Rapid identification of orexin receptor binding ligands using cell-based screening accelerated with magnetic beads
Xin
Qi
,
John
Astle
and
Thomas
Kodadek
*
Departments of Chemistry & Cancer Biology, The Scripps Research Institute, Scripps Florida, 130 Scripps Way, #3A2, Jupiter, FL 33458, USA. E-mail: Kodadek@scripps.edu
First published on 29th September 2009
Abstract
We report here a simple and rapid method by which to screen one bead one compound libraries for highly specific ligands to cell surfaceproteins such as G protein-coupled receptors. This protocol, which harvests “hits” in a cell-based binding screen magnetically, eliminates the most tedious aspects of previously published bead screening techniques and allows millions of different compounds to be screened rapidly and cheaply. The method is demonstrated using the orexin receptor 1, which resulted in the isolation of moderate potency antagonists.
Introduction
Integral membrane proteins such as G protein-coupled receptors (GPCRs) constitute an important class of drug targets. Typically, agonists or antagonists of these proteins are discovered through high-throughput (HTP) screens in which thousands of compounds are tested for their ability to modulate receptor activity in some sort of functional experiment, such as a reporter geneassay . While effective, such screens are expensive and require significant infrastructure. Moreover, they leave open the question of the specificity of the “hits” for the receptor of interest. To address these limitations and to develop an approach with a potentially much higher throughput, an attractive alternative is to carry out binding assays in which cells carrying the receptor of interest are exposed to bead-displayed compounds from a one bead one compound (OBOC) library and beads that retain the receptor-expressing cells are identified and isolated.1,2 We recently reported a novel two-color assay of this sort that makes it particularly easy to segregate the desired receptor ligands from compounds that bind other cell surface molecules.3 This involves labeling cells that lack the target protein with green quantum dots that are internalized by endocytosis. The receptor of interest is then introduced into the same cell line and these cells are labeled with red quantum dots. The two cell lines , which differ only in the presence or absence of the target receptor, are then mixed together and exposed to hundreds of thousands of compounds displayed on hydrophilic beads. Specific ligands for the target receptor can be identified by the retention of red-labeled, but not green-labeled, cells on the much larger beads. We demonstrated the utility of this strategy by identifying five compounds in a library of more than 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 peptoids that proved to be highly specific ligands for the vascular endothelial growth factor receptor 2 (VEGFR2). This type of screen, which demands only highly specific binding to the target receptor, can be carried out quickly and cheaply. The few hits that arise from these assays are then tested for their ability to modulate receptor function without the need to develop highly robust HTP assays . In the VEGFR2 case, all five peptoids proved to be receptor antagonists and a dimeric derivative of one of the hits proved to be a potent inhibitor of angiogenesisin vivo.3
000 peptoids that proved to be highly specific ligands for the vascular endothelial growth factor receptor 2 (VEGFR2). This type of screen, which demands only highly specific binding to the target receptor, can be carried out quickly and cheaply. The few hits that arise from these assays are then tested for their ability to modulate receptor function without the need to develop highly robust HTP assays . In the VEGFR2 case, all five peptoids proved to be receptor antagonists and a dimeric derivative of one of the hits proved to be a potent inhibitor of angiogenesisin vivo.3
The major limitation of these types of assays is that the beads that bind the target cells are identified by visual inspection using a low power fluorescence microscope. This is feasible, though tedious, for up to a few hundred thousand beads, but clearly limits the application of the technique to much larger libraries or to the development of high-throughput efforts to target large numbers of GPCRs. Whereas some sort of flow sorting protocol could be employed to isolate the hits, the beads employed in the cell-based screening are much larger than cells and do not flow easily through standard fluorescence-activated cell sorters (unpublished observations).
Magnetic separation techniques are used commonly in molecular biology and have been applied in a few cases to sorting beads displaying chemical libraries.4 In this paper, we demonstrate that a magnetic isolation procedure can indeed be incorporated into our previously reported two-color cell-based screening protocol, providing a powerful, but rapid and inexpensive, method to screen large bead-displayed libraries for receptor agonists and antagonists.
