Of proteins and DNA—proteomic role in the field of chromatin research
Jean-Philippe
Lambert
,
Kristin
Baetz
* and
Daniel
Figeys
*
Ottawa Institute of Systems Biology (OISB), University of Ottawa and Department of Biochemistry, Microbiology and Immunology, University of Ottawa, 451 Smyth Road, Ottawa, Ontario, Canada K1H 8M5. E-mail: dfigeys@uottawa.ca; kbaetz@uottawa.ca
First published on 3rd September 2009
Abstract
To paraphrase Robert Burns’s poem To a Mouse, the best laid schemes of DNA–protein complexpurification often go awry. Chromatin with its heterogeneous and dynamic protein composition remains difficult to analyze. Still critical progress has been made in recent years in characterizing the interface between DNA and proteins due, in part, to significant advances in proteomic technologies. Proteomics has progressed to a point where affinity purification of soluble complexes and protein identification by mass spectrometry are routine. The new challenge for chromatin proteomics lies in studying proteins and protein complexes in their native environment, which is on chromatin. These novel types of data represent an additional layer of information that can be used to better characterize and understand cellular processes. This review will focus on the past contributions as well as on emerging mass spectrometry-based methodologies attempting to better define the complex relationship between proteins, protein complexes and DNA.
 Jean-Philippe Lambert | Jean-Philippe Lambert completed his Honours BSc degree in biochemistry at Concordia University. Jean-Philippe is currently a PhD candidate at the University of Ottawa in biochemistry and is a member of the Ottawa Institute of Systems Biology. His current research interests are in the characterization of chromatin in budding yeast using novel proteomics tools and mass spectrometry. |
 Kristin Baetz | Kristin Baetz is a professor in the Department of Biochemistry, Microbiology and Immunology, a member of the Ottawa Institute of Systems Biology and a Canada Research Chair in Chemical and Functional Genomics. Kristin obtained her PhD in Molecular Genetics at the University of Toronto, and did her postdoctoral studies at the University of British Columbia. Her laboratory explores how chromatin binding proteins, such as transcription factors and acetyltransferases, are regulating chromosome stability. |
 Daniel Figeys | Daniel Figeys is a professor in the Department of Biochemistry, Microbiology and Immunology, the Director of the Ottawa Institute of Systems Biology, and a Tier-1 Canada Research Chair in proteomics and systems biology. Daniel obtained a BSc and a MSc in chemistry from the Université de Montréal. He obtained a PhD in chemistry from the University of Alberta and did his postdoctoral studies at the University of Washington. Prior to his current position, Daniel was Senior VP of Systems Biology with MDS-Proteomics. From 1998 to 2000, he was a Research Officer at the NRC-Canada. Daniel’s research involves developing proteomics technology and their applications in systems biology. |
Introduction to chromatin
Proper maintenance and regulation of an organism’s genetic material are essential for its survival. It is thus unsurprising that all organisms possess a wide array of redundant mechanisms to not only prevent damage and mutations to their genetic material but also control access to it. Seminal work performed over thirty years ago demonstrated that DNA is organized around a core protein complex, termed the nucleosome.1Nucleosomes are octomeric protein complexes composed of two copies of the four different canonical histones (H2A, H2B, H3 and H4), the crystal structure of which has been determined at high resolution.2DNA is wrapped around nucleosomes to not only package or compact its unruly length but also regulate other biomolecules access to the genetic material. The histone-dependent restriction of access to DNA is opposed by the sequence specificity of transcription factors and other chromatin-associated proteins, such as nucleosome remodelers and modifiers, resulting in a well orchestrated regulation of gene expression.3Early on it was recognized that another abundant histoneprotein, histone H1, participated in increasing the chromatin compaction and formation of chromatin’s most condensed form, the chromosomes. Furthermore chromatin researchers observed that a wide array of histone variants existed in addition to the core nucleosome members. These histone variants are of great interest since many possess specialized roles and functions.4–6 Numerous proteins and protein complexes have been implicated in modifying the composition of core nucleosomes with histone variants at specific genomic loci, at centromeres for instance, or under specific cellular conditions, such as mitosis or DNA damage. In addition to the exchange of core histone for their variants, chromatin is also controlled by an intricate number of histonepost-translational modifications (PTM).7 A wide array of enzymes including lysineacetyltransferases (KAT), lysine methyltransferases (KMT), kinases, E3 ubiquitin ligases and many more are known to modify one or many residues of histoneproteins. This plethora of modifications act as signals, or anchors, for chromatin binding factors and this phenomenon is termed the histone code.8 To truly understand the dynamic and complex nature of the genome will require a detailed understanding of the combinatorial role of histone variants and modifications along with the chromatin-associated proteins they recruit.
The field of chromatin research has rapidly progressed in recent years due to the development of novel technologies. Advances in molecular biology, genomic sciences, crystallography and proteomics enabled the field to grow to its current state. This review will focus on the role of proteomic technologies in the study of chromatin as well as discussing emerging methods to study chromatin-associated proteins.
Chromatin foundation—the histoneproteins
Histones are small basic proteins that are present in all eukaryotes that can be grouped as either core histones (H2A, H2B, H3 and H4) or linker histones (H1 and H5). Histones were discovered more than a hundred years ago by Albrecht Kossel as an acid soluble class of proteins associated with nucleic acid; this is some of the work for which he received the Nobel Prize of Physiology and Medicine in 1910. The fact that histones are soluble in acidic solutions has been exploited for a long time to enable their purification (see ref. 9 for an overview of the acid extraction). Early work often used electrophoresis methods to study the acid extracted histones but in recent years mass spectrometry has become the method of choice to analyze histones and their post-translational modifications (reviewed in ref. 10). For instance, a number of groups have embarked on the cataloguing of histones present in various organisms using mass spectrometry. Bonenfant et al. performed such a study to better characterize the histone H2A and H2B isoforms present in Jurkat cells.11 By using both top-down (intact proteins) and bottom-up (enzymatically digested proteins) approaches, they were able to identify nine histone H2A and eleven histone H2B variants. In addition, numerous post-translational modifications, such as phosphorylation and acetylation, were detected on some of these variants. Another similar study was performed by Thomas et al. in which they used top-down mass spectrometry to study the histone H3 isoforms from HeLa cells.12 The authors were able to easily distinguish three histone H3 isoforms containing a wide number of post-translational modifications. It was also observed that acid extracted histone H3.1 from colchicine treated cells was a heterogeneous population containing zero, one or two phosphate moieties on serine 10 and 28. From these top-down studies, and other work, it becomes quite apparent that a single histone molecule can contain a diversity of post-translational modifications at once. This was further reinforced by the work of two independent groups which used mass spectrometry to study histoneprotein–PTM relationship. Jiang et al. analyzed acid extracted histone H3 from budding yeast following their enzymatic digestion with the endoproteinase Glu–C.13 This enzyme digests protein into long peptides since it selectively cuts peptide bonds at the C-terminal of glutamic acid. Glu–C digestion of histone H3 produces a large peptide of 50 amino acids containing its N-terminal tail which was known to be heavily modified. Analysis of this peptide demonstrated that certain modifications are linked, i.e. present simultaneously on the peptide. In particular, the authors observed that Set1-dependent methylation of H3 lysine four (H3K4) concurrently occurred with acetylation of K9, K14 and K23. Taverna et al. performed a similar study in the ciliated protozoan Tetrahymena thermophilia.14 Acid extracted histone H3 was resolved on a PolyCAT A column, digested by Glu–C and analyzed by a mass spectrometer capable of performing electron transfer dissociation fragmentation and proton transfer charge reduction. The use of this new mass spectrometer enabled the first fifty amino acids of histone H3 to be fully sequenced in a single scan. Using this approach, the diversity of PTMs on histone H3 was discerned and a clear connection between certain methylation and acetylation modifications established. These highly significant technical achievements overshadow the fact that it remains extremely difficult to analyze histones and their PTMs. For instance, many of the histone H2B isoforms present in human cells identified by Bonenfant et al. only differ from one another by a single or a few amino acids.11 This means that distinguishing these isoforms in shotgun proteomic experiments is extremely difficult in most cases and impossible in some. Furthermore, the detection of some large PTM on histones, such as ubiquitylation, or labile PTM, such as phosphorylation, by mass spectrometry requires forethought and experimental planning. As such, the field of chromatin proteomics still requires innovation in both the instruments and methodologies in order to fully grasp the complexities and subtleties of histone biology.The chromatin neighborhood—protein complexes and networks
Mass spectrometry has had a deep impact on the field of chromatin research by enabling researchers to better observe the diversity in both the histone composition and their modifications. But mass spectrometry driven proteomic research also enabled a better understanding of the composition of chromatin itself. An early report by Shiio et al. attempted to better characterize the protein content of chromatin with and without the expression of the oncogene Myc.15 This was achieved by first isolating nuclei from P493-6 human B lymphocytes and subsequently isolating the chromatin fraction prior to ICATlabeling and mass spectrometric analysis. This study resulted in the identification of 64 nuclear proteins and their relative quantitation upon Myc induction. While this study was small in scope, it introduced the notion that proteomics is not only valuable for the identification of chromatin bound proteins but can also be exploited to study the dynamic nature of chromatinprotein networks. Below we highlight innovative proteomic methods that are currently being used to characterize the diverse and dynamic chromatin environment.In addition to helping define histones, their variants and their PTMs, proteomic technologies have been key in the characterization of numerous protein complexes that regulate chromatin. An early example is the use of conventional and tandem affinity purification for the elucidation of the Set1 histone methyltransferase containing COMPASS protein complex from budding yeast.16 Other examples include the purification of histoneproteins with their histone chaperones in both yeast17 and human cells.18 This work, and many other reports, helped characterize core protein complexes that are both soluble and resistant to washing. Interestingly, none of these methods attempted to purify these protein complexes while they were associated with chromatin and its network of proteins.
