 Open Access Article
Open Access ArticleMeta-analysis of genome regulation and expression variability across hundreds of environmental and genetic perturbations in fission yeast†‡
Vera
Pancaldi
,
Falk
Schubert
and
Jürg
Bähler
*
Department of Genetics, Evolution and Environment and UCL Cancer Institute, University College London, Darwin Building, Gower Street, London, UK WC1E 6BT. E-mail: j.bahler@ucl.ac.uk
First published on 15th December 2009
Abstract
Genome-wide gene expression is re-programmed in response to external or internal factors such as environmental stress or genetic mutation, respectively, or as a function of endogenous processes such as cell proliferation or differentiation. Here we integrate expression profiling data that have been collected by our laboratory since 2001 and that interrogate more than 900 different experimental conditions. We take advantage of this large data set to rank all genes based on their variability in gene expression across the different conditions. The most variable genes were enriched for functions such as stress response, carbohydrate metabolism and trans-membrane transport, and these genes were underrepresented for introns and tended to be close to telomeres. We then compared how overall gene regulation and variability of gene expression across conditions is affected by environmental or genetic perturbations, and by endogenous programmes. Meiotic differentiation and environmental perturbations led to substantially greater gene expression variability and overall regulation than did genetic perturbations and the transcriptional programme accompanying cell proliferation. We also used the integrated data to identify gene regulation modules using two different clustering approaches. Two major clusters, containing growth- and metabolism-related genes on one hand and stress- and differentiation-related genes on the other, were reciprocally regulated across conditions. We discuss these findings with respect to other recent reports on the regulation and evolution of gene expression.
Background
Genome-wide gene expression data from a wide range of biological conditions can be collected using DNA microarrays and other high-throughput approaches. To obtain insight into gene regulation, cells are often perturbed in different ways. These experimental perturbations can involve environmental factors such as various external stresses, toxins, drugs, or nutrient levels. Other useful perturbations involve intrinsic factors such as genome manipulations to delete, change, or overexpress specific genes. Alternatively, some endogenous gene expression programmes, including those driving cell proliferation or development, do not require any stimulus and can be studied under special conditions, e.g. after cell synchronization.We define gene regulation as the amount by which a gene is up- or down-regulated averaged over different conditions. With the word variability we indicate the width of the distribution of these values, expressed by the standard deviation. The two measures provide different information as a gene could have a high regulation, in the case where it is found to be always differentially expressed, while having low variability if the differential expression is always in the same direction and of similar amount. Little is known from a global perspective whether and how the overall regulation and variability of gene expression differ in response to environmental or genetic perturbations and during endogenous programmes.
In a pioneering study Hughes at al.,1 determined expression signatures in budding yeast in response to 300 mutations or chemical treatments, and this data compendium in turn allowed to predict the roles of uncharacterized perturbations and drug targets. These authors did not make a systematic comparison of any global differences in gene regulation between environmental or genetic perturbations, Luscombe et al.uncovered intriguing differences in transcriptional networks in budding yeast from two types of experiments: internal cell-cycle regulation, an endogenous programme, and response to different environmental perturbations, reflecting exogenous conditions.2 No data from genetic perturbations were used for this analysis. Recent studies indicate that genes up-regulated during stress show more variable expression, which reflects their promoter structure and could be advantageous under changing environments and in turn promote evolvability of gene regulation.3,4–8
Noisy gene expression can thus provide a driving force for phenotypic variation and evolutionary innovation and is itself subject to natural selection.3,9–16
Meta-analyses of genome-wide expression data can help to get the most from the huge amount of information available. Various clustering methods have been popular to tease out and visualize functional gene groups within microarray data sets.17–20
Simple correlation of gene expression has been applied to extract biologically meaningful information from gene expression data21 as well as to reverse-engineer genetic networks.22,23
Here we take advantage of microarray data sets from the fission yeast Schizosaccharomyces pombe that have been collected by our laboratory over the past eight years using a wide range of biological conditions. We explore how different types of perturbations (environmental vs. genetic) and endogenous programmes affect the variability and overall regulation of gene expression on a global scale. We also describe major regulatory clusters emerging from these data.
Results
Overview of experiments and conditions
The Bähler laboratory, in collaboration with many other groups, has collected a large amount of genome-wide expression data for fission yeast using the same custom-spotted DNA microarray platform.24 These data represent a wide range of experimental conditions, including global gene regulation in response to environmental stresses such as oxidants, heavy metals, heat shock, high osmolarity, ionizing radiation, changes in growth media such as iron, copper, and zinc levels, and exposure to drugs such as cisplatin, methylmethane sulfonate, and 3-aminotriazole.25–34Other experiments have interrogated intrinsic gene regulation programmes during cell proliferation and meiotic differentiation.35–38 Most of these data have been sampled using time courses to study the dynamic changes of gene expression during the different biological processes. In addition, many of these gene expression programmes have also been studied after different genetic perturbations such as deletion, mutation, or overexpression of genes. Gene expression signatures of numerous genetic perturbations have also been analyzed in steady-state conditions compared to wild-type cells.14,39–55 Furthermore, in the analyses below we also include several unpublished microarray data sets. This large collection of data is unique in that all the experiments were performed in the same laboratory using standardized conditions. In total, the data set encompasses 1272 microarray hybridizations (including replicate hybridizations), which provide 188 different steady-state experiments and 153 different timecourse experiments, encompassing 778 time points (Fig. 1). The raw microarray data are normalized as described in ref. 56 and gene expression levels are relative to wild-type reference samples.
 | ||
| Fig. 1 Microarray experiment summary showing the number of conditions in different experimental categories. | ||
Ranking and functional analysis of genes based on expression variability
We first ranked the S. pombe genes based on their variability in relative expression levels across conditions. For this analysis, the different time points of time course experiments were treated as different conditions, but the main results were validated including only a single time point for each time course. In some time course experiments, the first time point corresponds to untreated wild-type cells (identical to reference), and these time points were not used as they may bias the analysis of gene variability. The distributions of the logarithms of relative gene expression levels for single genes across conditions were uni-modal and close to normal (Fig. S1, ESI).‡ We therefore calculated the standard deviations of these distributions as a simple measure for the variability in gene expression. Supplementary Table S1 (ESI)‡ provides a list of genes ranked by their expression variability across conditions.The transcripts showing the most variable expression across conditions included a sequence orphan (SPAC23H3.15c), small heat-shock protein genes (hsp16 and hsp9), the metallothionein gene zym1,57 the uracil regulatable gene urg131 and thiamine-regulatable gene nmt1.58 Two non-coding transcripts, prl65 and prl44, also showed highly variable expression levels, although these transcripts were only present in a later version of our microarrays and were therefore measured in fewer conditions. The range of relative expression levels for these highly variable transcripts was between 3200-fold for urg1 and > 64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400-fold for SPAC23H3.15c. This value was found for a few late time points of the same experiment and it corresponds to the dynamic range of the microarray scanner, suggesting that the relative values recorded for this gene in those conditions are even lower than the sensitivity of the instrument. The second most variable transcript was that of nmt1 (>53000-fold). In comparison, the least variable transcripts only ranged in relative expression levels between 3- to 6-fold across all conditions. Among the least variable genes were ppb1, encoding the calcineurin phosphatase catalytic subunit,59ssr4, encoding a SWI/SNF and RSC complex subunit,53usp106 and usp107, encoding U1 snRNP-associated proteins, and vps45 and snx3, with likely functions in protein sorting and secretion.60
400-fold for SPAC23H3.15c. This value was found for a few late time points of the same experiment and it corresponds to the dynamic range of the microarray scanner, suggesting that the relative values recorded for this gene in those conditions are even lower than the sensitivity of the instrument. The second most variable transcript was that of nmt1 (>53000-fold). In comparison, the least variable transcripts only ranged in relative expression levels between 3- to 6-fold across all conditions. Among the least variable genes were ppb1, encoding the calcineurin phosphatase catalytic subunit,59ssr4, encoding a SWI/SNF and RSC complex subunit,53usp106 and usp107, encoding U1 snRNP-associated proteins, and vps45 and snx3, with likely functions in protein sorting and secretion.60
We next pulled out lists of the 500 most and 500 least variable genes, followed by examination for Gene Ontology (GO) enrichments.61 The most variable genes were mainly enriched for terms relating to response to stress or stimulus (p = 10−38), carbohydrate catabolic processes (p = 10−9) and transmembrane transport (p = 10−6). The least variable genes included terms relating to mRNA metabolic processes (p = 10−11), mitochondrial translation and organization (p = 10−6), intracellular protein transport (p = 10−5), vesicle mediated transport and protein localization (p = 10−3) (Supplementary Tables S2 and S3, ESI).‡
The top-500 genes were also analysed for enrichments with gene lists produced in different microarray experiments as well as for different properties of genes or proteins. Notably, the most variable genes were significantly closer to the telomeres (two-sided Wilcoxon rank sum test, p < 10−8) than would be expected by chance. This bias does not reflect any general correlation across the chromosomes between expression variability and distance from telomeres, but rather a specific enrichment of variable genes close to chromosome ends (data not shown). Gene clusters close to chromosome ends are induced in environmental conditions such as nitrogen starvation and may be regulated by chromatin remodelling.35,40
The telomeric regions might thus be ‘hotspots’ of variability in gene expression, which could promote cell survival under changing conditions. Moreover, the most variable genes were significantly under-enriched for introns (two-sided Wilcoxon rank sum test, p < 10−8). This observation is consistent with the finding that highly regulated genes are intron-poor, possibly reflecting selection against introns in genes whose expression levels need rapid adjustment to external or internal challenges.62 As expected, the Core Environmental Stress Response (CESR) genes were highly enriched amongst the most variable genes (p < 10−8), with approximately 35% of the 500 genes being up-regulated and 10% being down-regulated as part of the CESR. Both environmental and genetic perturbations frequently lead to activation of the CESR (e.g.ref. 25,40,63).
Environmental perturbations lead to stronger gene expression variability than genetic perturbations
We divided our microarray data set into experiments that applied environmental perturbations, genetic perturbations, or both perturbations simultaneously (Fig. 1). For this analysis, we separately analyzed the time course experiments interrogating the cell cycle or meiotic differentiation as they reflect endogenous programmes that can run independently of environmental or genetic perturbations. We included steady-state experiments with mutants of genes involved in these functions as these are treated simply as genetic perturbations. Fig. 2a shows a comparison of average gene regulation in response to genetic and environmental perturbations. Gene regulation between these two situations was correlated, although environmental conditions seemed to lead to overall stronger differences in relative expression levels, especially for up-regulated genes. The majority of the genes tended to be up-regulated rather than down-regulated, especially in the environmental perturbations. The variability in gene expression was also correlated between genetic and environmental perturbations, but the environmental perturbations produced more variability than the genetic perturbations (Fig. 2b). | ||
| Fig. 2 Comparison of gene regulation in response to environmental or genetic perturbations. (a) Plot of the mean regulation of genes (log2 of expression ratios) across all genetic and environmental perturbations (Spearman correlation = 0.69). Red: 121 genes that were manipulated for genetic perturbations; yellow: 11 genes that were manipulated for simultaneous environmental and genetic perturbations. (b) Plot of the variability of gene expression across conditions in response to environmental or genetic perturbations (Spearman correlation = 0.83). Coloured genes as in (a). Experiments including simultaneous environmental and genetic perturbations were excluded from the analysis in (a) and (b). (c) Comparison of standard deviation of all genes within one type of perturbation: genetic, environmental, or both. 50% of all conditions are included in the box (interquartile range), and whiskers extend to 1.5 times of it. (d) Same analysis as (c) but including only a single time point per time course experiment. | ||
A possible bias in assessing the variability of gene expression under genetic perturbations is represented by the limited number of genes that have been perturbed in total (121 genes). These genes have been chosen for their known or suggested roles in the biological processes of interest. Accordingly, they are enriched for GO terms related to regulatory mechanisms, chromatin modification, and transcriptional control. The mutated genes do not behave atypically in the scatter plots of Fig. 2, indicating that the total number of experiments in which these genes are not perturbed is sufficiently large so that the perturbed genes do not create any bias. Of the 121 perturbed genes, 11 genes were also used as genetic perturbations in combination with environmental perturbations. As a control, we have repeated parts of the analysis eliminating the conditions where mutants of these 11 genes are exposed to environmental perturbations and found no difference in the results (Supplementary Fig. S2, ESI).‡
Fig. 2c and 2d show gene variability within conditions belonging to the three perturbation types (genetic, non-genetic or both), using the distribution of the standard deviation of regulation over all genes for each perturbation. Whereas in Fig. 2c all the data from time courses was used in the analysis, Fig. 2d shows the results of including in the analysis only a single time point per experiment. The aim was to avoid possible biases due to the correlation between the different time points within each experiment. This analysis confirms that environmental perturbations cause a larger gene expression response than genetic ones.
The distribution of the log-ratios of transcript levels averaged over all conditions for each gene was approximately normal and centred around zero (Fig. 3a), the latter being a consequence of the normalization procedure applied to the data.56 This property allowed us to use the standard deviation of this distribution as a measure for the variability of gene expression under the different conditions. Environmental perturbations led to overall more consistent regulation, i.e. in the same direction, in gene expression across conditions than genetic perturbations (Fig. 3a), meaning that either genetic regulation triggered less regulation or it led to more random changes between conditions, resulting in an average regulation closer to zero for most genes. The differing standard deviations show that environmental conditions tended to affect many genes consistently across conditions (either up- or down-regulation), whereas genetic modifications produced less consistent regulation. Combining genetic and environmental perturbations reinforced this bias leading to a wider distribution. This effect was even more pronounced in meiotic differentiation, which led to effects similar to a combination of genetic and environmental perturbations. Cell cycle progression, however, induced little overall regulation.
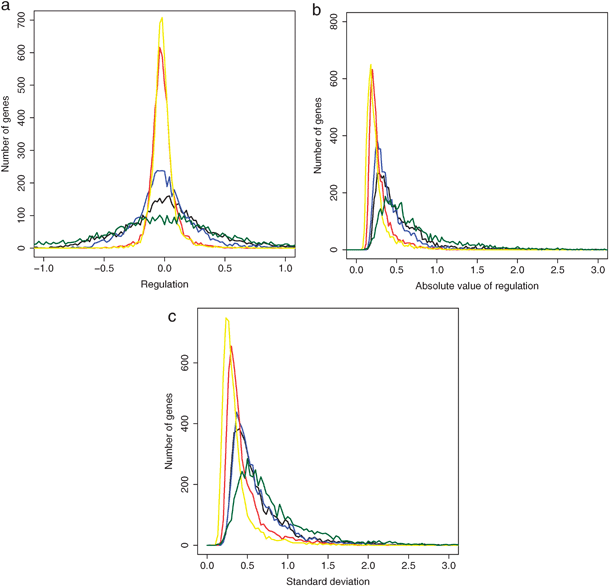 | ||
| Fig. 3 Comparison of different sets of conditions: genetic perturbations (red), environmental perturbations (blue), both environmental and genetic perturbations (black), cell cycle (yellow) and meiotic differentiation (green). (a) Distribution of average regulation over all conditions. (b) Distribution of average of absolute value of regulation over all conditions. (c) Distribution of the standard deviation of regulation over all conditions (gene variability). | ||
Fig. 3b shows the distribution of the average of the absolute value of regulation, revealing that the low regulation for genetic perturbations and cell cycle was due to generally low regulation and not to alternating large positive and negative values compensating each other. Environmental perturbations produced more regulation than genetic ones, while cell cycle progression produced little regulation and was similar to the genetic conditions. Combining genetic and environmental perturbations produced slightly more regulation than environmental perturbations alone. Finally, meiotic conditions produced the most regulation.
Fig. 3c shows the distribution of the standard deviation of gene regulation across conditions, reflecting gene expression variability. The environmental perturbations produced a greater variability than the genetic ones and meiotic differentiation produced the largest variability. Combining genetic and environmental perturbations produced a variability comparable to the environmental perturbations alone. When only the cell cycle progression was taken into account, the variability in regulation was slightly less than for genetic perturbations. The high variability induced by environmental perturbations was due to both an increase of single-gene variability (Fig. 3c) and to an increase in the number of genes that were regulated (Fig. 2a). Notably, the experiments interrogating the cell cycle, which reflect endogenous regulatory programmes, showed a similar low expression variability as did the genetic perturbations, and accordingly showed much less variability than the environmental perturbations. We conclude that environmental perturbations lead to stronger gene regulation and greater variability in gene expression across conditions than genetic perturbations or endogenous programmes. Meiosis, as an internal programme induced by external conditions, behaves more like a strong environmental perturbation.
Identification of gene regulation modules
We next applied clustering to identify biologically relevant gene regulation modules within our list of the top-500 most variable genes. Different results can be obtained depending on the clustering method. In some cases, clustering performs better when the number of expected clusters is decided a priori, as would be the case for methods such as k-means64 or Self Organizing Maps.65 However, the emphasis in our analysis was exploratory in nature so that the chosen clustering method did not require any assumptions on the structure of the data. We present here two types of clustering: a hierarchical clustering of the genes across conditions and a clustering of the gene correlation matrix. For each type, we clustered all conditions together and also separately for genetic and environmental perturbations.Fig. 4 shows a hierarchical clustering applied to the top-500 most variable genes, using all the conditions tested. The conditions were grouped by the main types of experiments. A GO term enrichment analysis was performed based on major gene clusters. Depending on the size of the clusters considered, different enrichments were obtained. A division between two main clusters was evident: one was enriched for stress (p < 10−5) and meiotic differentiation (p < 10−3), generally up-regulated genes, and the other for biosynthesis (p < 10−8) and metabolism (p < 10−2), generally down-regulated genes. We also notice how these two sets of genes are broadly regulated in opposite directions, reinforcing the idea that the stress response and maximal growth programmes are mutually exclusive in the cell.3 In the conditions tested, the perturbations are likely to stimulate stress response and limit cell growth, consistent with what is observed. More detailed cluster descriptions are provided in Fig. 4 and in Supplementary Table S5 (ESI).‡
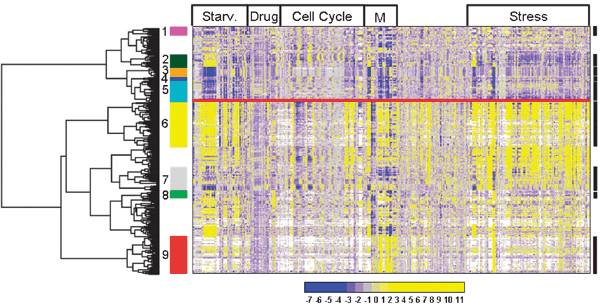 | ||
| Fig. 4 Hierarchical clustering of top-500 most variable genes including all microarray experiments. The colour legend shows the entire range of observed regulation (log2 of expression ratios). Selected GO categories that were enriched in clusters are highlighted as follows. Cluster 1 (magenta): iron related functions, cluster 2 (dark green): cytokinesis, cluster 3 (orange): translation, cluster 4 (blue): vitamins and thiamine, cluster 5 (cyan): metabolism, cluster 6 (yellow): stress response, cluster 7 (grey): protein folding, cluster 8 (green): conjugation, and cluster 9 (red): meiosis. The experimental conditions are divided in major groups as indicated on top: starvation, treatment with drugs, cell-cycle, meiosis and stress. The unassigned conditions are from various other experiments, mainly addressing chromatin modification, transcription, and mRNA decay. See Supplementary Table S5 (ESI)‡ for more details. The clusters with no number did not present any biological significant enrichment. | ||
The same procedure was carried out using only the genetic perturbations, only the environmental perturbations, or combined genetic and environmental perturbations (Supplementary Fig. S3, ESI).‡ Clusters enriched for similar GO terms were evident in all three cases, partially overlapping with what was found with the combined analysis of Fig. 4. Similarly, we observed two well separated, major clusters: genes involved in stress response and meiotic differentiation on one hand, and genes involved in metabolism and transport on the other. The different perturbations provided different information on the regulatory systems. In general, a higher number of conditions helped to produce richer and more detailed regulatory modules.
A second approach for the identification of biological modules involves clustering applied to the gene correlation matrix. For each gene pair among the top-500 most variable genes, the Pearson correlation over all the 956 experimental conditions was calculated. This value was taken as a measure of the ‘regulatory relatedness’ between different gene pairs. When clustering the matrix of all the gene-to-gene correlations, we grouped together the genes that showed similar correlation to all the other genes in the list, thus identifying gene clusters that showed similar correlation profiles (Fig. 5). By definition, all elements on the diagonal of the matrix are equal to unity, as they represent the correlation of a set of values with itself. Similarly, the appearance of bright yellow squares along the diagonal indicates clusters of genes with similar profiles across the experimental conditions. The dark red areas, on the other hand, show clusters of genes that are negatively correlated with each other, that is they tend to be regulated in opposite directions. As before, we included a GO enrichment analysis (Fig. 5). Again, we see a distinction between genes related to stress (p = <10−2) and meiotic differentiation (p < 10−4) opposed to biosynthesis (p < 10−7) and metabolism (p < 10−3). More detailed cluster descriptions are provided in Fig. 5 and in Supplementary Table S6 (ESI).‡
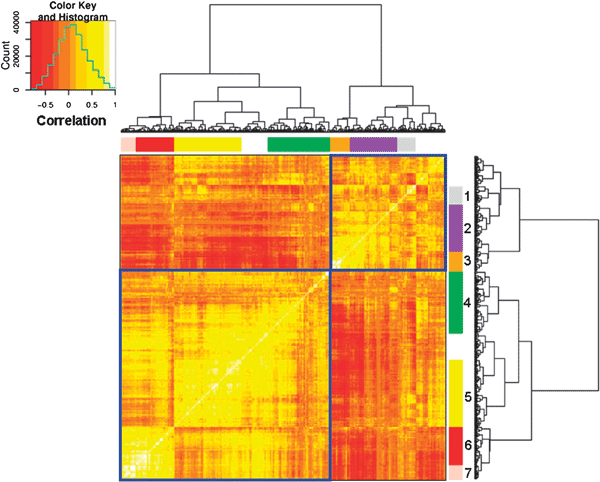 | ||
| Fig. 5 Hierarchical clustering of the correlation matrix of the top-500 most variable genes including all experimental data. Selected GO categories that were enriched in clusters are highlighted as follows. Cluster 1 (grey): protein folding, cluster 2 (purple): amino acid biosynthesis and nitrogen-related terms, cluster 3 (orange): translation, primary metabolism and biosynthesis, cluster 4 (green): conjugation, cluster 5 (yellow): response to stress, cluster 6 (red): meiosis and cell cycle, cluster 7 (pink): cell differentiation and sporulation. Note that the figure is symmetric on one diagonal, and the bright yellow squares reflect regulatory modules. The colour legend shows the correlation values from inverse (dark red) to positive (bright yellow) along with the distribution of the matrix values. The clusters with no number did not present any biological significant enrichment. See Supplementary Table S6 (ESI)‡ for more details. | ||
We also clustered genes separately using only genetic or environmental perturbations (Supplementary Fig. S4, ESI).‡
Combining all the available conditions in the calculation of the gene-to-gene correlations produced more clusters with clear enrichments for GO terms. The clusters obtained were similar to the ones obtained for hierarchical gene clustering. Many apparent regulatory modules were not enriched for GO terms, which could reflect connections between sets of genes that are not accurately covered by the GO ontology or heterogeneous in function. It would be interesting to investigate some of these unknown regulatory modules to further tease out the biological meaning of the structure of the correlation matrix.
Discussion
We present an overview of fission yeast gene regulation across more than 900 experimental conditions. The results suggest that environmental perturbations produce a larger variability in gene regulation between different conditions than genetic perturbations. The endogenous cell-cycle programme involves even less variation than genetic perturbations. The combination of external and genetic perturbations leads to stronger gene regulation, while the variability in gene expression is comparable to the purely environmental perturbations. The extent and variability in regulation are highest during meiotic differentiation, even higher than in combined genetic and environmental perturbations. Meiotic differentiation itself is triggered by a strong environmental stress (nitrogen starvation), followed by an endogenous programme that culminates in stress-resistant spores. Meiotic differentiation in yeast can therefore be considered as a sophisticated stress response. The similarity between the overall gene expression patterns during meiotic differentiation and stress responses is consistent with the suggestion that the stress response is a primordial process for the evolution of cellular differentiation.3Whereas in experiments involving environmental perturbations the cells are monitored as they are being exposed to the threat, in the case of a genetic perturbation the newly created strains have time to adapt to the new genetic condition over a few generations. A further assessment should be undertaken as soon as cells have undergone a genetic modification, to capture the transient gene expression response as was done for the environmental perturbations. We predict that this transient response would be much stronger, similar to responses to environmental perturbations. Hence, the large difference in gene regulation observed between genetic and environmental perturbations could reflect the difference in the experimental timing (transient response vs. steady-state after multiple generations) rather than the nature of the perturbations themselves.
However, we can explain these observations in the light of previous studies. Although genetic perturbations are clearly more deeply imposed on the organism, as they are inheritable, they seem to disrupt the cellular expression programme less than external factors. The sub-division between genetic and environmental perturbations analyzed here may be related to the distinction between endogenous and exogenous conditions introduced in ref. 2. These authors observe that two types of conditions elicit the activation of different parts of the regulatory network. When dealing with endogenous conditions, the response is based on a highly combinatorial control of multiple transcription factors that regulate few targets, creating a sub-network with high in-degrees, long path lengths and high cluster coefficients. In contrast, the response to exogenous conditions involves a sub-network with high out-degrees, short path lengths and low levels of clustering. Biologically, this difference might represent a rapid large-scale response to external perturbations opposed to a carefully coordinated rearrangement for internal programmes.
If we assume a parallel between endogenous conditions and genetic perturbations on one side with exogenous conditions and environmental perturbations on the other, we can compare the topological network features and the variability and regulation of gene expression measured here. The higher levels of variability in gene expression observed for environmental perturbations and meiotic differentiation partially reflect an increase in the number of genes being regulated. A faster and more extensive gene expression response, propagating through a less tightly controlled network, could explain such patterns. Meiotic differentiation leads to gene expression patterns more similar to a strong environmental perturbation than to an endogenous programme. Just like the conditions where genetic and environmental perturbations are combined, meiotic differentiation may combine both exogenous and endogenous aspects of the regulatory network, thus leading to strong changes in the expression programmes.
We speculate that there are two ways in which cells respond to challenges and threats to their survival. If the threat comes from an internal genetic perturbation that will endure over generations, the cells compensate and prepare a permanent adjustment of the regulatory network, helping them cope with the disruption in the long term. This adaptation, which may follow stronger short-term gene expression responses similar to those triggered by environmental perturbations, is probably optimized to involve only the minimal necessary changes, as the endogenous sub-network of the cell is bound to be tightly regulated. A drastic and permanent change in the expression programme would possibly jeopardize the state of dynamic equilibrium inside the cell, leading to compromised growth or even death. In the case of external challenges, for example in the form of potentially damaging changes to the cells’ environment, the response is immediate but transient to deal with the emergency. If the stress persists, however, global gene regulation will also adjust to new steady-state levels that are closer to, but distinct from the situation in unstressed cells.25,66 This steady-state condition may then be similar to cells living with a genetic perturbation.
Environmental challenges are likely to stimulate gene expression variability between single cells within the population through noisy gene regulation, which can promote survival of some cells that “get it right”.3,10,12,14 An interesting question is how these large rearrangements in gene expression are compatible with the observed high levels of robustness and cell survival. An analysis of the possible origins of this robustness from an evolutionary point of view is presented in ref. 15, where robustness is defined as how likely a system is to undergo random changes without impairment in its function. Two types of robustness are distinguished: robustness to genetic change such as random mutations in the genome, and robustness to non-genetic changes such as noise in cellular processes and changes in the environment.67Ref. 15 argues that robustness to genetic change is not an adaptation to genetic mutation but a secondary effect of acquired robustness to non-genetic changes, which are more ubiquitous and have stronger effects in the variation of phenotypes.68 Robustness against non-genetic change also increases fitness against genetic change and, importantly, it seems to be inheritable. It is therefore possible that cellular adaptation to internal noise and changing environmental conditions is at the origin of cells’ robustness to the genetic modifications. Although it has been suggested that noise has been selected against, possibly constraining the evolvability of gene expressionfor dosage sensitive genes,69 noise in development is seen as a fundamental factor in explaining the increase of organisms’ robustness through evolution.70
We ranked genes based on their expression variability across conditions. The most variable genes were related to stress response, meiotic differentiation, and metabolism. Two clustering approaches were used to identify biological modules within the most variable genes. In both cases, the GO term enrichment analysis highlights a general sub-division of the genome into two large gene clusters that are reciprocally regulated: genes related to stress response and meiotic differentiation on the one hand, and genes related to biosynthesis, metabolism, and translation on the other. This finding reflects the bipolar transcriptome (growth vs. stress response) that needs to balance between rapid cell proliferation, but relative stress sensitivity, or maximal stress resistance, but slow growth or quiescence.3–6,63,71,72
Ideally, the clusters obtained should be analyzed in a broader framework that is less restrictive than the GO categories. Recently, an alternative classification scheme was suggested where identified gene modules were found to be biologically highly relevant, although they were not sharing GO annotation.73 Some clusters, especially among the ones obtained by clustering the gene correlation matrix, remain biologically unexplained. This could either point to new biology which is not adequately covered by the current GO annotation, or to functionally heterogeneous regulatory modules.
One shortcoming of the presented approach is the assumption that time points within time course experiments can be treated as independent conditions. A more rigorous approach would entail measuring gene correlation through the time course in different experiments. Current work is being devoted to the development of an adaptation of the algorithm presented in ref. 74, which could be used to measure gene-to-gene correlation. Preliminary results show that this technique could identify targets of the same transcription factor.
To conclude, the method presented can be easily used to mine large sets of gene expression data or, as it becomes more and more available, to RNA-seq data75 and is immediately applicable to other organisms as well as to entire ecosystems.76
Experimental
Data preprocessing
Gene expression was measured with two-colour DNA microarrays for fission yeast cells in different experimental conditions. All experiments used isogenic control strains as a reference. The raw ratios of signals were normalised to wild type as described in ref. 56. The assumption behind this normalization step is that the raw values are normally distributed, which is true in most cases and widely assumed. The up- or down-regulation of genes is assessed as a relative measure compared to wild type. A better method to introduce an external control would have been to use spikes of known mRNA value in the arrays but this was not done for all of the arrays and hence could not be used across the whole dataset.For most time courses, all data is normalized to time point 0 of the isogenic reference; these time point 0 measurements were discarded as they were normalised to themselves yielding a high number of values equal to 1, which would create a bias for expression variability in the subdivision between genetic and non-genetic conditions. All the available biological replicates and dye swaps were averaged, a total of 1272 hybridizations. The logarithm of the normalized ratios was taken and no further scaling was performed. The data was then filtered to leave only experiments where more than 85% of the genes were measured and only genes that were measured in at least 80 conditions, eliminating zero time points in time courses, leaving 4939 genes and 956 conditions.
Genes ranking
The genes were ranked based on the standard deviation of the fold changes across conditions. The standard deviation was calculated for all genes (5166). The lists of most and least variable genes were analysed with the GO term finder,61 choosing the category biological process and a background distribution given by the whole genome. The range was also calculated for each gene, taking the difference between the maximum and minimum value of relative expression through all conditions. These two measures are both affected by the number of missing values in the data, which might affect the data for the genes that were only recently included in the arrays and therefore have measurements in fewer conditions. Whereas the range of expression values would be very high for genes that are particularly regulated in just a single condition, the standard deviation would consider highly variable only those genes that are considerably regulated in a large number of conditions. Thus we choose the standard deviation as a measure of variability.Global test
A pairwise global test77 was employed to check whether the gene expression patterns of the three subgroups (genetic, environmental, both) were significantly different (Fig. 2c,d). The analysis was repeated using only one time point per time course to ensure the statistical independence of the measurements. The test was performed in an implementation of R (version 2.90), based on a logistic model and 10000 permutations.
Comparison between genetic and environmental perturbations.
Frequency histograms were generated to show how many genes have a certain regulation value averaged over all conditions. Moreover, the distribution of the absolute regulation values for all genes was calculated. The statistics of the distributions in Fig. 3a are as follows (std = standard deviation): mesiosis, mean = 0.029, std = 0.61; genetic and environmental: mean = 0.065, std = 0.43; environmental: mean = 0.008, std = 0.25; genetic: mean = −0.006, std = 0.11; cell cycle: mean = −0.006, std = 0.10. The statistics of the distributions in Fig. 3b are as follows: mesiosis, mean = 0.72, std = 0.45; genetic and environmental: mean = 0.53, std = 0.33; environmental: mean = 0.43, std = 0.22; genetic: mean = 0.29, std = 0.15; cell cycle: mean = 0.26, std = 0.15. The statistics of the distributions in Fig. 3c are as follows: mesiosis, mean = 0.82, std = 0.48; genetic and environmental: mean = 0.65, std = 0.37; environmental: mean = 0.62, std = 0.33; genetic: mean = 0.47, std = 0.25; cell cycle: mean = 0.37, std = 0.22.Biological modules
The Agnes (agglomerative hierarchical clustering) function was used in R to perform hierarchical clustering of the data.78 The Euclidean distance and the Ward method were applied. The Ward method minimizes the sum of squares of two clusters at each step of the clustering procedure.79 Clustering was first applied to the 500 most variable genes. The lists of genes in each cluster were analysed with the GO term finder described in ref. 61 using the list of top-500 genes as a background distribution. Clustering was also performed after separating genetic and environmental conditions.Pearson correlation
Pearson correlation was calculated across all the experimental conditions (956) considering each time point in time course experiments as a separate condition. The values were stored in a correlation matrix. It must be noted that for this analysis all the conditions were merged, including steady-state and time course time points. This raises the issue of applying the Pearson correlation to time course data. The statistical assumptions underlying this method, namely the independence of the time points as single conditions, are not satisfied in this case. Aware of this methodological problem, we seek to investigate whether making the simplifying assumption can lead to biologically interesting results.Although there are more advanced methods to treat this type of data,80 we decided for now to make this simplifying assumption, and we plan to address the issue in further work.
Clustering of the correlation matrix
The symmetric correlation matrix of the top-500 most variable genes, where correlation is measured by Pearson correlation, was clustered using the Euclidean distance measure and the Ward method as above.Acknowledgements
The authors would like to acknowledge the contribution of Amy Li, Sergei Manakov, Oliver Stegle, Valerie Wood for help with data integration and analysis, and Luis López-Maury for providing unpublished data. We also thank Samuel Marguerat, Luis López-Maury, Martin Převorovský and two anonymous referees for comments on the manuscript. This research has been funded by Cancer Research UK grant number C9546/A6517.Notes and references
- T. R. Hughes, M. J. Marton, A. R. Jones, C. J. Roberts, R. Stoughton, C. D. Armour, H. A. Bennett, E. Coffey, H. Dai, Y. D. He, M. J. Kidd, A. M. King, M. R. Meyer, D. Slade, P. Y. Lum, S. B. Stepaniants, D. D. Shoemaker, D. Gachotte, K. Chakraburtty, J. Simon, M. Bard and S. H. Friend, Cell, 2000, 102, 109–126 CrossRef CAS.
- N. M. Luscombe, M. M. Babu, H. Yu, M. Snyder, S. A. Teichmann and M. Gerstein, Nature, 2004, 431, 308–312 CrossRef CAS.
- L. Lopez-Maury, S. Marguerat and J. Bähler, Nat. Rev. Genet., 2008, 9, 583–593 CrossRef CAS.
- S. Stern, T. Dror, E. Stolovicki, N. Brenner and E. Braun, Mol. Syst. Biol., 2007, 3, 106.
- A. D. Basehoar, S. J. Zanton and B. F. Pugh, Cell, 2004, 116, 699–709 CrossRef CAS.
- S. J. Zanton and B. F. Pugh, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16843–16848 CrossRef CAS.
- W. J. Blake, M. Kaern, C. R. Cantor and J. J. Collins, Nature, 2003, 422, 633–637 CrossRef CAS.
- W. J. Blake, G. Balázsi, M. A. Kohanski, F. J. Isaacs, K. F. Murphy, Y. Kuang, C. R. Cantor, D. R. Walt and J. J. Collins, Mol. Cell, 2006, 24, 853–865 CrossRef CAS.
- J. Gerhart and M. Kirschner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8582–8589 CrossRef CAS.
- N. Barkai and B. Z. Shilo, Mol. Cell, 2007, 28, 755–760 CrossRef CAS.
- J. M. Raser and E. K. O’Shea, Science, 2004, 304, 1811–1814 CrossRef CAS.
- C. R. Landry, B. Lemos, S. A. Rifkin and Dickinson, Science, 2007, 118 Search PubMed.
- H. B. Fraser, A. E. Hirsh, G. Giaever, J. Kumm and M. B. Eisen, PLOS Biology, 2004, 2, e137 CrossRef.
- N. Maheshri, M. Schonbrun, D. Laor, L. López-Maury, J. Bähler, M. Kupiec and R. Weisman, Mol. Cell Biol., 2009 Search PubMed.
- A. Wagner, Robustness and Evolvability in Living Systems Andreas Wagner, 2005 Search PubMed.
- M. Kaern, T. C. Elston, W. J. Blake and J. J. Collins, Nat. Rev. Genet., 2005, 6, 451–464 CrossRef CAS.
- S. Datta and S. Datta, Bioinformatics, 2003, 19, 459–466 CrossRef CAS.
- P. D’haeseleer, Nat. Biotechnol., 2005, 23, 1499 CrossRef CAS.
- A. Prelic, S. Bleuler, P. Zimmermann, A. Wille, P. Bühlmann, W. Gruissem, L. Hennig, L. Thiele and E. Zitzler, Bioinformatics, 2006, 22, 1122–1129 CAS.
- J. Tamames, D. Clark, J. Herrero, J. Dopazo, C. Blaschke, J. M. Fernandez, J. C. Oliveros and A. Valencia, J. Biotechnol., 2002, 98, 269–283 CrossRef CAS.
- A. Almudevar, L. B. Klebanov, X. Qiu, P. Salzman and A. Y. Yakovlev, Journal of American Society for Experimental Neuro Therapeutics, 2006, 3, 384–395 Search PubMed.
- M. Bansal, V. Belcastro, A. Ambesi-Impiombato and D. di Bernardo, Mol. Syst. Biol., 2007, 3 Search PubMed.
- A. V. Werhli, M. Grzegorczyk and D. Husmeier, Bioinformatics, 2006, 22, 2523–2531 CrossRef CAS Epub 2006 Jul 2514.
- R. Lyne, G. Burns, J. Mata, C. J. Penkett, G. Rustici, D. Chen, C. Langford, D. Vetrie and J. Bähler, BMC Genomics, 2003, 4, 27 CrossRef.
- D. Chen, W. M. Toone, J. Mata, R. Lyne, G. Burns, K. Kivinen, A. Brazma, N. Jones and J. Bähler, Mol. Biol. Cell, 2003, 14, 214–229 CrossRef CAS.
- D. Chen, C. R. Wilkinson, S. Watt, C. J. Penkett, W. M. Toone, N. Jones and J. Bähler, Mol. Biol. Cell, 2008, 19, 308–317 CAS.
- A. Watson, J. Mata, J. Bähler, A. Carr and T. Humphrey, Mol. Biol. Cell, 2004, 15, 851–860 CAS.
- L. Gatti, D. Chen, G. L. Beretta, G. Rustici, N. Carenini, E. Corna, D. Colangelo, F. Zunino, J. Bähler and P. Perego, Cell Mol. Life Sci., 2004 Search PubMed; L. Gatti, D. Chen, G. L. Beretta, G. Rustici, N. Carenini, E. Corna, D. Colangelo, F. Zunino, J. Bähler and P. Perego, Cell Mol. Life Sci., 2004, 61, 2253–2263 CAS.
- M. A. Rodríguez-Gabriel, G. Burns, W. H. McDonald, V. Martin, J. R. Yates, III, J. Bähler and P. Russell, Embo. J., 2003, 22, 6256–6266 CrossRef CAS.
- G. Rustici, H. van Bakel, D. Lackner, F. Holstege, C. Wijmenga, J. Bähler and A. Brazma, GenomeBiology, 2007, 8, R73 CrossRef.
- S. Watt, J. Mata, L. López-Maury, S. Marguerat, G. Burns and J. Bähler, PLoS ONE, 2008, 3, e1428 CrossRef.
- W. Reiter, S. Watt, D. Dawson, C. L. Lawrence and J. Bähler, J. Biol. Chem., 2008, 283, 9945–9956 CrossRef CAS.
- T. Udagawa, N. Nemoto, C. R. M. Wilkinson, J. Narashimhan, L. Jiang, S. Watt, A. Zook, N. Jones, R. C. Wek, J. Bähler and K. Asano, J. Biol. Chem., 2008, 283, 22063–22075 CrossRef CAS.
- S. J. Dainty, C. A. Kennedy, S. Watt, J. Bähler and S. Whitehall, Eukaryotic Cell, 2008, 7, 454–464 CrossRef CAS.
- J. Mata, R. Lyne, G. Burns and J. Bähler, Nat. Genet., 2002, 32, 143–147 CrossRef CAS.
- G. Rustici, J. Mata, K. Kivinen, P. Lio, C. J. Penkett, G. Burns, J. Hayles, A. Brazma, P. Nurse and J. Bähler, Nat. Genet., 2004, 36, 809–817 CrossRef CAS.
- J. Mata and J. Bähler, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15517–15522 CrossRef CAS.
- J. Mata, A. Wilbrey and J. Bähler, GenomeBiology, 2007, 8, R217 CrossRef.
- S. L. Sanders, M. Portoso, J. Mata, J. Bähler, R. C. Allshire and T. Kouzarides, Cell Mol. Life Sci., 2004, 119, 603–614 CAS.
- K. R. Hansen, G. Burns, J. Mata, T. A. Volpe, R. A. Mrtienssen, J. Bähler and G. Thon, Mol. Cell. Biol., 2005, 25, 590–601 CrossRef CAS.
- J. G. Mandell, J. Bähler, T. A. Volpe, R. A. Martienssen and T. R. Cech, GenomeBiology, 2005, 6, R1.
- C. Harrison, S. Katayama, S. Dhut, D. Chen, N. Jones, J. Bähler and T. Toda, EMBO J., 2005, 24, 599–610 CrossRef CAS.
- K. M. Lee, I. Miklos, H. Du, S. Watt, Z. Szilagyi, J. E. Saiz, R. Madabushi, C. J. Penkett, M. Sipiczki, J. Bähler and R. P. Fisher, Mol. Biol. Cell, 2005, 16, 2734–2745 CrossRef CAS.
- C. C. Jenkins, J. Mata, R. F. Crane, B. Thomas, A. Akoulitchev, J. Bähler and C. J. Norbury, Eukaryotic Cell, 2005, 4, 1840–1850 CrossRef CAS.
- F. Bachand, D. H. Lackner, J. Bähler and P. A. Silver, Mol. Cell. Biol., 2006, 26, 1731–1742 CrossRef CAS.
- V. Martin, M. A. Rodríguez-Gabriel, W. H. Mc Donald, S. Watt, J. R. Yates III, J. Bähler and P. Russell, Mol. Biol. Cell, 2006, 17, 1176–1183 CAS.
- M. A. Rodríguez-Gabriel, S. Watt, J. Bähler and P. Russell, Mol. Cell. Biol., 2006, 26, 6347–6356 CrossRef CAS.
- N. Sharma, S. Marguerat, S. Mehta, S. Watt and J. Bähler, Mol. Genet. Genomics, 2006, 276, 545–554 CrossRef CAS.
- M. Gordon, D. Holt, A. Panigrahi, B. Wilhelm, H. Erdjument-Bromage, P. Tempst, J. Bähler and B. Cairns, Mol. Cell. Biol., 2007, 27, 4058–4069 CrossRef CAS.
- I. Miklos, Z. Szilagyi, S. Watt, E. Zilahi, G. Batta, Z. Antunovics, K. Enczi, J. Bähler and M. Sipiczki, Mol. Genet. Genomics, 2008, 279, 225–238 CrossRef CAS.
- S. W. Wang, A. L. Stevenson, S. E. Kearsey, S. Watt and J. Bähler, Mol. Cell. Biol., 2008, 28, 656–665 CrossRef CAS.
- A. Mercier, S. Watt, J. Bähler and S. Labbe, Eukaryotic Cell, 2008, 7, 493–508 CrossRef CAS.
- B. J. Monahan, J. Villén, S. Marguerat, J. Bähler, S. P. Gygi and F. Winston, Nat. Struct. Mol. Biol., 2008, 15, 873–880 CrossRef CAS.
- A. Anders, S. Watt, J. Bähler and K. E. Sawin, Yeast, 2008, 25, 913–925 CrossRef CAS.
- D. Helmlinger, S. Marguerat, J. Villén, S. P. Gygi, J. Bähler and F. Winston, Genes Dev., 2008, 22, 3184–3195 CrossRef CAS.
- R. Lyne, G. Burns, J. Mata, C. J. Penkett, G. Rustici, D. Chen, C. Langford, D. Vetrie and J. Bähler, BMC Genomics, 2003, 4, 27 CrossRef.
- G. P. M. Borrelly, M. D. Harrison, A. K. Robinson, S. G. Cox, N. J. Robinson and S. J. Whitehall, J. Biol. Chem., 2002, 277, 30394–30400 CrossRef CAS.
- K. Maundrell, The Journal of Biological Chemistry, 1990, 265, 10857–10864 Search PubMed.
- T. Yoshida, T. Toda and M. Yanagida, Journal of Cell Science, 1994, 107, 1725–1735 CAS.
- M. Miyatake, T. Kuno, A. Kita, K. Katsura, K. Takegawa, S. Uno, T. Nabata and R. Sugiura, Genetics, 2007, 175, 1695–1705 CrossRef CAS.
- E. I. Boyle, S. Weng, J. Gollub, H. Jin, D. Botstein, J. M. Cherry and G. Sherlock, Bioinformatics, 2004, 20, 3710–3715 CrossRef CAS.
- D. J. Jeffares, C. J. Penkett and J. Bähler, Trends Genet., 2008, 8, 375–378 CrossRef.
- A. P. Gasch, Yeast, 2007, 11, 961–976 CrossRef.
- S. Tavazoie, J. D. Hughes, M. J. Campbell, R. J. Cho and G. M. Church, Nat. Genet., 1999, 22, 281–285 CrossRef CAS.
- P. Tamayo, D. Slonim, J. Mesirov, Q. Zhu, S. Kitareewan, E. Dmitrovsky, E. S. Lander and T. R. Golub, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2907–2912 CrossRef CAS.
- A. P. Gasch, P. T. Spellman, C. M. Kao, O. Carmel-Harel, M. B. Eisen, G. Storz, D. Botstein and P. O. Brown, Mol. Biol. Cell, 2000, 11, 4241–4257 CAS.
- S. Ciliberti, O. C. Martin and A. Wagner, PLoS Comput. Biol., 2007, 3, e15 Search PubMed.
- M. Lynch, Genet. Res., 1988, 51, 137–148 CrossRef CAS.
- B. Lehner, Mol. Syst. Biol., 2008, 4 Search PubMed.
- K. Kaneko, PLoS ONE, 2007, 2, e434 CrossRef.
- W. J. Blake, I. Tirosh, A. Weinberger, M. Carmi and N. Barkai, Nat. Genet., 2006, 38, 830–834 CrossRef.
- M. J. Brauer, C. Huttenhower, E. M. Airoldi, R. Rosenstein, J. Matese, D. Gresham, V. M. Boer, O. G. Troyanskaya and D. Botstein, Mol. Biol. Cell, 2008, 19, 352–367 CAS.
- D. Dotan-Cohen, S. Ltovsky, A. A. Melkman and S. Kasif, PLoS ONE, 2009, 4, e5313 CrossRef.
- O. Stegle, K. Denby, D. L. Wild, Z. Ghahramani and K. M. Borwardt, Lecture Notes in Computer Science (RECOMB), 2009 Search PubMed.
- S. Marguerat and J. Bähler, Cell Mol. Life Sci., 2009 Search PubMed.
- J. Raes and P. Bork, Nat. Rev. Microbiol., 2008, 6, 693–699 Search PubMed.
- J. J. Goeman, S. A. van de Geer, F. de Kort and H. C. van Houwelingen, Bioinformatics, 2004, 20, 93–99 CrossRef CAS.
- J. A. Hartigan, Clustering Algorithms, Wiley and Sons, 1975 Search PubMed.
- J. H. Ward, J. Am. Stat. Assoc., 1963, 58, 236–244.
- Z. Bar-Joseph, Bioinformatics, 2004, 20, 2493–2503 CrossRef CAS.
Footnotes |
| † This article is part of a Molecular BioSystems themed issue on Computational and Systems Biology. |
| ‡ Electronic supplementary information (ESI) available: Supplementary Figures S1–S4 and Tables S1–S6. See DOI: 10.1039/b913876p |
| This journal is © The Royal Society of Chemistry 2010 |