Results
The orexin receptor 1 (OxR1), a GPCR involved in a number of interesting metabolic events and a potential drug target for insomnia, diabetes and drug addiction,5–16 was employed as the target receptor for these studies. Chinese hamster ovary (CHO) cells, which do not express OxR1, were labeled with Qtracker 565 (green) and CHO cells that overexpress OxR1 were labeled with Qtracker 655 (red). The cells were then mixed and exposed to about three million peptoid-displaying TentaGel beads from an OBOC library created by split and pool synthesis using the sub-monomer chemistry first described by Zuckermann and co-workers (see following Experimental section for a detailed protocol).17,18 The peptoid library had the general structure shown in Fig. 1 and was created with the amines also shown in Fig. 1, providing a theoretical diversity of one million compounds. After thorough washing, the beads were exposed to anti-OxR1 polyclonal antibody from rabbit and, after further washing, iron oxide particles (Dynabeads, Invitrogen) coated with sheep anti-rabbit IgG secondary antibody (Fig. 2). The suspension was then placed in a conical tube and, after gentle shaking, was placed in a holder that positioned a powerful magnet at the side of the tube. TentaGel beads that retained the magnetic Dynabeads were held there, while those that did not settled to the bottom of the tube and were removed with a pipette. This protocol should result in the magnetic retention of TentaGel beads that display peptoids able to bind to OxR1 in a way that does not compete with antibody binding (Fig. 2). After removal of all of the unmagnetized beads, those retained by the magnet were spread in a Petri dish and examined under a low power fluorescence microscope (Fig. 2c). This visual inspection was necessary because we observed that some beads that do not display OxR1-binding peptoids are physically trapped in the magnet-associated fraction. Of course, the number of beads analyzed at this point is a small fraction of the original library, so this step is quite straightforward. Moreover, beads that bind a significant amount of the green-stained cells and thus display either non-specific ligands or ligands against other cell surface targets other than the orexin receptor can easily be eliminated at this stage. Several beads were observed that bound only red cells (see Fig. 2c for an example). Two of these were picked using a micropipette and the sequences of the peptoids they displayed were determined by MALDI TOF-TOF mass spectrometry after release of the peptoid from the solid support by treatment with cyanogen bromide, which cleaves the peptoids from the beads adjacent to methionine residue (Fig. 1). To reconfirm that these peptoids are indeed true “hits”, these peptoids, called XQ1 and XQ2, were re-synthesized in bulk on TentaGel beads and the cell binding experiment was repeated with this new homogenous population of beads. After the beads were exposed to labeled cells that did or did not express OxR1, anti-OxR1 polyclonal antibody from rabbit and then Dynabeads coated with anti-rabbit secondary antibody, only red quantum dot-labeled cells (OxR1-expressing) bound to the beads (see Fig. 3b for beads displaying XQ1). A negative control experiment was performed using anti-VEGFR2antibody from rabbit in place of the anti-OxR1antibody. The TentaGel beads displaying XQ1 were no longer retained by the magnet (not shown).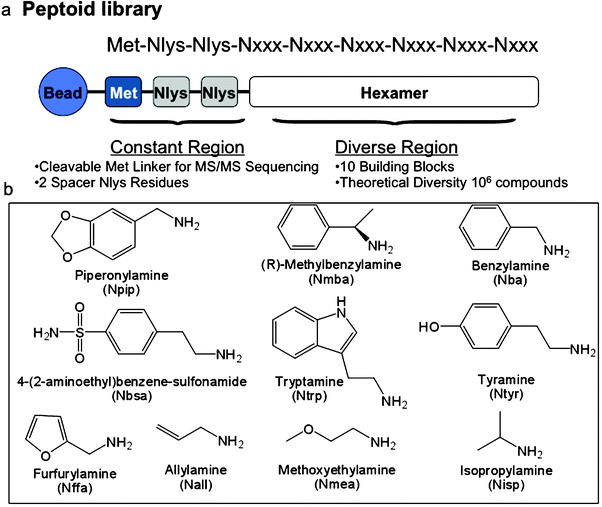 | ||
