and M. F. Anjum, Genome scale reconstruction of a Salmonella metabolic model, J. Biol. Chem., 2009, 284, 29480–29488 CrossRef CAS.
- N. C. Duarte, S. A. Becker, N. Jamshidi, I. Thiele, M. L. Mo, T. D. Vo, R. Srivas and B. O. Palsson, Global reconstruction of the human metabolic network based on genomic and bibliomic data, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1777–1782 CrossRef CAS.
- A. M. Feist and B. O. Palsson, The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli, Nat. Biotechnol., 2008, 26, 659–667 CrossRef CAS.
- M. G. Poolman, L. Miguet, L. J. Sweetlove and D. A. Fell, A genome-scale metabolic model of Arabidopsis and some of its properties, Plant Physiol., 2009, 151, 1570–1581 CrossRef CAS.
- M. A. Oberhardt, B. O. Palsson and J. A. Papin, Applications of genome-scale metabolic reconstructions, Mol. Syst. Biol., 2009, 5, 320.
- K. Raman and N. Chandra, Flux balance analysis of biological systems: applications and challenges, Briefings Bioinf., 2009, 10, 435–440 Search PubMed.
- E. P. Gianchandani, A. K. Chavali and J. A. Papin, The application of flux balance analysis in systems biology, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2009, 2, 372–382 Search PubMed.
- J. H. Park, K. H. Lee, T. Y. Kim and S. Y. Lee, Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7797–7802 CrossRef CAS.
- S. J. Lee, D. Y. Lee, T. Y. Kim, B. H. Kim, J. Lee and S. Y. Lee, Metabolic engineering of Escherichia coli for enhanced production of succinic acid, based on genome comparison and in silico gene knockout simulation, Appl. Environ. Microbiol., 2005, 71, 7880–7887 CrossRef CAS.
- J. D. McKinney, Z. Honer, E. Munoz, A. Miczak, B. Chen, W. T. Chan, D. Swenson, J. C. Sacchettini, W. R. Jacobs and D. G. Russell, Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase, Nature, 2000, 406, 735–738 CrossRef CAS.
- E. J. Munoz-Elias and J. D. McKinney, Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence, Nat. Med., 2005, 11, 638–644 CrossRef CAS.
- M. S. Glickman, J. S. Cox and W. R. Jacobs, A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis, Mol. Cell, 2000, 5, 717–727 CrossRef CAS.
- L. A. Keating, P. R. Wheeler, H. Mansoor, J. K. Inwald, J. Dale, R. G. Hewinson and S. V. Gordon, The pyruvate requirement of some members of the Mycobacterium tuberculosis complex is due to an inactive pyruvate kinase: implications for in vivo growth, Mol. Microbiol., 2005, 56, 163–174 CrossRef CAS.
- E. J. Munoz-Elias, A. M. Upton, J. Cherian and J. D. McKinney, Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence, Mol. Microbiol., 2006, 60, 1109–1122 CrossRef CAS.
- A. D. Baughn, S. J. Garforth, C. Vilchze and W. R. Jacobs, An anaerobic-type α-ketoglutarate ferredoxin oxidoreductase completes the oxidative tricarboxylic acid cycle of Mycobacterium tuberculosis, PLoS Pathog., 2009, 5, e1000662 Search PubMed.
- J. Tian, R. Bryk, M. Itoh, M. Suematsu and C. Nathan, Variant tricarboxylic acid cycle in Mycobacterium tuberculosis: identification of α-ketoglutarate decarboxylase, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10670–10675 CrossRef CAS.
- D. J. V. Beste, J. Peters, T. Hooper, C. Avignone-Rossa, M. E. Bushell and J. McFadden, Compiling a molecular inventory for Mycobacterium bovis BCG at two growth rates: evidence for growth rate-mediated regulation of ribosome biosynthesis and lipid metabolism, J. Bacteriol., 2005, 187, 1677–1684 CrossRef CAS.
- E. Eylert, V. Herrmann, M. Jules, N. Gillmaier, M. Lautner, C. Buchrieser, W. Eisenreich and K. Heuner, Isotopologue profiling of Legionella pneumophila, J. Biol. Chem., 2010, 285, 22232–22243 CrossRef CAS.
- A. Gotz, E. Eylert, W. Eisenreich and W. Goebel, Carbon metabolism of enterobacterial human pathogens growing in epithelial colorectal adenocarcinoma (caco-2) cells, PLoS One, 2010, 5, e10586 CrossRef.
- E. Eylert, J. Schar, S. Mertins, R. Stoll, A. Bacher, W. Goebel and W. Eisenreich, Carbon metabolism of Listeria monocytogenes growing inside macrophages, Mol. Microbiol., 2008, 69, 1008–1017 CrossRef CAS.
- K. Raman, P. Rajagopalan and N. Chandra, Flux balance analysis of mycolic acid pathway: targets for anti-tubercular drugs, PLoS Comput. Biol., 2005, 1, e46 Search PubMed.
- R. Schuetz, L. Kuepfer and U. Sauer, Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli, Mol. Syst. Biol., 2007, 3, 119.
- D. Beste, T. Hooper, G. Stewart, B. Bonde, C. Avignone-Rossa, M. Bushell, P. Wheeler, S. Klamt, A. Kierzek and J. McFadden, GSMN-TB a web-based genome-scale network model of Mycobacterium tuberculosis metabolism, GenomeBiology, 2007, 8, R89 CrossRef.
- N. Jamshidi and B. O. Palsson, Investigating the metabolic capabilities of Mycobacterium tuberculosis H37rV using the in silico strain iNj661 and proposing alternative drug targets, BMC Syst. Biol., 2007, 1, 26 CrossRef.
- C. M. Sassetti, D. H. Boyd and E. J. Rubin, Genes required for mycobacterial growth defined by high density mutagenesis, Mol. Microbiol., 2003, 48, 77–84 CrossRef CAS.
- W. Bishai, Lipid lunch for persistent pathogen, Nature, 2000, 406, 683–685 CrossRef CAS.
- C. V. Smith, V. Sharma and J. C. Sacchettini, TB drug discovery: addressing issues of persistence and resistance, Tuberculosis, 2004, 84, 45–55 CrossRef.
- D. J. V. Beste, M. Espasa, B. Bonde, A. M. Kierzek, G. R. Stewart and J. McFadden, The genetic requirements for fast and slow growth in mycobacteria, PLoS One, 2009, 4, e5349 CrossRef.
- K. R. Patil and J. Nielsen, Uncovering transcriptional regulation of metabolism by using metabolic network topology, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2685–2689 CrossRef CAS.
- B. K. Bonde, D. J. V. Beste, E. Laing, A. M. Kierzek and J. McFadden, Differential producibility analysis (DPA) of transcriptomic data in the context of genome scale metabolic reaction networks: deconstructing the metabolic response of M. tuberculosis to the host environment, PLoS Comput. Biol. Search PubMed , submitted.
- T. Shlomi, M. N. Cabili, M. J. Herrgard, B. O. Palsson and E. Ruppin, Network-based prediction of human tissue-specific metabolism, Nat. Biotechnol., 2008, 26, 1003–1010 CrossRef CAS.
- C. Colijn, A. Brandes, J. Zucker, D. S. Lun, B. Weiner, M. R. Farhat, T. Y. Cheng, D. B. Moody, M. Murray and J. E. Galagan, Interpreting expression data with metabolic flux models: predicting Mycobacterium tuberculosis mycolic acid production, PLoS Comput. Biol., 2009, 5, e1000489 Search PubMed.
- I. Thiele, N. Jamshidi, R. M. T. Fleming and B. O. Palsson, Genome-scale reconstruction of Escherichia coli's transcriptional and translational machinery: a knowledge base, its mathematical formulation, and its functional characterization, PLoS Comput. Biol., 2009, 5, e1000312 Search PubMed.
- N. Chandra, Computational systems approach for drug target discovery, Expert Opin. Drug Discovery, 2009, 4, 1221–1236 Search PubMed.
- K. Raman, K. Yeturu and N. Chandra, TargetTB: a target identification pipeline for Mycobacterium tuberculosis through an interactome, reactome and genome-scale structural analysis, BMC Syst. Biol., 2008, 2, 109.
- T. Y. Kim, H. U. Kim and S. Y. Lee, Metabolite-centric approaches for the discovery of antibacterials using genome-scale metabolic networks, Metab. Eng., 2010, 12, 105–111 CrossRef CAS.
- M. V. B. Dias, F. Ely, M. S. Palma, W. F. De Azevedo, L. A. Basso and D. S. Santos, Chorismate synthase: an attractive target for drug development against orphan diseases, Curr. Drug Targets, 2007, 8, 437–444 Search PubMed.
- F. Ely, J. Nunes, E. Schroeder, J. Frazzon, M. Palma, D. Santos and L. Basso, The Mycobacterium tuberculosis Rv2540c dna sequence encodes a bifunctional chorismate synthase, BMC Biochem., 2008, 9, 13 CrossRef.
- T. Parish and N. G. Stoker, The common aromatic amino acid biosynthesis pathway is essential in Mycobacterium tuberculosis, Microbiology, 2002, 148, 3069–3077 CAS.
- K. Raman, R. Vashisht and N. Chandra, Strategies for efficient disruption of metabolism in Mycobacterium tuberculosis from network analysis, Mol. BioSyst., 2009, 5, 1740–1751 RSC.
- X. Fang, A. Wallqvist and J. Reifman, A systems biology framework for modeling metabolic enzyme inhibition of Mycobacterium tuberculosis, BMC Syst. Biol., 2009, 3, 92 CrossRef.
- C. B. Milne, P.-J. Kim, J. A. Eddy and N. D. Price, Accomplishments in genome-scale in silico modeling for industrial and medical biotechnology, Biotechnol. J., 2009, 4, 1653–1670 Search PubMed.
- E. Fischer and U. Sauer, A novel metabolic cycle catalyzes glucose oxidation and anaplerosis in hungry Escherichia coli, J. Biol. Chem., 2003, 278, 46446–46451 CrossRef CAS.
- N. Zamboni, S. M. Fendt, M. Ruhl and U. Sauer, C-13-based metabolic flux analysis, Nat. Protoc., 2009, 4, 878–892 Search PubMed.
- M. L. Mo, N. Jamshidi and B. O. Palsson, A genome-scale, constraint-based approach to systems biology of human metabolism, Mol. BioSyst., 2007, 3, 598–603 RSC.
| This journal is © The Royal Society of Chemistry 2010 |