A major shift in the perception of protein–protein interactions has started to emerge in the recent literature. In particular, the view of protein complexes has been extended to increasingly include a macromolecular, or network, view of the interconnections between protein complexes. This is in part derived from the emergence of systems biology, which attempts to identify and quantify the biological connections present in an organism.19 Progress toward this goal has been fueled by technical improvements in the analysis of these connections whether they are between protein, DNA, RNA or other types of biomolecules. For instance, a recent bioinformatic study20 re-analyzing the Saccharomyces cerevisiaeTAPprotein interaction datasets21,22 produced ComplexNet, a network of interactions between proteins and protein complexes. This effort exposed novel associations, such as a physical link between a poorly characterized protein complex composed of Yer071c and Yir003w, and the yeast actin-capping heterodimer. Other examples of complex–complex interactions have also been reported in the literature. For example, Sardiu et al. reported an interaction network around Tip49a and Tip49b using 27 reciprocal affinity purifications.23 Due to the density of information present in their dataset they could effectively derive the association not only between proteins but also between complexes. These selected examples expose the growing interest in not only mapping protein–protein interactions but also placing these interactions in the larger context of a living organism.
Another approach to study protein networks and their individual associations is the yeast-two hybrid (Y2H) method. Y2H has been used for over a decade and applied in many organisms greatly improving our understanding of the interconnection between proteins. Recently, Yu et al. re-tested a significant fraction of the budding yeast S. cerevisiae proteome by Y2H.24 This work resulted in a significant increase in the number of known binary interactions in this model organism. But more importantly, it enabled them to gather some critical insight in the type of interactions detected by Y2H. It was highlighted in an accompanying article that interactions derived from Y2H, AP-MS and protein complementation assay (PCA) are fundamentally different but complementary in nature.25 In particular, Jensen and Bork showed that Y2H and AP-MS are both efficient at detecting interaction between nuclear proteins.25 Thus these two methods appear to be the best candidates for the analysis of chromatin and its meshwork of interactions between proteins and DNA.
Despite these recent advances, defining the interaction networks of chromatin-associated proteins remains challenging using conventional methodologies. This problem is exemplified by the various protein complexes involved in transcription. For instance, RNA polymerase II (RNAPII) has been extensively studied in many organisms.26,27 Yet despite all of these efforts, novel RNAPII-associated proteins were only discovered once technical improvements were made to alleviate the challenge posed by the presence of DNA.28,29 A common view in the field of chromatin proteomic is that the presence of DNA hinders the effective purification of protein complexes, thus DNA should be excluded from the sample under study. This was first recognized many years ago in a seminal study by Lai and Herr which demonstrated that the common laboratory reagent ethidium bromide (EtBr) could be used to disrupt DNA–protein interactions.30 The authors effectively demonstrated that their affinity purification of the transcription factor Oct-2 from 293 cells suffered from non-specific association of some proteins with the DNA that was co-purified. Removal of DNA by chemical treatment with EtBr, or with enzymatic digestion of DNA by micrococcal nuclease (MNase), was sufficient to abolish these non-specific associations. This study was critical since it described a fast and efficient way to determine whether a protein–protein interaction was dependent on the presence of DNA or not. But more importantly, it also showed that some DNA–protein associations could be enriched in the laboratory. The importance of this fact is only starting to be recognized in the literature.31
The practice of removing DNA prior to affinity purification remains common to this day in many laboratories. The reasons for this are multiple and include a reduced viscosity of the samples, improved solubility of large complexes and streamlined sample preparation. A good example of this strategy was reported by Foltz et al. for the study of CENP-A, a centromeric histone H3 variant, in human cells.32 The authors used MNase treatment to drastically reduce DNA size in their cellular extracts prior to two step purification of CENP-A and histone H3. The resulting mass spectrometric analysis of protein networks enabled the identification of numerous new CENP-A interaction partners which were shown to possess critical roles in the maintenance of chromosome stability.32 A similar procedure was used by Du et al. to study H2A.X, a histone variant critical for efficient DNA damage response.33 The authors used DNase I treatment to completely remove DNA from their cell extract prior to purification of H2A.X-FLAG and subsequent analysis by mass spectrometry. This type of procedure enabled H2A.X interaction partners to be specifically observed under DNA damage conditions and not under control conditions. Benzonase, an endonuclease digesting both DNA and RNA, is another reagent that is commonly used to degrade chromatin and thus preventing DNA–protein association from being observed.34 Reagents affecting protein–DNA interaction are often used for validating previously observed protein–protein interaction. For instance, a report studying the transcription factor Oct-4 observed an interaction with the estrogen receptor beta which was validated to be a direct interaction, and not to be mediated through chromatin, since it was insensitive to both benzonase and EtBr.35 While these studies are successful at purifying protein complexes off chromatin, they forgo information in doing so, particularly about the chromatin environment surrounding the baits. Thus, researchers attempting to obtain a more holistic view of chromatin must use different protocols in which the DNA remains fully or partially intact.
Early attempts at performing functional proteomics studies of chromatin-associated proteins often used protocols not designed especially for this unique class of proteins; hence, these studies suffered from both sensitivity and specificity issues. A good example of this problem is the use of the well established chromatin immunoprecipitation (ChIP) method which permits the identification of a protein’s DNA binding site.36ChIP is typically composed of a few key steps: (1) stabilization of protein–DNA interaction through the use of chemical crosslinkers, such as formaldehyde; (2) DNA shearing to improve the solubility of the protein–DNA complexes; (3) affinity purification of the complex of interest using antibodies; (4) amplification and analysis of the DNA of interest by some means (PCR, ChIP, etc.). At first glance the general idea of ChIP appears directly applicable to proteomics studies, but one major problem needs to be addressed, the impossibility of amplifying protein samples prior to their analysis. Thus, direct transfer of the ChIP protocol to proteomic studies produces less than optimal results, mainly due to low sample recovery and the heterogeneity of the sample.37 In short, researchers attempting to study chromatin networks need to purify large amount of material, of high purity, at a reasonable cost. No small feat indeed. Still, numerous laboratories are tackling this challenge through innovation and a selection of these new methods will be discussed in details below.
In a new report Déjardin and Kingston presented a new method termed proteomics of isolated chromatin segments (or PICh) for the analysis of proteins associated with specific chromatin loci.38 The PICh method relies on nucleic acidprobes that recognize specific genomic loci which are then enriched for with their associated proteins (Fig. 1). The procedure begins by fixing approximately 3 × 109cells with formaldehyde which stabilizes both protein–protein and protein–DNA interactions. The cells were then lysed and the chromatin was solubilized by sonication. To specifically purify the genomic loci of interest, a 25 base pairprobe made of locked nucleic acid (which possesses a higher melting temperature than a regular nucleic acid) linked to a desthiobiotin moiety was used. The probe was efficiently hybridized with the chromatin samples under stringent detergent conditions and then subsequently purified using streptavidin beads and eluted with excess biotin. The purified proteins were resolved on a SDS-PAGE gel and identified by mass spectrometry. This new technique was first applied for the purification of proteins associated with telomeres. Telomeres were selected since they were abundant (∼100 copies per cell) which reduced the amount of material needed per experiment. The authors used a probe directed at telomere and a probe with the same nucleic acid composition but in a randomized order as a control. Three cell lines were studied, two of which had telomerase activity, the enzyme responsible for maintaining telomere length, and one cell line defective in telomerase activity which served as a comparative control. The authors purified approximately 200 proteins associated with telomerechromatin, but not with the scrambled probe, in the different cell lines and approximately half these hits were shared between cell lines. 33 proteins observed by mass spectrometry following PIChpurifications, such as Apollo and Ku70, were previously reported to be present at telomere, supporting the specificity of the method. Colocalization of putative telomereproteins with the telomeric protein Rap1 by indirect immunofluorescence along with ChIP experiments was used to validate the PICh datasets. For most novel proteins purified by PICh with telomeres, the secondary validations supported their telomeric localization and clearly demonstrated the strength of the new method. One drawback of the PICh procedure is the amount of starting material needed per experiment. By targeting a specific DNA sequence which is present at a few copies per cell, it becomes extremely difficult to purify sufficient associated proteins for mass spectrometry analysis. While PICh represents a significant step forward in the study of chromatin-associated protein complexes it remains to be seen how widely applicable the method will become.
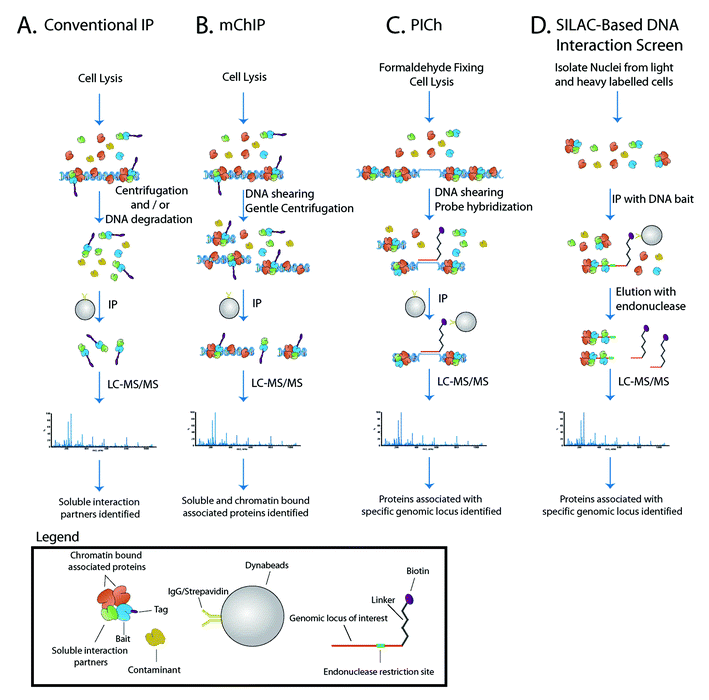 | ||
| Fig. 1 Proteomic schemes for the study of chromatin bound proteins. (A) Conventional IP circumvents the technical hurdles posed by chromatin by high centrifugation and/or DNA degradation prior to proteinpurification. (B) The mChIP method maintains the native protein–DNA interaction to purify protein networks associated with chromatin. (C) The PICh method uses a DNAprobe to selectively purify proteins associated with a genomic locus. (D) The SILAC-based DNA interaction screen uses quantitative proteomics and in vitro reconstituted protein–DNA interaction to identify proteins associated with a genomic locus. | ||
A different approach was taken by Schultz-Norton et al. to detect chromatin-associated protein complexes with the estrogen receptor alpha (ERα) in an in vitro assay.39 The assay was designed to study chromatin bound protein complexes by reconstituting them in vitro prior to mass spectrometry identification, thus bypassing some of the technical issues previously discussed. Their assay was conducted as follows: first HeLa nuclear cell extract and recombinant ERα were prepared. Then, DNA oligos containing the Xenopus laevis A2 estrogen responsive element sequence and its flanking elements were left at room temperature to anneal. The double stranded DNA was then incubated with the purified ERα and subsequently with HeLa cell nuclear extract. The resulting protein complexes associated with chromatin were then isolated from the unbound material using an agarose gel. The gel section corresponding to the ERα containing DNA bound complexes was excised, its protein content concentrated and analyzed by mass spectrometry. Follow-up work on some of these newly discovered proteins associated with ERα revealed that they impacted the transcriptional function of ERα. This new procedure reported by Schultz-Norton et al. represents an attractive approach to study chromatin bound protein complexes but might be difficult to widely utilize as knowledge of DNA binding sites of the protein being studied is required. Finally, the assay is performed on nucleosome-free DNA which does not mimic chromatin perfectly.
The use of DNAprobes for the enrichment of protein complexes was also used by Rubio et al. to study the CCTC-binding factor CTCF in Jurkat cells.40 The authors used 163 base pairDNAprobe corresponding to either WT or mutant c-myc insulator sequences coupled with biotin. Insulator sequences participate in proper gene expression by preventing inappropriate enhancers’ action as well as by restricting heterochromatin spreading.41 The probes were immobilized on magnetic beads coated with streptavidin enabling efficient immunopurification to be performed once the probes were mixed with nuclear extracts from Jurkat cells. By Western blotting, the authors showed a strong enrichment of CCTC-binding factor CTCF with the WTprobe over the mutant one, reproducing previously shown data and validating their enrichment method. The next step was to analyze the proteins associated with their probes by mass spectrometry. To better distinguish real hits from background contamination, the ICAT method was used to quantify the proteins associated with the probes. Putative interaction partners were easily identified since they were enriched with the WTprobes over the mutant c-myc insulator. This method showed a significant enrichment of the cohesion subunit Scc3/SA1 with the WTprobe. Subsequent validation experiments showed that the CCTC-binding factor CTCF and Scc3/SA1 physical distribution on chromosome overlap often. This represented a novel role for CCTC-binding factor CTCF in cohesion which may have clinical implications.
This type of approach was shown by Mittler et al. to be compatible with SILAC, a quantitative proteomics method.42 The method used by the authors is similar to the examples previously described but with a few key differences in their sample preparation and probe design (Fig. 1). The probes used in this study were designed to contain 40 base pairs encompassing a binding site for the transcription factor AP2 and its surrounding sequences, a restriction site for an endonuclease and a biotin moiety. A control probe containing a 2 point mutation in the AP2 binding site was also prepared to act as a negative control in the immunoprecipitation experiments. The nuclear extracts used in this paper were prepared from HeLa-S3 cells grown in suspension from media containing either light lysine (lysine-d0) or heavy 2H4-lysine (lysine-d4). The light extracts (unlabeled) were used in the control immunoprecipitation and the heavy one with the wild-type probe. Following the elution of the protein complexes by the addition of an endonuclease, the purified proteins from immunopurification were combined, run on a SDS-PAGE gel and analyzed by mass spectrometry. This protocol resulted in the identification of more than 250 proteins, a small fraction of which were specifically enriched with the wild-type probe. Among these proteins, TFAP2A was found to be enriched with the wild-type probe over the mutated probe by more than 6 fold which demonstrates the specificity of the purification. In a subsequent experiment, the authors tested whether their new method could specifically enrich proteins associated with methylated DNA. To do so, they synthesized two new probes containing a CpG island found ahead of the MAT2 loci with and without methylation. Using the experimental protocol described earlier, the authors were successful at efficiently enriching proteins associated with the methylated probe over the unmodified one. The works described by Mittler et al. and Rubio et al. clearly demonstrate the feasibility of performing efficient proteomics studies of DNA–protein interactions.40,42 The use of biotinylated DNAprobes, a relatively inexpensive reagent, was able to purify proteins in sufficient quantity for mass spectrometric analysis. In both cases though, the methods suffered from significant background contaminations but the authors were still able to detect specific interactions through the use of quantitative proteomic methods. These reports provide a basis for more elaborate work which could study single-nucleotide polymorphism and other genomic variations from a proteomic perspective.
As the previous examples have demonstrated, the use of DNAprobes in immunoprecipitation enables protein networks to be purified and characterized by mass spectrometry. While this kind of work is bound to positively affect the proteomic field, it does pose the unusual constraint of designing experiments around a genomic locus rather than a protein. Thus to better understand the function of a given chromatin bindingprotein, a different approach is needed. Recently we reported a novel method of purifying chromatin-associated networks termed modified chromatin immunopurification (mChIP).43 The mChIP method consists of a single affinity purification step, whereby chromatin bound protein networks are isolated from mildly sonicated and gently clarified cellular extracts using magnetic beads coated with antibodies (Fig. 1). The method was first developed for the purification of histones and their associated protein networks on chromatin in the yeast S. cerevisiae. We observed that the mChIP method enabled not only the purifications of large protein network associated with a bait of interest but also the DNA bound by it. Liquid chromatography-tandem mass spectrometry analysis of the mChIP purified proteins enabled us to detect different protein networks associated with the canonical histone H2A and its less abundant variant H2A.Z. These mChIPpurifications were demonstrated to be efficient at enriching specific chromatinprotein networks associated with the different bait proteins used. To insure that chromatin was not binding non-specifically to the magnetic beads, two different control experiments were performed. In one case, Htz1-TAP was purified by mChIP in swr1Δ strains. Swr1 is the enzyme responsible for the deposition of Htz1 in nucleosome and its deletion results in Htz1 being excluded from chromatin.44–46 In the other case, the Htz1-TAP sample was prepared following a traditional protocol (no DNA shearing, sample clarification through heavy centrifugation) prior to the affinity purification. Both control experiments showed a drastic reduction of proteins associated with Htz1-TAP. Moreover, the proteins still associated with Htz1-TAP in a swr1 strain or following the traditional sample preparation were not known to associate with chromatin. To further characterize the role of chromatin in mChIPpurifications, a set of experiments were designed to directly determine the impact of chromatin size (i.e. length) on the protein network associated with Htz1-TAP. To achieve this a Htz1-TAP extract was split into three equal aliquots; one aliquot was sonicated (DNA size 500–1500 base pair), another one was treated with MNase (DNA size 150–450 base pair) and the last one with DNase I (no DNA detected). Immunopurification of Htz1-TAP from each aliquot showed that as the DNA size was reduced, the associated protein networks were also reduced without affecting the amount of purified bait. Thus DNA size can be used as a tool to study the protein environment of a chromatin-associated bait. The new mChIP technique was also used to purify three different non-histonechromatin-associated proteins of different functions with success. For example, mChIP of Lge1-TAP enabled the detection of a large protein network. This result contrasts strongly with genome wide proteomics studies performed on Lge1 which only revealed a single interaction partner for Lge1. We observed a large enrichment in Lge1mChIP data for proteins associated or linked to the spindle pole body, which could in part explain some of the cell size phenotypes associated with this gene. Efforts are ongoing to expand the work presented in this initial report by performing high-throughput mChIPpurifications of diverse classes of chromatin-associated proteins. It is our goal to obtain a clearer view of the chromatin environment surrounding a significant fraction of the enzymes known to affect chromatin in S. cerevisiae.
Summary
The study of chromatin through mass spectrometry based proteomic methods remains challenging. Fortunately, the recent technological advances based on both nucleic acid and proteinprobes announced a bright future for chromatin proteomic. Furthermore, mass spectrometry remains an effective tool for the characterization of histone and of their PTM that will surely continue to play a critical role in chromatin research. It is conceivable that shortly, protein networks associated with chromatin at specific loci or with particular histone PTM will be routinely studied. This could prove critical in understanding both basic cellular processes as well as human pathologies which are often rooted in chromatin itself.Acknowledgements
This work was supported by operating grants to D.F. from the Canada Research Chair program, the Canadian Institute of Health Research (CIHR), MDS Inc., the Ontario Genomics Institute and the University of Ottawa and operating grants to K.B. from the National Cancer Institute of Canada through funds raised by the Terry Fox Research Foundation and an Early Researcher Award from the Ontario Government. D.F. is a Canada Research Chair in Proteomics and Systems Biology. K.B. is a Canada Research Chair in Chemical and Functional Genomics. J.-P.L. is supported by an Ontario Graduate Scholarship.References
- R. D. Kornberg, Chromatin structure: a repeating unit of histones and DNA, Science, 1974, 184, 868–871 CrossRef CAS.
- K. Luger, A. W. Mader, R. K. Richmond, D. F. Sargent and T. J. Richmond, Crystal structure of the nucleosome core particle at 2.8 Å resolution, Nature, 1997, 389, 251–260 CrossRef CAS.
- G. G. Wang, C. D. Allis and P. Chi, Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling, Trends Mol. Med., 2007, 13, 373–380 CrossRef CAS.
- M. Boulard, P. Bouvet, T. K. Kundu and S. Dimitrov, Histone variant nucleosomes: structure, function and implication in disease, Subcell. Biochem., 2007, 41, 71–89 Search PubMed.
- A. Izzo, K. Kamieniarz and R. Schneider, The histone H1 family: specific members, specific functions, Biol. Chem., 2008, 389, 333–343 CrossRef CAS.
- A. Loyola and G. Almouzni, Marking histone H3 variants: how, when and why?, Trends Biochem. Sci., 2007, 32, 425–433 CrossRef CAS.
- S. D. Taverna, H. Li, A. J. Ruthenburg, C. D. Allis and D. J. Patel, How chromatin-binding modules interpret histone modifications lessons from professional pocket pickers, Nat. Struct. Mol. Biol., 2007, 14, 1025–1040 CrossRef CAS.
- B. D. Strahl and C. D. Allis, The language of covalent histone modifications, Nature, 2000, 403, 41–45 CrossRef CAS.
- D. Shechter, H. L. Dormann, C. D. Allis and S. B. Hake, Extraction, purification and analysis of histones, Nat. Protoc., 2007, 2, 1445–1457 Search PubMed.
- B. A. Garcia, J. Shabanowitz and D. F. Hunt, Characterization of histones and their post-translational modifications by mass spectrometry, Curr. Opin. Chem. Biol., 2007, 11, 66–73 CrossRef CAS.
- D. Bonenfant, M. Coulot, H. Towbin, P. Schindler and J. van Oostrum, Characterization of histone H2A and H2B variants and their post-translational modifications by mass spectrometry, Mol. Cell Proteomics, 2006, 5, 541–552 CAS.
- C. E. Thomas, N. L. Kelleher and C. A. Mizzen, Mass spectrometric characterization of human histone H3: a bird’s eye view, J. Proteome Res., 2006, 5, 240–247 CrossRef CAS.
- L. Jiang, J. N. Smith, S. L. Anderson, P. Ma, C. A. Mizzen and N. L. Kelleher, Global assessment of combinatorial post-translational modification of core histones in yeast using contemporary mass spectrometry. LYS4 trimethylation correlates with degree of acetylation on the same H3 tail, J. Biol. Chem., 2007, 282, 27923–27934 CrossRef CAS.
- S. D. Taverna, B. M. Ueberheide, Y. Liu, A. J. Tackett, R. L. Diaz, J. Shabanowitz, B. T. Chait, D. F. Hunt and C. D. Allis, Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2086–2091 CrossRef CAS.
- Y. Shiio, R. N. Eisenman, E. C. Yi, S. Donohoe, D. R. Goodlett and R. Aebersold, Quantitative proteomic analysis of chromatin-associated factors, J. Am. Soc. Mass Spectrom., 2003, 14, 696–703 CrossRef CAS.
- T. Miller, N. J. Krogan, J. Dover, H. Erdjument-Bromage, P. Tempst, M. Johnston, J. F. Greenblatt and A. Shilatifard, COMPASS a complex of proteins associated with a trithorax-related SET domain protein, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12902–12907 CrossRef CAS.
- E. Luk, N. D. Vu, K. Patteson, G. Mizuguchi, W. H. Wu, A. Ranjan, J. Backus, S. Sen, M. Lewis, Y. Bai and C. Wu, Chz1, a nuclear chaperone for histone H2AZ, Mol. Cell, 2007, 25, 357–368 CrossRef CAS.
- H. Tagami, D. Ray-Gallet, G. Almouzni and Y. Nakatani, Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis, Cell, 2004, 116, 51–61 CrossRef CAS.
- H. Kitano, Systems biology: a brief overview, Science, 2002, 295, 1662–1664 CrossRef CAS.
- H. Wang, B. Kakaradov, S. R. Collins, L. Karotki, D. Fiedler, M. Shales, K. M. Shokat, T. Walther, N. J. Krogan and D. Koller, A complex-based reconstruction of the S. cerevisiae interactome, Mol. Cell Proteomics, 2009, 8, 1361–1381 CrossRef CAS.
- A. C. Gavin, P. Aloy, P. Grandi, R. Krause, M. Boesche, M. Marzioch, C. Rau, L. J. Jensen, S. Bastuck, B. Dumpelfeld, A. Edelmann, M. A. Heurtier, V. Hoffman, C. Hoefert and K. Klein, et al., Proteome survey reveals modularity of the yeast cell machinery, Nature, 2006, 440, 631–636 CrossRef CAS.
- N. J. Krogan, G. Cagney, H. Yu, G. Zhong, X. Guo, A. Ignatchenko, J. Li, S. Pu, N. Datta, A. P. Tikuisis, T. Punna, J. M. Peregrin-Alvarez, M. Shales, X. Zhang and M. Davey, et al., Global landscape of protein complexes in the yeast Saccharomyces cerevisiae, Nature, 2006, 440, 637–643 CrossRef CAS.
- M. E. Sardiu, Y. Cai, J. Jin, S. K. Swanson, R. C. Conaway, J. W. Conaway, L. Florens and M. P. Washburn, Probabilistic assembly of human protein interaction networks from label-free quantitative proteomics, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1454–1459 CrossRef CAS.
- H. Yu, P. Braun, M. A. Yildirim, I. Lemmens, K. Venkatesan, J. Sahalie, T. Hirozane-Kishikawa, F. Gebreab, N. Li, N. Simonis, T. Hao, J. F. Rual, A. Dricot, A. Vazquez and R. R. Murray, et al., High-quality binary protein interaction map of the yeast interactome network, Science, 2008, 322, 104–110 CrossRef CAS.
- L. J. Jensen and P. Bork, Biochemistry. Not comparable, but complementary, Science, 2008, 322, 56–57 CrossRef CAS.
- H. Cho, G. Orphanides, X. Sun, X. J. Yang, V. Ogryzko, E. Lees, Y. Nakatani and D. Reinberg, A human RNA polymerase II complex containing factors that modify chromatin structure, Mol. Cell Biol., 1998, 18, 5355–5363 CAS.
- E. Maldonado, R. Shiekhattar, M. Sheldon, H. Cho, R. Drapkin, P. Rickert, E. Lees, C. W. Anderson, S. Linn and D. Reinberg, A human RNA polymerase II complex associated with SRB and DNA-repair proteins, Nature, 1996, 381, 86–89 CrossRef CAS.
- O. Aygun, J. Svejstrup and Y. Liu, A RECQ-RNA5 polymerase II association identified by targeted proteomic analysis of human chromatin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8580–8584 CrossRef CAS.
- C. Jeronimo, D. Forget, A. Bouchard, Q. Li, G. Chua, C. Poitras, C. Therien, D. Bergeron, S. Bourassa, J. Greenblatt, B. Chabot, G. G. Poirier, T. R. Hughes, M. Blanchette and D. H. Price, et al., Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme, Mol. Cell, 2007, 27, 262–274 CrossRef CAS.
- J. S. Lai and W. Herr, Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 6958–6962 CrossRef CAS.
- N. Rusk, Reverse ChIP, Nat. Methods, 2009, 6, 187 CrossRef CAS.
- D. R. Foltz, L. E. Jansen, B. E. Black, A. O. Bailey, J. R. Yates, 3rd and D. W. Cleveland, The human CENP-A centromeric nucleosome-associated complex, Nat. Cell Biol., 2006, 8, 458–469 CrossRef CAS.
- Y. C. Du, S. Gu, J. Zhou, T. Wang, H. Cai, M. A. Macinnes, E. M. Bradbury and X. Chen, The dynamic alterations of H2AX complex during DNA repair detected by a proteomic approach reveal the critical roles of Ca(2+)/calmodulin in the ionizing radiation-induced cell cycle arrest, Mol. Cell. Proteomics, 2006, 5, 1033–1044 CrossRef CAS.
- G. I. Chen and A. C. Gingras, Affinity-purification mass spectrometry (AP-MS) of serine/threonine phosphatases, Methods, 2007, 42, 298–305 CrossRef CAS.
- D. L. van den Berg, W. Zhang, A. Yates, E. Engelen, K. Takacs, K. Bezstarosti, J. Demmers, I. Chambers and R. A. Poot, Estrogen-related receptor beta interacts with Oct to positively regulate Nanog gene expression, Mol. Cell. Biol., 2008, 28, 5986–5995 CrossRef CAS.
- C. E. Massie and I. G. Mills, ChIPping away at gene regulation, EMBO Rep., 2008, 9, 337–343 CrossRef CAS.
- R. M. Ricke and A. K. Bielinsky, Easy detection of chromatin binding proteins by the histone association assay, Biol. Proced. Online, 2005, 7, 60–69 Search PubMed.
- J. Déjardin and R. E. Kingston, Purification of proteins associated with specific genomic Loci, Cell, 2009, 136, 175–186 CrossRef CAS.
- J. R. Schultz-Norton, Y. S. Ziegler, V. S. Likhite, J. R. Yates and A. M. Nardulli, Isolation of novel coregulatory protein networks associated with DNA-bound estrogen receptor alpha, BMC Mol. Biol., 2008, 9, 97 CrossRef.
- E. D. Rubio, D. J. Reiss, P. L. Welcsh, C. M. Disteche, G. N. Filippova, N. S. Baliga, R. Aebersold, J. A. Ranish and A. Krumm, CTCF physically links cohesin to chromatin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8309–8314 CrossRef CAS.
- J. Komura, H. Ikehata and T. Ono, Chromatin fine structure of the c-MYC insulator element/DNase I-hypersensitive site I is not preserved during mitosis, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15741–15746 CrossRef CAS.
- G. Mittler, F. Butter and M. Mann, A SILAC-based DNA protein interaction screen that identifies candidate binding proteins to functional DNA elements, Genome Res., 2009, 19, 284–293 CAS.
- J. P. Lambert, L. Mitchell, A. Rudner, K. Baetz and D. Figeys, A Novel Proteomics Approach for the Discovery of Chromatin-Associated Protein Networks, Mol. Cell. Proteomics, 2009, 8, 870–882 CrossRef CAS.
- M. S. Kobor, S. Venkatasubrahmanyam, M. D. Meneghini, J. W. Gin, J. L. Jennings, A. J. Link, H. D. Madhani and J. Rine, A protein complex containing the conserved Swi2/Snf2-related ATPase Swr1p deposits histone variant H2A.Z into euchromatin, PLoS Biol., 2004, 2, e131 CrossRef.
- N. J. Krogan, K. Baetz, M. C. Keogh, N. Datta, C. Sawa, T. C. Kwok, N. J. Thompson, M. G. Davey, J. Pootoolal, T. R. Hughes, A. Emili, S. Buratowski, P. Hieter and J. F. Greenblatt, Regulation of chromosome stability by the histone H2A variant Htz1, the Swr1 chromatin remodeling complex, and the histone acetyltransferase NuA4, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13513–13518 CrossRef CAS.
- G. Mizuguchi, X. Shen, J. Landry, W. H. Wu, S. Sen and C. Wu, ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex, Science, 2004, 303, 343–348 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |