DOI:
10.1039/B917346N
(Paper)
J. Mater. Chem., 2010,
20, 83-89
Reaction of water-soluble fullerenes with O2˙− and other reactive radical species†
Received
21st August 2009
, Accepted 2nd October 2009
First published on
12th November 2009
Abstract
In this work we focused on the reaction of water-soluble fullerene derivatives with various organic (i.e., methyl radical and t-butyl radical) and inorganic (i.e., superoxide radical and azide radical) radicals to mimic their superoxide dismutase (SOD) activity. Importantly, with the help of time-resolved pulse radiolysis and steady-state gamma radiolysis measurements all of the assays were conducted in aqueous solutions. Our fully-fledged spectroscopic and kinetic investigations leave no doubt about a diffusion-controlled addition mechanism (1010 M−1 s−1) by which superoxide radical reacts with fullerenes to yield (C60-O2)˙−. Notable is that the formations of (C60-CH3)˙, (C60-(CH2)(CH3)2COH)˙, and (C60-N3)˙ are much slower and proceed with activation-controlled rate constants less than 109 M−1 s−1. The major deactivation path of (C60-O2)˙− is a pH dependent protonation (1010 M−1 s−1). Despite lacking unambiguous evidence for the formation of C60˙− – via a direct impact or an indirect dissociation mechanism – (C60-O2)˙− is still redoxactive. The latter has been confirmed by an activation-controlled reduction (108 M−1 s−1) of a series of p-benzoquinones that display different electron affinities. In other words, (C60-O2)˙− – but not C60˙− – is likely to emerge as a key intermediate in the SOD activity of fullerenes. Its slow protonation is beneficial toward an efficient dismutation to form H2O2.
Introduction
The great excitement commencing with the advent of fullerenes and their large scale production was the inception to a wealth of scientific projects focused on two major disciplines – material chemistry1 and bio-medical applications.2 Research and development of bio-medical applications was, however, penalized by the necessity to work with soluble and biocompatible materials. Still, the outstanding physico-chemical features of fullerenes in general and [60]fullerene in particular, together with their unique shape, renders this class of carbon nanostructures as a candidate par excellence for biological applications.3 Its ability, for example, to uptake electrons4 causes fullerenes to act as a very appropriate radical sponge, which is unequivocally reflected in its neuroprotective action.5,6 Photoexcitation of fullerenes, on the other hand, produces selectively reactive oxygen species that are known to be cytotoxic.
The desire to utilize this unique reactivity also in a polar, especially aqueous, environment has triggered numerous successful attempts to modify the hydrophobic fullerene core by functionalization. Covalent attachment7 of addends across the double bonds located at the junctions of two hexagons8,9 afforded novel and innovative materials with appealing characteristics ranging from excellent water solubility to drug delivery10 and advanced electronic devices.11 Enhancement of water-solubility has been achieved, for example, by mono-functionalization of C60 with negatively charged carboxylic,12,13 positively charged quaternary ammonium,14,15 and non-charged ethylene glycol groups.16 However, strong adsorption forces between the fullerene cores still remain and lead to an instantaneous and irreversible formation of micellar clusters in aqueous solution.17 As a consequence of this aggregation, for example, the lifetime of the excited triplet state is reduced by orders of magnitude,18 and negatively charged fullerene clusters are effectively shielded against reduction by electron transfer.19 One possibility to overcome these cluster-specific problems was to encapsulate the respective monomer species in suprastructures such as gamma-cyclodextrin20 or surfactants.21 Another promising approach is by increasing the number of addends at the fullerene core via controlled bis-,22 tris-,23 or poly-functionalization.24,25 The associated reduction of the fullerene's hydrophobic surface is expected to prevent formation of colloidal C60 clusters. One possibility to overcome these cluster-specific problems is the use of dendritic fullerene derivatives.26–28
Some of the water-soluble C60 derivatives have been shown to exhibit the following therapeutic benefits linked to their antioxidant activities: (1) they locate preferentially to mitochondria and can reconstitute mitochondrial superoxide dismutase (SOD) protection from superoxide radicals in SOD2 genetically deficient mice;29 (2) they are highly potent neuroprotective agents, preventing cell death across a variety of different neuronal types in disease models as diverse as Parkinson's, Alzheimer's, ALS, excitotoxicity, macular degeneration and stroke;30–33 (3) they protect cells from the damaging effects of UV and gamma-irradiation;34,35 (4) they protect from morbidity and mortality in the presence of overwhelming infection with both Gram-positive and Gram-negative organisms;36–38 (5) they protect a variety of different cell types, including leukocytes, hepatocytes and renal tubular epithelial cells from oxidative injury associated with chemical and biological agents.39,40
In this work, particular emphasis is placed on elucidating the modus operandi, by which oxygen, especially reactive oxygen species such as superoxide radical anion (O2˙−), and fullerene and/or fullerene derivatives react with each other in aqueous solutions. Two opposing possibilities are outlined in the literature. Firstly, singly reduced fullerene radical anions are susceptible to react with molecular oxygen.41,42 In accordance with the thermodynamics, formation of superoxide radical anion is likely to be involved. Secondly, it has been proposed that superoxide radical anion reduces some C60 derivatives depending on their reduction potentials. Subsequently, they react with another O2˙− and in a protonation assisted step hydrogen peroxide is formed.43 Common to all work is the lack of spectroscopic identification of reactive intermediates as well as final products and/or the lack biological media. To shed light onto the true mechanism we have investigated the reaction of two water-soluble C60 derivatives in situ with several radicals – organic (i.e., methyl radicals and hydroxyl-t-butyl radicals) and inorganic (i.e., azide radicals and superoxide radicals) – and hydrated electrons. We took advantage of radiolytic techniques – time-resolved (i.e., pulse radiolysis) and steady-state (i.e., gamma radiolysis) – providing an exclusive means to probe practically any desired transient intermediate including reduced/oxidized states, radical adducts, excited states, etc., with high spectral and kinetic precision.
Experimental
Synthesis
The synthesis of dendrofullerenes 1 and 2 (Fig. 1) has already been described.28,39
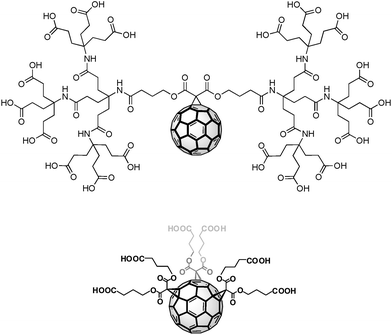 |
| | Fig. 1 Structures of dendrofullerenes 1 (top) and 2 (bottom). | |
Results and discussions
In light of the reductive pathway, namely that the superoxide radical anion might reduce C60, we initially focused on reducing conditions. All reductive investigations were conducted in N2-saturated, dilute aqueous solutions containing 10 vol% 2-propanol. Such conditions lead to the production of three highly reactive species, namely ˙H, ˙OH, eaq−, beside the molecular products Haq, H2, H2O2. The function of 2-propanol is to efficiently scavenges two of the radical species, namely ˙OH and ˙H radicals, via hydrogen abstraction. The main resulting radical, (CH3)2˙COH, is a powerful reductant that has been shown to rapidly reduce, for example, pristine C60. Likewise eaq− is expected to reduce C60.| | H2O ![[long arrow, wavy then straight]](https://www.rsc.org/images/entities/char_e0f6.gif) ˙OH + ˙H + eaq− ˙OH + ˙H + eaq− | (1) |
| | | ˙OH + (CH3)2CH(OH) → H2O + (CH3)2˙C(OH) | (2) |
| | | ˙H + (CH3)2CH(OH) → H2 + (CH3)2˙C(OH) | (3) |
| | 1/2 + (CH3)2˙C(OH) → 1˙−/2˙− + (CH3)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + H+ O + H+ | (5) |
The reaction of the fullerene core with eaq− was directly monitored via the decay of its absorption at 700 nm, which was found to parallel the grow-in of a new transient absorption with a diagnostic near-infrared transition band – see Fig. S1 and S2.† In the present cases, 1˙− and 2˙−, formed in the radiolysis, show pronounced absorption maxima at 1030 nm. The similarity of the transient maxima observed for the water-soluble 1 and 2 with the corresponding water-insoluble precursors specifically points to the successful fullerene reduction. From the time absorption profiles we derived rate constants for the formation of 1˙− (2.0 × 1010 M−1 s−1) and 2˙− (2.1 × 1010 M−1 s−1). These results are in good agreement with previously reported data.47
Under the described conditions, radiolysis leads also to the formation of a second type of reducing species, namely (CH3)2˙C(OH) radicals. Based on its lower reduction potential (−1.39 V versus NHE)48 compared to hydrated electrons (−2.9 V versus NHE),48 the fullerene reduction by (CH3)2˙C(OH) radicals is substantially slower with rate constants typically around 8 × 108 M−1 s−1.15 The formation of 1˙− and 2˙− accordingly occurs in two distinguishable processes, that is, a fast eaq−-induced reduction followed by a slower (CH3)2˙C(OH)-induced reduction. Normalized differential absorption changes recorded ca. 100 μs after the pulse are superimposable with those recorded after the completion of the reaction with eaq− (i.e., ca. 2 μs after the pulse) and thus confirm the reduction of 1 and 2 – see Fig. 2, S1, and S2.†
 |
| | Fig. 2 Time absorption profile (black) at 1030 nm of 1 (5 × 10−5 M) upon pulse radiolysis in N2 saturated aqueous solution pH = 7.2 upon adding 10 vol% 2-propanol – and corresponding kinetic simulation of the time absorption profile (red) using ACUCHEM49 with the rate constants (C60˙− formation – blue; eaq− decay – brown) stated above. | |
In stark contrast to the wealth of information on the features of C60˙− much less is known about fullerene radical adducts, especially in aqueous solutions. Previous work by Dimitrijevic et al., Priyadarsini et al., and Guldi et al. suggests that the formation of the radical adduct is linked to a transient absorption with a pronounced maximum in the visible region of the spectrum.50–54
To establish the transient absorption characteristics of a fullerene radical adduct we first probed methyl radicals, which are expected to form only radical adducts with C60. Methyl radicals were generated radiolytically following the common strategy by adding 5% DMSO to an N2O saturated aqueous solution and irradiating them.55,56 This results in the quantitative scavenging of eaq− and conversion of the latter into ˙OH. Finally, ˙OH reacts with DMSO by releasing ˙CH3 radicals.
| | | eaq− + H2O + N2O → ˙OH + OH− + N2 | (6) |
| | | (CH3)2SO + ˙OH → (CH3)2S(O)OH˙ | (7) |
| | | (CH3)2S(O)OH˙ → ˙CH3 + CH3S(O)OH | (8) |
| | | 1/2 + ˙CH3 → (1-CH3)˙/(2-CH3)˙ | (9) |
The visible part of the transient optical absorption spectrum, obtained upon pulse radiolysis of an N2O purged, aqueous solution of 1 700 μs after the electron pulse, is shown in Fig. 3, while that for 2 is displayed in Fig. S3.† It exhibits an absorption band at 440 nm. These spectral changes are thus indicative for a reaction with the fullerene core. Our assignment to the formation of an adduct radical – (1-CH3)˙/(2-CH3)˙ – is further supported by comparison with results on the addition ˙CH3 radicals to pristine C60. This reaction was observed upon radiolysis in aprotic media, and revealed a transient adduct spectrum of comparable shape and a transient maximum around 370 nm.57
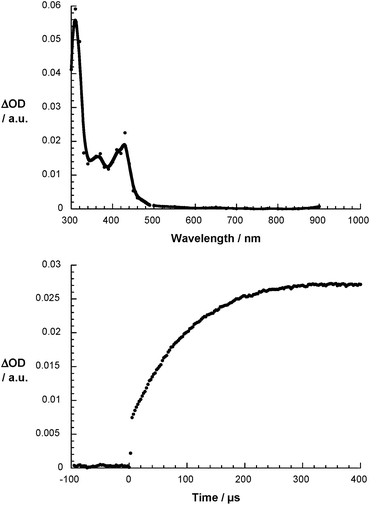
 |
| | Fig. 3 Differential absorption spectrum of 1 (5 × 10−5 M) upon pulse radiolysis in N2O saturated aqueous solution pH = 7.2 upon adding 5 vol% DMSO 700 μs after the electron pulse (upper part) and the corresponding time absorption profile at 440 nm (lower part). | |
With increasing concentration of 1 and 2 over the entire experimentally covered range from 2.5 × 10−5 M to 2 × 10−4 M. The respective first-order rate constant for the formation of (1-CH3)˙/(2-CH3)˙ versus concentration plots exhibit linear relationships, from which bimolecular rate constants of 1.6 × 108 and 4.8 × 107 M−1 s−1 were derived for 1 (Fig. S4†) and 2 (Fig. S5†), respectively.
A similar strategy was pursued for testing the reaction of hydroxyl-t-butyl radicals with 1 and 2. In particular, 5% t-butanol was added to an N2O saturated aqueous fullerene solution and irradiated.58,59 Like before the only reactive form is ˙OH, which abstracts a hydrogen from one of the methyl groups to form (CH3)2˙(CH2)COH. In contrast to the reductive nature of (CH3)2˙C(OH), (CH3)2˙(CH2)COH is redoxinactive and is thought tentatively to react with 1 or 2via addition:
| | | (CH3)3COH + ˙OH → (CH3)2˙(CH2)COH + H2O | (10) |
| | | 1/2 + (CH3)2˙(CH2)COH → (1-(CH2)(CH3)2COH)˙/(2-(CH2)(CH3)2COH)˙ | (11) |
Differential absorption changes all throughout the visible and near-infrared region recorded upon pulse radiolysis of 1 or 2 confirm, indeed, the formation of the radical adducts, namely (1-(CH2)(CH3)2COH)˙/(2-(CH2)(CH3)2COH)˙. Most importantly, the maxima at 430 nm for 1 and at 410 nm for 2 – Fig. S6 and S7† – are exact matches of the spectral features registered for the analog (1-CH3)˙/(2-CH3)˙ adducts.
Variation of the concentration impacted the formation of (1-(CH2)(CH3)2COH)˙/(2-(CH2)(CH3)2COH)˙ in a linear manner. The underlying rate was found to be dependent on the concentration with rate constants – 7.6 × 107 M−1 s−1 for 1 and 4.5 × 107 M−1 s−1 for 2 (Fig. S8 and Fig. S9†) – that are only slightly slower than those found for the formation of (1-CH3)˙/(2-CH3)˙. A reasonable rationale for this slow down is the larger size of (CH3)2˙(CH2)COH versus ˙CH3, which impacts the diffusion. Steric hindrance is also expected to contribute to the difference in rate constant.
Similar observations were made when the reaction of, for example, 1 with N3˙ radicals was investigated. N3˙ radical formation requires a sufficient amount of sodium azide (10−3 M). Here, a reaction of N3− with ˙OH leads to the desired N3˙ radicals:60
| | | N3− + ˙OH → N3˙ + OH− | (12) |
| | | 1/2 + N3˙ → (1-N3)˙/(2-N3)˙ | (13) |
Again, the transient absorption spectrum of (1-N3)˙ gave rise to a maximum at 440 nm – see Fig. S10.† The rate constant for the reaction of 1 with N3˙ was determined by varying the initial fullerene concentration and plotting the obtained pseudo first-order rate constant for the formation of (1-N3)˙ obtaining the second-order rate constant from the slope of the linear fit – see Fig. S11.† The second-order rate constant was 6.9 × 108 M−1 s−1 for 1.
In the next step we turned to the reaction of 1 and 2 with the superoxide radical. The superoxide radical is easily formed radiolytically in aqueous media that are purged with a mixture of N2O and O2 and that contain 5 × 10−3 M formate.61–63 Initially, reducing CO2˙− radicals are formed in N2O saturated aqueous solutions containing 0.1 M HCOONa. Under these conditions, ˙OH radicals – directly and indirectly formed via scavenging eaq− with N2O – react primarily with formate to yield CO2˙−. The latter reduces O2 to O2˙−:
| | | HCOO− + ˙OH → CO2˙− + H2O | (15) |
| | | CO2˙− + O2 → CO2 + O2˙− | (16) |
| | | 1/2 + O2˙− → (1-O2)˙−/(2-O2)˙− | (17) |
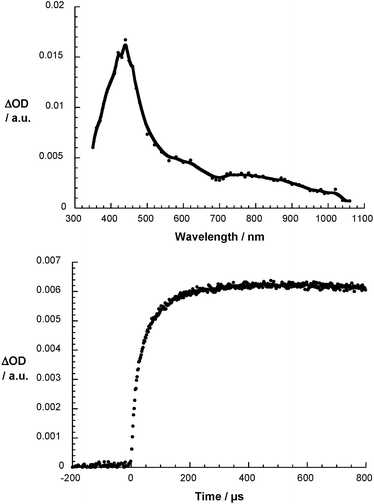
Fig. 4 illustrates the differential absorption changes recorded upon the reaction of O2˙− with 1. In particular, the rapid formation of a new transient emerges. For 1, a distinct maximum is seen at 440 nm. Dendrofullerene 2, on the other hand, gives rise to a transient maximum at 430 nm – Fig. S12.† Notable is in both cases the lack of the diagnostic near-infrared transition that would imply the formation of 1˙− and 2˙−. This lack implies the absence of any charge transfer reactions to form 1˙− and 2˙− either directly – via a direct impact mechanism – or indirectly – via a dissociative mechanism – and the presence of radical addition reactions affording (1-O2)˙− and (2-O2)˙−.
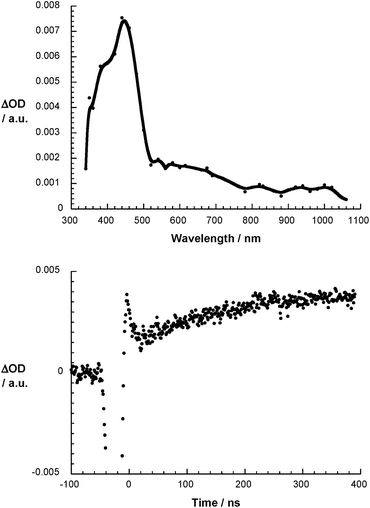 |
| | Fig. 4 Differential absorption spectrum of 1 (3 × 10−4 M) upon pulse radiolysis in N2O/O2 saturated aqueous solution pH = 7.2 upon adding 5 × 10−3 M HCOONa 300 ns after the electron pulse (upper part) and the corresponding time absorption profile at 430 nm (lower part). | |
Interesting is the fact that the analyses of the Stern–Volmer plots gave values for the addition reactions of 1.3 × 1010 M−1 s−1 (1 – Fig. 5) and 1.7 × 1010 M−1 s−1 (2 – Fig. S13†). Considering that the reactions were performed in aqueous solutions, we conclude that the addition of O2˙− to 1 and 2 is essentially diffusion-controlled.
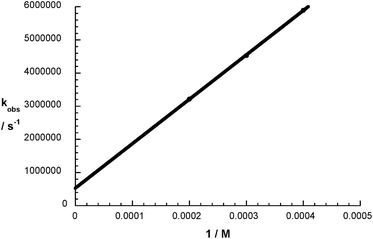 |
| | Fig. 5 Stern–Volmer plot of 1 in aqueous N2O and O2 saturated buffer solution pH = 7.2 with an addition of HCOONa (5 × 10−3 M). | |
To test the stability of the rapidly formed (1-O2)˙−/(2-O2)˙− some of the experimental parameters – pH, fullerene concentration and radiation dose – were varied systematically. Interestingly, when lowering the pH from 7.2 to 6.2, the (1-O2)˙− lifetime is changed from an initial 150 μs to a final 20 μs. Further variations of the pH such as going beyond 6.2 were not practical due to fast protonation of O2˙−.63 Taking this pH dependence, we derived a rate constant of 1010 M−1 s−1. The underlying protonation reactions induced a decay of the initial products and a growth of new products. For instance, as (1-O2)˙−/(2-O2)˙− decay, new absorption maxima developed at 420 nm for 1 (Fig. 6) and at 400 nm for 2 (Fig. S14†). The insolubility of 2 at a pH lower than 8.2 hampered a meaningful investigation. Once formed, (1-O2H)˙ turned out to be stable without showing a significant decay up to 80 ms, which is the upper time limit of the pulse radiolysis set-up. Similarly, changing the fullerene concentration from, for example, 5 × 10−5 to 2 × 10−4 M, while keeping the pH at 7.2, also affected the (1-O2)˙− lifetime: from 800 μs down to 150 μs. Only the radiation dose and subsequently the (1-O2)˙−/(2-O2)˙− concentration exerted no influence on the decay kinetics. Based on this observation we discard the pathway, in which the (1-O2)˙−/(2-O2)˙− may set up an equilibrium with its dimer, etc. Such an equilibrium has been proposed in the general reaction mechanism of (C60-CH3)˙:51
| | | (1-O2)˙−/(2-O2)˙− + H+ → (1-O2H)˙/(2-O2H)˙ | (18) |
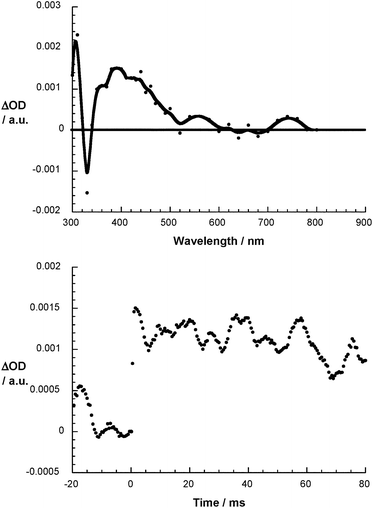 |
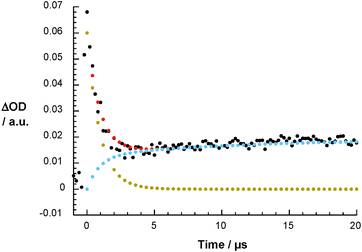
| | Fig. 6 Differential absorption spectrum of 1 (10−5 M) upon pulse radiolysis in N2O/O2 saturated aqueous solution pH = 7.2 upon adding 5 × 10−3 M HCOONa 80 ms after the electron pulse (upper part) and the corresponding time absorption profile at 420 nm (lower part). | |
To shed further light onto the stability of the (1-O2H)˙ intermediate the time-resolved pulse radiolysis studies were completed by steady-state gamma radiolysis. Here, with increasing irradiation times a new species – absorbing around 420 nm – was formed. Generating the differential absorption spectrum helps to confirm the identity of the transient, namely being identical to that product seen on the 80 ms time scale of the pulse radiolysis experiments – please compare Fig. 6 and Fig. 7.
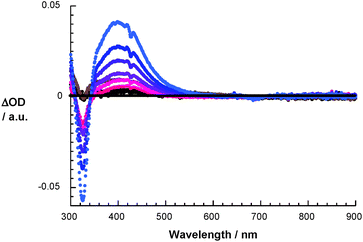 |
| | Fig. 7 Differential absorption spectra of 1 (10−5 M) upon 60Co gamma radiation in N2O/O2 saturated aqueous solution pH = 7.2 upon adding 5 × 10−3 M HCOONa after different irradiation times. | |
In the final assays, (1-O2)˙− was reacted with several electron acceptors. In this context, we used a series of p-benzoquinones, that is, p-benzoquinone and 2,5-dimethyl-p-benzoquinone, that have different electron affinities/reduction potentials. Particular emphasis was placed on avoiding direct reduction of the p-benzoquinones by CO2˙− and/or O2˙−. Please note that for the direct reduction of p-benzoquinone by CO2˙− and O2˙−, rate constants of 6.6 × 109 M−1 s−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 64 and 9.0 × 108 M−1 s−1 have been established.65 As a matter of fact, we note that (1-O2)˙− reacts with p-benzoquinones involving the formation of the one-electron reduced radical semiquinone, BQ˙−. Fig. 8 provides spectroscopic and kinetic evidence for this hypothesis, namely that the decay of the (1-O2)˙− transient with its characteristic features at 360/440 nm is linked to the BQ˙− formation.66 For BQ˙− a differential absorption spectrum is registered that is in perfect agreement with previously published results.67 From a concentration dependence we derived the second-order rate constants for the reaction of (1-O2)˙− with p-benzoquinone and 2,5-dimethyl-p-benzoquinone of 6.5 × 108 and 1.4 × 108 M−1 s−1, respectively. Importantly, the difference in rate constants – 6.5 × 108versus 1.4 × 108 M−1 s−1 – scales well with the differences in driving force for the underlying bimolecular electron transfer reactions, that is, p-benzoquinone having a higher electron affinity than 2,5-dimethyl-p-benzoquinone.
64 and 9.0 × 108 M−1 s−1 have been established.65 As a matter of fact, we note that (1-O2)˙− reacts with p-benzoquinones involving the formation of the one-electron reduced radical semiquinone, BQ˙−. Fig. 8 provides spectroscopic and kinetic evidence for this hypothesis, namely that the decay of the (1-O2)˙− transient with its characteristic features at 360/440 nm is linked to the BQ˙− formation.66 For BQ˙− a differential absorption spectrum is registered that is in perfect agreement with previously published results.67 From a concentration dependence we derived the second-order rate constants for the reaction of (1-O2)˙− with p-benzoquinone and 2,5-dimethyl-p-benzoquinone of 6.5 × 108 and 1.4 × 108 M−1 s−1, respectively. Importantly, the difference in rate constants – 6.5 × 108versus 1.4 × 108 M−1 s−1 – scales well with the differences in driving force for the underlying bimolecular electron transfer reactions, that is, p-benzoquinone having a higher electron affinity than 2,5-dimethyl-p-benzoquinone.
| | | (1-O2)˙− + BQ → 1 + O2 + BQ˙− | (19) |
Conclusions
In summary, our fully-fledged spectroscopic and kinetic investigations leave no doubt about a diffusion-controlled addition mechanism (1010 M−1 s−1) by which superoxide radical reacts with 1 and 2 – and most likely with other fullerenes/fullerene derivatives in aqueous solutions (!) – to yield (C60-O2)˙−. The major deactivation path of (C60-O2)˙− is a pH dependent protonation (1010 M−1 s−1). Despite lacking unambiguous evidence for the formation of C60˙− – via a direct impact or an indirect dissociation mechanism41,42,68 – (C60-O2)˙− is still redoxactive. As a matter of fact, the reaction of (C60-O2)˙− with several p-benzoquinones proceeds via the formation of the one-electron reduced radical semiquinone and, in turn, corroborates its function as the key intermediate of the SOD activity of fullerenes. On the other hand, the formation of, for example, (C60-CH3)˙ is activation-controlled resembling an earlier established trend in organic solvents.57 Moreover, the formation rate is seen to depend on the available double bonds in the C60 derivatives, that is, mono- (1) versus tris-adduct (2).
Acknowledgements
We like to thank Prof. Brede and his group from the Leipzig University, Leipzig, Germany and the Notre Dame Radiation Lab, Notre Dame, Indiana USA for the support during the pulse radiolysis and gamma radiolysis measurements. We also appreciate fruitful discussions with Prof. Ivana Ivanovic-Burmazovic. This work was supported by the Deutsche Forschungsgemeinschaft through SFB583.
References
- A. D. Darwish, Annu. Rep. Prog. Chem., Sect. A, 2008, 104, 360 RSC.
- R. Tenne, Nat. Nanotechnol., 2006, 1, 103 CrossRef CAS.
- M. Satoh and I. Takayanagi, J. Pharmacol. Sci., 2006, 100, 513 CrossRef CAS.
- M. Prato and M. Maggini, Acc. Chem. Res., 1998, 31, 519 CrossRef CAS.
- M. Maggini, G. Scorruno, M. Prato, G. Brusatin, P. Innocenzi, M. Guglielrni, A. Renier, R. Signorini, M. Meneghetti and R. Bozio, Adv. Mater., 1995, 7, 404 CrossRef CAS.
- J. R. Morton, F. Negri and K. F. Preston, Acc. Chem. Res., 1998, 31, 63 CrossRef CAS.
- F. Diederich, Pure Appl. Chem., 1997, 69, 395 CrossRef CAS.
- F. Djojo, A. Herzog, I. Lamparth, F. Hampel and Andreas Hirsch, Chem.–Eur. J., 1996, 2, 1537 CrossRef CAS.
- M. Prato, V. Lucchini, M. Maggini, E. Stimpfl, G. Scorrano, M. Eiermann, T. Suzuki and Fred Wudl, J. Am. Chem. Soc., 1993, 115, 8479 CrossRef CAS.
- M. Gallo, A. Favila and D. Glossman-Mitnik, Chem. Phys. Lett., 2007, 447, 105 CrossRef CAS.
- D. Bonifazi, O. Enger and F. Diederich, Chem. Soc. Rev., 2007, 36, 390 RSC.
- I. Lamparth and A. Hirsch, J. Chem. Soc., Chem. Commun., 1994, 1727 RSC.
- M. Bergamin, T. Da Ros, G. Spalluto, A. Boutorine and M. Prato, Chem. Commun., 2001, 17 RSC.
- T. Itoh, M. Mishiro, K. Matsumoto, S. Hayase, M. Kawatsura and M. Morimoto, Tetrahedron, 2008, 64, 1823 CrossRef CAS.
- D. M. Guldi, J. Phys. Chem. B, 2000, 104, 1483 CrossRef CAS.
- D. Vitalini, E. Spina, S. Dattilo, P. Mineo and E. Scamporrino, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2145 CrossRef CAS.
- D. M. Guldi, H. Hungerbühler and K.-D. Asmus, J. Phys. Chem., 1995, 99, 13487 CrossRef CAS.
- D. M. Guldi, H. Hungerbühler and K.-D. Asmus, J. Phys. Chem. B, 1999, 103, 1444 CrossRef CAS.
- D. M. Guldi, H. Hungerbühler and K.-D. Asmus, J. Phys. Chem. A, 1997, 101, 1783 CrossRef CAS.
- K. I. Priyadarsini, H. Mohan, A. K. Tyagi and J. P. Mittal, J. Phys. Chem., 1994, 98, 4756 CrossRef CAS.
- A. F. Clements, J. E. Haley, A. M. Urbas, A. Kost, R. D. Rauh, J. F. Bertone, F. Wang, B. M. Wiers, D. Gao, T. S. Stefanik, A. G. Mott and D. M. Mackie, J. Phys. Chem. A, 2009, 113, 6437 CrossRef CAS.
- F. Djojo and A. Hirsch, Chem.–Eur. J., 1998, 4, 344 CrossRef CAS.
- F. Beuerle and A. Hirsch, Chem.–Eur. J., 2009, 15, 7434 CrossRef CAS.
- F. Beuerle and A. Hirsch, Chem.–Eur. J., 2009, 15, 7447 CrossRef CAS.
- P. Witte, F. Hörmann and A. Hirsch, Chem.–Eur. J., 2009, 15, 7423 CrossRef CAS.
- P. Witte, F. Beuerle, U. Hartnagel, R. Lebovitz, A. Savouchkina, S. Sali, D. Guldi, N. Chronakisd and A. Hirsch, Org. Biomol. Chem., 2007, 5, 3599 RSC.
- R. Partha, M. Lackey, A. Hirsch, S. W. Casscells and J. L. Conyers, J. Nanobiotechnol., 2007, 5, 6 CrossRef.
- M. Brettreich and A. Hirsch, Tetrahedron Lett., 1998, 39, 2731 CrossRef CAS.
- S. S. Ali, J. I. Hardt, K. L. Quick, J. S. Kim-Han, B. F. Erlanger, T.-T. Huang, C. J. Epstein and L. L. Dugan, Free Radical Biol. Med., 2004, 37, 1191 CrossRef CAS.
- A. A. Corona-Morales, A. Castell, A. Escobar, R. Drucker-Colin and L. Zhang, J. Neurosci. Res., 2003, 71, 121 CrossRef CAS.
- H.-M. Huang, H.-C. Oua, S.-J. Hsieha and Long-Yong Chiang, Life Sci., 2000, 66, 1525 CrossRef CAS.
- I. Y. Podolski, Z. A. Podlubnaya, E. A. Kosenko, E. A. Mugantseva, E. G. Makarova, L. G. Marsagishvili, M. D. Shpagina, Y. G. Kaminsky, G. V. Andrievsky and V. K. Klochkov, J. Nanosci. Nanotechnol., 2007, 7, 1479 CrossRef CAS.
- S.-F. Tzeng, J.-L. Lee, J.-S. Kuo, C.-S. Yang, P. Murugan, L. A. Tai and Kuo Chu Hwang, Brain Res., 2002, 940, 61 CrossRef CAS.
- L. Xiao, K. Matsubayashi and N. Miwa, Arch. Dermatol. Res., 2007, 299, 245 CrossRef CAS.
- B. Daroczi, G. Kari, M. F. McAleer, J. C. Wolf, U. Rodeck and A. P. Dicker, Clin. Cancer Res., 2006, 12, 7086 CrossRef CAS.
- D. Pantarotto, A. Bianco, F. Pellarini, A. Tossi, A. Giangaspero, I. Zelezetsky, J.-P. Briand and M. Prato, J. Am. Chem. Soc., 2002, 124, 12543 CrossRef CAS.
- N. Tsao, T.-Y. Luh, C.-K. Chou, J.-J. Wu, Y.-S. Lin and H.-Y. Lei, Antimicrob. Agents Chemother., 2001, 45, 1788 CrossRef CAS.
- N. Tsao, P. P. Kanakamma, T.-Y. Luh, C.-K. Chou and H.-Y. Lei, Antimicrob. Agents Chemother., 1999, 43, 2273 CAS.
- F. Beuerle, P. Witte, U. Hartnagel, R. Lebovitz, C. Parng and A. Hirsch, J. Exp. Nanosci., 2007, 2, 147 Search PubMed.
- C. Y. Usenko, S. L. Harper and R. L. Tanguay, Toxicol. Appl. Pharmacol., 2008, 229, 44 CrossRef CAS.
- Y. Yamakoshi, S. Sueyoshi, K. Fukuhara and N. Miyata, J. Am. Chem. Soc., 1998, 120, 12363 CrossRef CAS.
- Y. Yamakoshi, N. Umezawa, A. Ryu, K. Arakane, N. Miyata, Y. Goda, T. Masumizu and T. Nagano, J. Am. Chem. Soc., 2003, 125, 12803 CrossRef CAS.
- G.-F. Liu, M. Filipovic, I. Ivanovi -Burmazovi, F. Beuerle, P. Witte and A. Hirsch, Angew. Chem., Int. Ed., 2008, 47, 3991 CrossRef CAS.
- O. Brede, H. Orthner, V. Zubarev and R. Hermann, J. Phys. Chem., 1996, 100, 7097 CrossRef CAS.
- R. H. Schuler, L. K. Patterson and E. Janata, J. Phys. Chem., 1980, 84, 2088 CrossRef CAS.
- G. L. Hug, Y. Wang, C. Schöneich, P.-Y. Jiang and R. W. Fessenden, Radiat. Phys. Chem., 1999, 54, 559 CrossRef.
- R. V. Bensasson, M. Brettreich, J. Frederiksen, H. Göttinger, A. Hirsch, E. L. Land, S. Leach, D. J. McGravey and H. Schönberger, Free Radical Biol. Med., 2000, 29, 26 CrossRef CAS.
- P. Wardman, J. Phys. Chem. Ref. Data, 1989, 18, 1637 CAS.
- W. Braun, J. T. Herron and D. K. Kahaner, Int. J. Chem. Kinet., 1988, 20, 51 CrossRef CAS.
- N. M. Dimitrijevic, Chem. Phys. Lett., 1992, 194, 457 CrossRef.
- N. M. Dimitrijevic, P. V. Kamat and R. W. Fessenden, J. Phys. Chem., 1993, 97, 615 CrossRef.
- K. I. Priyadarsini, H. Mohan, P. R. Birkett and J. P. Mittal, J. Phys. Chem., 1996, 100, 501 CrossRef CAS.
- D. Guldi, H. Hungerbühler, E. Janata and K.-D. Asmus, J. Chem. Soc., Chem. Commun., 1993, 84 RSC.
- D. M. Guldi and K.-D. Asmus, Radiat. Phys. Chem., 1999, 56, 449 CrossRef CAS.
- C. B. Gilbert, R. O. C. Norman and R. C. Sealy, J. Chem. Soc., Perkin Trans. 2, 1975, 303 RSC.
- D. J. Nelson, J. Phys. Chem., 1978, 82, 1400 CrossRef CAS.
- D. M. Guldi, H. Hungerbühler, E. Janata and K.-D. Asmus, J. Phys. Chem., 1993, 97, 11258 CrossRef CAS.
- R. Livingston and H. Zeldes, J. Am. Chem. Soc., 1966, 88, 4333 CrossRef CAS.
- M. Simic, P. Neta and E. Hayon, J. Phys. Chem., 1969, 73, 3794 CrossRef CAS.
- E. Hayon and M. Simic, J. Am. Chem. Soc., 1970, 92, 7486 CrossRef CAS.
- G. E. Adams and R. L. Willson, Trans. Faraday Soc., 1969, 65, 2981 RSC.
- B. H. J. Bielski and H. W. Richter, J. Am. Chem. Soc., 1977, 99, 3019 CrossRef CAS.
- B. H. J. Bielski, D. E. Cabeli and R. L Arudi, J. Phys. Chem. Ref. Data, 1985, 14, 1041 CAS.
- L. R. Willson, Trans. Faraday Soc., 1971, 67, 3020 RSC.
- M. Simic and E. Hayon, Biochem. Biophys. Res. Commun., 1973, 50, 364 CrossRef CAS.
- S. Fukuzumi, Y. Ono and T. Keii, Bull. Chem. Soc. Jpn., 1973, 46, 3353 CAS.
- A. Maroz and O. Brede, Radiat. Phys. Chem., 2003, 67, 275 CrossRef CAS.
- K. Ohkubo, H. Kitaguchi and S. Fukuzumi, J. Phys. Chem. A, 2006, 110, 11613 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Transient absorption spectra of 1˙−/2˙−. Transient absorption spectra and time absorption profiles of (2-CH3)˙, (1-(CH2)(CH3)2COH)˙/(2-(CH2)(CH3)2COH)˙, (2-O2)˙− and (1-N3)˙ as well as Stern–Volmer plots for the formation of (1-CH3)˙/(2-CH3)˙, 1-(CH2)(CH3)2COH)˙/(2-(CH2)(CH3)2COH)˙, (2-O2)˙− and (1-N3)˙. See DOI: 10.1039/b917346n |
| ‡ Current address: Department of Chemical and Biological Engineering/Physical Chemistry, Chalmers University of Technology, SE-412 96, Göteborg, Sweden. Fax: +46 31 772 38 58; Tel: +46 31 772 38 55; |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.