| Fig. 1 Structure of the peptoid library employed in the screen. (a) General structure of the compounds in the library, including three fixed residues at the C-terminus and the remaining six diversified residues. (b) The ten amines employed to make the library using sub-monomer synthesis.17 | ||
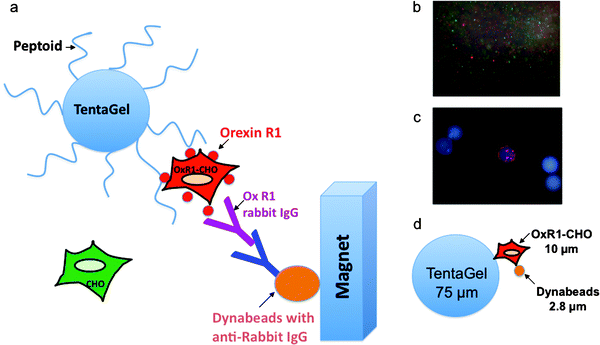 | ||
| Fig. 2 A two-color, cell-based magnetic screen to identify OxR1-binding ligands. (a) Schematic representation of the assay . This protocol results in the magnetic retention of the TentaGel beads that display peptoids able to bind to OxR1. (b) Cells labeled with quantum dots were visualized under a fluorescent microscope equipped with a DAPI filter to verify labeling efficiency. The small red circles are Qtracker 655-labeled OxR1-CHO cells and the green cells are Qtracker 565 labeled CHO parental cells. (c) Fluorescence microscopic image of select beads after magnetic screening (100× total magnification, DAPI filter). The bead observed to bind only red-stained cells represents one of our hits out of 3000000 beads. (d) Size comparison of TentaGel beads, living cells and Dynabeads. | ||
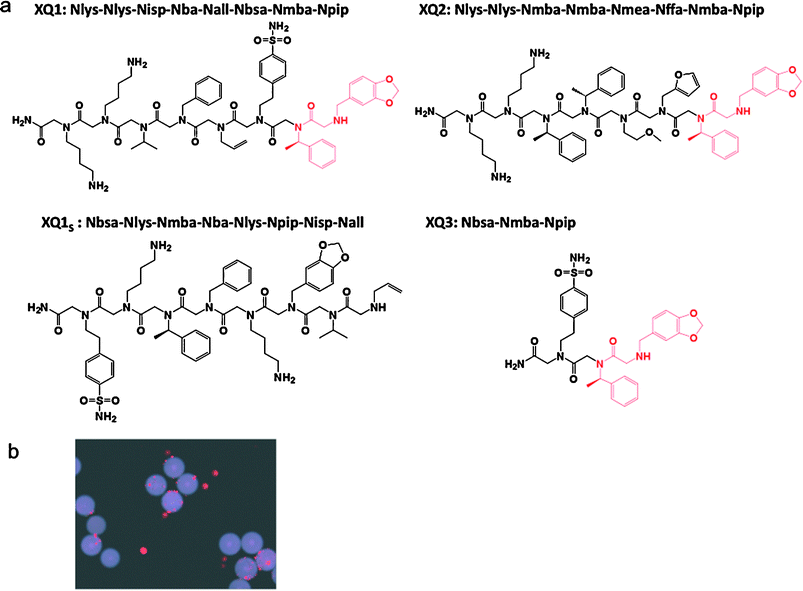 | ||
| Fig. 3 The hits identified from the screen and evaluation of their binding specificity. (a) Chemical structures of two of the hits isolated in the screen (XQ1 and XQ2) in soluble form as C-terminal primary amides. A scrambled version of the XQ1 sequence as XQ1s was synthesized as a negative control. A truncated derivative of XQ1, named as XQ3, was created that contains the putative pharmacophore. (b) Evaluation of the binding specificity of two of the hits identified from the screen. XQ1 and XQ2 were re-synthesized on TentaGel beads and incubated with OxR1–CHO and parental CHO cells labeled with red and green quantum dots, respectively. Only red-labeled cells bound to the beads as shown here for the beads displaying XQ1. | ||
We then proceeded to ask if these peptoids could modulate OxR1 function. In addition to XQ1 and XQ2, two other peptoids were included in these experiments. XQ1 and XQ2 have the same two N-terminal residues , suggesting that these might be critical for binding to OxR1. Therefore, a truncated derivative of XQ1, called XQ3, was created that contains these two residues, as well as one additional monomer C-terminal to the putative pharmacophore. As a negative control, a scrambled version of the XQ1 sequence was created. All of these peptoids were synthesized in soluble form as C-terminal primary amides and employed in cellular reporter geneassays (see Fig. 3a).
OxR1-expressing HEK293 cells co-transfected with an orexin-responsive firefly luciferase reporter plasmid as well as an orexin-unresponsive Renillaluciferase reporter gene were treated with peptoid compounds in the range of 26 nM to 10 mM for half an hour followed by addition of 300 nM orexin A for another six hours. After washing, luciferaseassays were conducted to characterize the level of expression of each reporter gene. These data (Fig. 4) showed that XQ1 and XQ2 are antagonists of OxR1, with modest potencies in the mid-μM range. XQ3 behaved similarly in this assay , confirming that only the N-terminal region of the peptoid is essential for activity (a more thorough analysis of the pharmacophore in other OxR1-antagonizing peptoids also supports this conclusion19). The scrambled control peptoid, XQ1s, had little or no effect on orexin-dependent luciferase reporter gene expression. These data show that the magnetic bead isolation protocol is capable of capturing even modest affinity hits in the primary screening step, probably due to multiple peptoid–receptor contacts at the bead–cell interface.
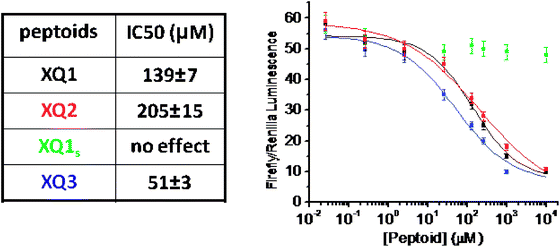 | ||
| Fig. 4 Dual-luciferase reporter assay to test the ability of the isolated peptoids to antagonize receptor function. OxR1-expressing HEK293 cells were co-transfected with an orexin-responsive firefly luciferase reporter plasmid p3CRE-Luc as well as an orexin-unresponsive Renilla luciferase reporter gene pRLuc. The cells were then treated with peptoid compounds in the range of 26 nM to 10 mM for half an hour followed by addition of 300 nM orexin A for another 6 hours. Activity of p3CRE-Luc was normalized to the activity of pRLuc transfection control and represented as relative luciferase activity in the ratio of Firefly–Renilla luminescence. | ||
In summary, we present here a simple protocol that significantly decreases the amount of time and effort required to screen very large one bead one compound libraries for integral membrane receptor ligands. Moreover, we characterized hit peptoid compounds that bind OxR1 and evaluated their potency for inhibition of OxR1 activity. The example reported here employed peptoid libraries, which have been shown previously to be a good source of receptor ligands,20–23 but this method would be applicable to any OBOC library where the identity of the compound can be determined directly or is encoded.24 Given the inexpensive and rapid nature of this screen, plus the fact that the source of the receptor target was merely an expression vector, one can imagine that this screening technology could be applied to isolation of many GPCR ligands in a relatively high-throughput fashion.
Experimental section
Peptoid library design and synthesis
A “one bead one compound” peptoid library was constructed by split and pool synthesis on TentaGel beads using methods described previously.17,18,25 A linker sequence including the amino acid methionine preceded the peptoid library, allowing compounds to be removed from the beads with cyanogen bromide post-screening to facilitate characterization by MALDI TOF-TOF sequencing. The two positively charged spacer Nlys residues repel the peptoids from each other on the bead surface and help avoid peptoid aggregation. Ten amines were used to construct the variable region. The theoretical diversity of the library was 106 (one million compounds) (Fig. 1). The 9-mer library was synthesized on TentaGel beads (75 μm, Rapp Polymere, HL 12902, capacity 0.49 mmol g−1, 4 × 106 beads per gram, 750 mg beads were used for screening 3000000 beads) using a microwave (1000 W) assisted synthesis protocol. TentaGel beads were swelled in dimethylformamide (DMF) in synthesis reaction vessels and were treated with Fmoc-Met-OH, HBTU and NMM followed by piperidine deprotection. The beads were then treated with 2 M bromoacetic acid and 2 M diisopropylcarbodiimide (DIC) in anhydrous DMF (2 M is starting reagent concentration). The reaction was performed in a microwave oven set to 10% power (2 × 15 s). After washing the beads with DMF, they were treated with primary amine (2 M) in the microwave oven as described above. The beads were washed and distributed equally into ten peptide synthesis reaction vessels after adding two Nlys spacers (mono-Boc-protected 1,4-diaminobutane). The coupling procedure was repeated until the desired length was achieved and a “one bead one compound” peptoid library was constructed by split and pool synthesis. After completion of the library synthesis, the beads were treated with a 95% TFA, 2.5% triisopropylsilane, and 2.5% water mixture for 2 h to remove side chain protecting groups and then were washed with dichloromethane, dried and stored at 4 °C.
On-bead cell binding assay for combinatorial peptoid library screening accelerated with magnetic beads
1. About 100000 beads with Met-Nlys-Nlys-(Nxxx)6 were equilibrated in 3% BSA containing DMEM media for 2 h in 1.5 ml microcentrifuge tubes (30 tubes for screening 3000000 beads).
2. HCRTR1-CHO-K1 DA cells and CHO parental cells (Invitrogen) were removed from culture plates by treating with 4 ml GIBCO cell dissociation buffer (enzyme free) each for 10 min at 37 °C. Cells were washed and resuspended in DMEM media.
3. The cells were counted and distributed at 1 × 106 cells per tube for each cell line in 1.5 ml microcentrifuge tubes, then labeled with Q-dots as follows: (1) pre-mix 1 μl each of Qtracker reagent A and B in a 1.5 ml microcentrifuge tube. Incubate for 5 min at room temperature. (2) Add 0.2 ml of fresh growth media to each tube and vortex for 30 s. (3) Add 1 × 106 cells in 800 μl of media to each tube containing the labeling solution and incubate at 37 °C for 45 min. (4) Wash the cells twice with DMEM media and resuspend the cells in each tube in 1 ml of 3% BSA-containing DMEM media. (5) HCRTR1-CHO-K1 DA cells labeled with Qtracker 655 evinced an intense red fluorescence and CHO parental cells labeled with Qtracker 565 evinced an intense green fluorescence when examined under the fluorescence microscope through a DAPI filter.
4. The incubation media was removed from the beads. To the beads in each tube, 1 ml of Qtracker 655 labeled HCRTR1-CHO-K1 DA cells and 1 ml of Qtracker 565 labeled CHO parental cells were added. Then the entire suspension was transferred to a 6 cm culture plate and incubated at 37 °C with gentle shaking at 60 rpm for 30 min.
5. Beads were washed gently twice with DMEM media and collected from the tissue culture plate, which should remove all non bead-bound cells. 10 μl of a solution (2 μg ml−1) of anti-orexin R1/2 antibody (obtained from Santa Cruz Biotechnology , sc-28936, rabbit polyclonal IgG, 200 μg ml−1) in 1 ml media. The solution was incubated for 1 h.
6. The beads were washed twice with DMEM media and added to Dynabeads M-280 sheep anti-rabbit IgG (50 μl pre-washed with media, cat. 112.03D, Invitrogen). This mixture was incubated for another 30 min. All of the beads were then pooled in a conical tube, which was placed in a holder that positioned a powerful magnet at the side of the tube. Any hit beads that retained the magnetic Dynabeads were held there at the side of the tube, while any “negative” beads will remain at the bottom of the tube. These were removed with a pipette. If desired, these “negatives” can be remixed with more magnetic beads and the procedure repeated to isolate hits that may have been missed in the first pass.
7. The beads retained on the side of the tube (several hundred) were transferred to a 6 cm culture plate in a small amount of buffer and examined under a fluorescent microscope (Olympus BX-51) using a DAPI filter (100× total magnification). We used an Olympus U-MNU2 microscope equipped with a DAPI filter (excitation 360–370 nm, emission 420 nm) and the beam splitter at 400 nm. Beads binding only to red-labeled cells were picked manually using a 20 μl pipette.
8. The “hit” beads were washed, then boiled in 1% SDS for 30 min to remove cells and any other bead-bound debris from the screening steps. Under a dissecting microscope, individual beads were then picked and transferred, one by one, into the wells of a 96-well plate. 20 μl of a cyanogen bromide solution (made by adding 30 mg of CNBr to 500 μl acetonitrile, 400 μl glacial acetic acid and 100 μl water) were then added to each well. The plates were covered and incubated overnight at room temperature. The plates were then uncovered and the solution allowed to evaporate. The cleaved molecules were dissolved in 20 μl of 1 : 1 CH3CN–H2O. MALDImass spectrometry-based sequencing was then carried out.
Re-synthesis of soluble peptoids
Resynthesis of peptoid ligands was conducted on Rink amide resin (Novabiochem). The general peptoid synthesis protocol as described in the library synthesis procedure was used to build the peptoid portion. After the synthesis, peptoids were cleaved off the resin using a 95% TFA, 2.5% triisopropylsilane, and 2.5% water mixture for 2 h and purified using HPLC. The purified peptoids were employed in cellular reporter geneassays .Dual-luciferase reporter geneassay
OXR1-expressing HEK293 cells were plated in DMEM with 1 mM D-glucose supplemented with 10% FBS, 2 mM glutamine, 100 U mL−1penicillin, and 100 μg mL−1streptomycin and were grown to 60% confluency. p3CRE-Luc was transiently co-transfected with pRLuc into these cells using lipofectamine (Invitrogen). After 24 h the cells were replaced in DMEM with 0.1% FBS and starved for 4 h. The cells were then treated with peptoid compounds in the range of 26 nM to 10 mM for half an hour followed by treatment with 300 nM orexin A for another 6 h. Cells were washed in PBS prior to dual luciferaseassay . Activity of p3CRE-Luc was normalized to the activity of pRLuc transfection control and represented as relative luciferase activity in the ratio of Firefly–Renilla luminescence.Acknowledgements
We thank Prof. Masashi Yanagisawa (University of Texas Southwestern) for providing HEK293 cells stably transfected with the orexin receptor 1. We also thank Dr Gomika Udugamsooriya and Dr Bettina Proneth for their helpful discussion. This work was supported by the National Institutes of Health (1 P01-DK58398)References
- O. H. Aina, T. C. Sroka, M. L. Chen and K. S. Lam, Biopolymers, 2002, 66, 184–199 CrossRef CAS.
- L. Peng, R. Liu, J. Marik, X. Wang, Y. Takada and K. S. Lam, Nat. Chem. Biol., 2006, 2, 381–389 CrossRef CAS.
- D. G. Udugamasooriya, S. P. Dineen, R. A. Brekken and T. Kodadek, J. Am. Chem. Soc., 2008, 130, 5744–5752 CrossRef CAS.
- B. H. Hu, M. R. Jones and P. B. Messersmith, Anal. Chem., 2007, 79, 7275–7285 CrossRef CAS.
- C. Boss, C. Brisbare-Roch and F. Jenck, J. Med. Chem., 2009, 52, 891–903 CrossRef CAS.
- C. Brisbare-Roch, J. Dingemanse, R. Koberstein, P. Hoever, H. Aissaoui, S. Flores, C. Mueller, O. Nayler, J. van Gerven, S. L. de Haas, P. Hess, C. B. Qiu, S. Buchmann, M. Scherz, T. Weller, W. Fischli, M. Clozel and F. Jenck, Nat. Med. (N. Y.), 2007, 13, 150–155 CrossRef CAS.
- R. M. Chemelli, J. T. Willie, C. M. Sinton, J. K. Elmquist, T. Scammell, C. Lee, J. A. Richardson, S. C. Williams, Y. M. Xiong, Y. Kisanuki, T. E. Fitch, M. Nakazato, R. E. Hammer, C. B. Saper and M. Yanagisawa, Cell (Cambridge, Mass.), 1999, 98, 437–451 CrossRef CAS.
- Y. Date, Y. Ueta, H. Yamashita, H. Yamaguchi, S. Matsukura, K. Kangawa, T. Sakurai, M. Yanagisawa and M. Nakazato, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 748–753 CrossRef CAS.
- H. Funato, A. L. Tsai, J. T. Willie, Y. Kisanuki, S. C. Williams, T. Sakurai and M. Yanagisawa, Cell Metab., 2009, 9, 64–76 CrossRef CAS.
- J. A. Hollander, Q. Lu, M. D. Cameron, T. M. Kamenecka and P. J. Kenny, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19480–19485 CrossRef CAS.
- M. Hungs and E. Mignot, Bioessays, 2001, 23, 397–408 CrossRef CAS.
- L. Lin, J. Faraco, R. Li, H. Kadotani, W. Rogers, X. Y. Lin, X. H. Qiu, P. J. de Jong, S. Nishino and E. Mignot, Cell (Cambridge, Mass.), 1999, 98, 365–376 CrossRef CAS.
- T. Sakurai, A. Amemiya, M. Ishii, I. Matsuzaki, R. M. Chemelli, H. Tanaka, S. C. Williams, J. A. Richardson, G. P. Kozlowski, S. Wilson, J. R. S. Arch, R. E. Buckingham, A. C. Haynes, S. A. Carr, R. S. Annan, D. E. McNulty, W.-S. Liu, J. A. Terrett, N. A. Elshourbagy, D. J. Bergsma and M. Yanagisawa, Cell (Cambridge, Mass.), 1998, 92, 573–585 CrossRef CAS.
- T. E. Scammell, Curr. Biol., 2001, 11, R769–R771 CrossRef CAS.
- D. Sikder and T. Kodadek, Genes Dev., 2007, 21, 2995–3005 CrossRef CAS.
- J. T. Willie, R. M. Chemelli, C. M. Sinton and M. Yanagisawa, Annu. Rev. Neurosci., 2001, 24, 429–458 CrossRef CAS.
- G. M. Figliozzi, R. Goldsmith, S. C. Ng, S. C. Banville and R. N. Zuckermann, Methods Enzymol., 1996, 267, 437–447 CAS.
- P. G. Alluri, M. M. Reddy, K. Bachhawat-Sikder, H. J. Olivos and T. Kodakek, J. Am. Chem. Soc., 2003, 125, 13995–14004 CrossRef CAS.
- J. Lee, et al., submitted.
- R. N. Zuckermann, E. J. Martin, D. C. Spellmeyer, G. B. Stauber, K. R. Shoemaker, J. M. Kerr, G. M. Figliozzi, D. A. Goff, M. A. Siani, R. J. Simon, S. C. Banville, E. G. Brown, L. Wang, L. S. Richter and W. H. Moos, J. Med. Chem., 1994, 37, 2678–2685 CrossRef CAS.
- C. Montoliu, M. Humet, J. J. Canales, J. Burda, R. Planells-Cases, F. Sanchez-Baeza, T. Carbonell, E. Perez-Paya, A. Messeguer, A. Ferrer-Montiel and V. Felipo, J. Pharmacol. Exp. Ther., 2002, 301, 29–36 CrossRef CAS.
- I. Masip, N. Cortes, M. J. Abad, M. Guardiola, E. Perez-Paya, J. Ferragut, A. Ferrer-Montiel and A. Messeguer, Bioorg. Med. Chem., 2005, 13, 1923–1929 CrossRef CAS.
- E. Valera, P. M. Fernandez-Salguero, R. Planells-Cases, A. Messeguer, W. Van Den Nest, C. Carreno, A. Ferrer-Montiel and J. M. Merino, Neuromol. Med., 2002, 2, 271–280 Search PubMed.
- R. Liu, J. Marik and K. S. Lam, J. Am. Chem. Soc., 2002, 124, 7678–7680 CrossRef CAS.
- H. J. Olivos, P. G. Alluri, M. M. Reddy, D. Saloney and T. Kodadek, Org. Lett., 2002, 4, 4057–4059 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |