Deposition of intact tetrairon(III) single molecule magnet monolayers on gold: an STM, XPS, and ToF-SIMS investigation†
Francesco
Pineider
a,
Matteo
Mannini
ab,
Chiara
Danieli
c,
Lidia
Armelao
d,
Federica M.
Piras
e,
Agnese
Magnani
e,
Andrea
Cornia
*c and
Roberta
Sessoli
*a
aLa.M.M.-Department of Chemistry and INSTM research unit, University of Florence, Via della Lastruccia 3, 50019, Sesto Fiorentino (FI), Italy. E-mail: roberta.sessoli@unifi.it; Fax: +39-0554573372; Tel: +39-0554573268
bISTM-CNR, URT Firenze, Via della Lastruccia 3, 50019, Sesto Fiorentino (FI), Italy
cDepartment of Chemistry and INSTM research unit, University of Modena and Reggio Emilia, Via G. Campi 183, 41100, Modena, Italy. E-mail: acornia@unimore.it
dISTM-CNR and INSTM research unit, Department of Chemistry, University of Padova, Via Marzolo 1, 35131, Padova, Italy
eDepartment of Chemical and Biosystems Science and Technology, University of Siena, Via A. Moro 2, 53100, Siena, Italy
First published on 9th November 2009
Abstract
The ability of a tetranuclear iron(III) single-molecule magnet functionalized with thioacetyl-terminated ligands to form monolayers on gold has been investigated by a multitechnique approach based on STM, XPS and ToF-SIMS. We discuss in detail several aspects, which are relevant to the reported observation of a memory effect on the monolayer prepared from dichloromethane (Mannini et al., Nat. Mater., 2009, 8, 194). In particular we show that the adsorbate comprises intact surface-bound Fe4 clusters as opposed to microcrystals or multilayers. The influence of the solvent used for the self-assembly process on the morphology, composition and structure of the adsorbate is also studied for four different solvents (dichloromethane, n-hexane, toluene, 1,4-dioxane).
1 Introduction
The realization of basic functional units using molecular building blocks represents significantly the stride of fundamental research toward near-future technological applications. The need for completely new paradigms appears most clearly in the field of electronics, when considering that traditional top-down nanofabrication techniques are approaching their physical limits and will soon be unable to satisfy the quest for ever-smaller electronic devices. For this reason, several types of functional molecular materials have been studied to replace classical electronic units such as mass memory storage and logic gates.1 Among these, single-molecule magnets (SMMs) seem highly promising candidates in the field of magnetic storage and molecular spintronics2 because they show magnetic hysteresis3 at the molecular level in addition to rich quantum behaviour.4 Since many proposed applications require the addressing of individual molecules,5 our group and others have undertaken the organization of derivatives of the archetypal Mn12 SMM on gold6–8 and silicon surfaces.9–11 Although the morphological and compositional data obtained using STM and XPS, respectively, suggested the presence of intact SMMs at the gold surface, a number of subsequent experiments using MCD,12 XAS and XMCD13 have proven that Mn12 complexes are chemically unstable when bound to gold14,15 and are structurally too weak to maintain SMM behavior even at the surface of bulk samples.16 This case highlights the difficulties inherent to the investigation of polynuclear metal complexes at surfaces and the need for a multi-technique approach.A different class of iron(III)-based SMMs, namely Fe4 complexes, has been recently shown to exhibit the chemical and structural robustness required for deposition on surfaces. Their propeller-like structure17–19 (see Fig. 1) stabilizes a S = 5 ground spin state and generates an anisotropy barrier as high as 17 K.20 Furthermore, they can be functionalized at will to promote interaction with different substrates,21 such as gold surfaces22 and silicon substrates23 or carbon nanotubes.24,25
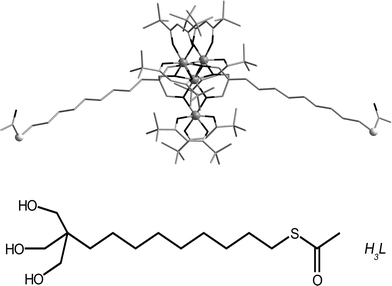 | ||
| Fig. 1 Top: molecular structure of the Fe4C9SAc complex with iron atoms drawn as large grey spheres, sulfur atoms as pale grey spheres, carbon as light grey sticks and oxygen as black sticks. Bottom: scheme of the sulfur-functionalized tripodal ligand H3L used in this work. | ||
Experiments at large-scale facilities using low-temperature XMCD22 have demonstrated that the electronic structure and magnetic properties of Fe4 molecules deposited on gold from dichloromethane (DCM) solution closely resemble those of bulk phases (antiferromagnetic intramolecular interactions, high-spin ground state, hysteresis loop). This important result confirms the intactness of Fe4 complexes at surfaces and, at the same time, provides a crucial proof-of-concept, i.e. that SMMs can be linked to metallic surfaces while not altering their characteristic functionality.
The Fe4 derivative specifically designed for grafting to gold surfaces, hereafter denoted as Fe4C9SAc, has formula Fe4(L)2(dpm)6, where Hdpm is dipivaloylmethane and H3L is the sulfur-functionalised triol ligand 11-(acetylthio)-2,2-bis(hydroxymethyl)undecan-1-ol (Fig. 1).21 Such ligand design is motivated by the strong affinity of sulfur toward gold; notice that acetyl-protected sulfur atoms are used instead of free thiols, because of the redox instability of iron(III) ions in the presence of –SH groups; notice also that acetyl-protected thiols have been used without deprotection by an exogenous base to generate self-assembled monolayers (SAMs).26–28 However it is still debated whether the acetyl group is retained when the Au–S bond is formed.27,28 The long alkyl chain was chosen as an insulating spacer, in order to minimise the interaction of free electrons of gold with the magnetic Fe4 core.
In this work we present a complete morphological, chemical and structural investigation of Fe4C9SAc adsorbates on gold. Four different solvents including DCM have been employed, as strong solvent effects have been already observed in monolayers of Mn12 complexes29 and simpler molecules.30,31 The solvents examined in this study are DCM (low viscosity, moderately polar), n-hexane (low-viscosity, apolar), toluene (moderately viscous, apolar and aromatic) and 1,4-dioxane (very viscous, polar). Information on the morphology of the surface, as revealed by STM imaging, has been complemented by semi-quantitative chemical composition data obtained using XPS spectroscopy and by structural information provided by ToF-SIMS. Monolayers obtained from these solvents, called Fe4C9SAc-exDCM, -exHex, -exTol and -exDiox, respectively, were characterised using the above-mentioned techniques, which demonstrated the intactness of Fe4 units in all samples but Fe4C9SAc-exDiox. Information was gained on the nature of the deposits, which are shown to largely consist of an array of surface-bound complexes rather than aggregates, microcrystals or multilayers. In addition, the arrangement of Fe4 units on the gold surface and the fate of acetyl groups were inferred.
2 Experimental
Sample preparation
Complex Fe4C9SAc was prepared by reacting the precursor [Fe4(OMe)6(dpm)6] and the tripodal ligand H3L, as detailed elsewhere.20,21 Anhydrous dichloromethane, 1,4-dioxane, n-hexane and toluene were purchased from Sigma-Aldrich and used as received. The cluster was found to be stable in the four solvents investigated for at least two days, see ESI† for the details on the aging experiments. Gold 5 mm × 5 mm substrates (Agilent Technologies) consisted of 150 nm thick layers of polycrystalline gold evaporated on mica; substrates were annealed with a hydrogen flame immediately before use in order to promote Au(111) reconstruction of the surface. To prepare monolayers of Fe4C9SAc, the gold substrates were immersed for 20 hours in a 0.3 mM DCM solution of the complex in the investigated solvents and were subsequently rinsed by immersion in the same pure solvent for 10 minutes. The sample of bulk Fe4C9SAc was prepared by drop casting 50 µl of a 0.3 mM solution of the complex on either gold or silicon surfaces, to afford reasonably flat layers with a thickness of a few hundreds of nanometers. All handling operations were carried out under the dry nitrogen atmosphere of a glove box.STM measurements
In-air STM characterisation was performed on an NT-MDT P47-pro instrument working with a custom made low current head and using 90:10 Pt/Ir tips obtained by mechanical sharpening. Tunneling parameters are reported for each scan in the text.XPS measurements
XPS measurements were performed on a Perkin-Elmer Φ 5600-ci spectrometer using a monochromatised (1486.6 eV) AlKα radiation (15 kV, 300 W). The sample analysis area was 800 µm in diameter, and the working pressure was lower than 10−9 mbar. The spectrometer was calibrated assuming the binding energy (BE) of the Au4f7/2 line at 83.9 eV with respect to the Fermi level. The standard deviation for the BEs was ±0.2 eV. Samples were mounted on steel holders under dry nitrogen environment in a portable glove bag which was then connected to the fast-entry lock system of the XPS analytical chamber, in order to minimise air exposure and atmospheric contamination. Survey scans were run in the 0–1300 eV range and detailed scans were recorded for the C1s, O1s, S2s, S2p, Fe2p and Au4f photopeaks. All samples were sufficiently electrically conductive at room temperature that no compensation for charging effects was required. The residual BE shifts (<0.5 eV) were corrected by assigning to the C1s peak associated with aliphatic and adventitious hydrocarbons a BE of 284.8 eV.32 The analysis involved Shirley-type background subtraction33 and, whenever necessary, spectral deconvolution, which was carried out by non-linear least-squares curve fitting, adopting a Gaussian-Lorentzian sum function. The atomic composition of the samples was calculated by peak integration, using sensitivity factors provided by the spectrometer manufacturer (Φ V5.4A software) and taking into account the geometric configuration of the apparatus. The experimental uncertainty on the reported atomic composition values does not exceed 5%.ToF-SIMS measurements
ToF-SIMS analysis was carried out with a TRIFT III time-of-flight secondary ion mass spectrometer (Physical Electronics, Chanhassen, MN, USA) equipped with a gold liquid-metal primary ion source. Positive and negative ion spectra were acquired with a pulsed, bunched 22 keV Au+ (SAMs) and Au3+ (drop cast samples) primary ion beam, by rastering the ion beam over a 100 µm × 100 µm sample area. The primary ion dose was kept below 1011 ions/cm2 to maintain static SIMS conditions.34 Positive mass spectra were calibrated to CH3+ (m/z 15.023), C2H3+ (m/z 27.023), C3H5+ (m/z 41.039), and C11H19O2Fe+ (m/z 239.073, [Fe + dpm]+); negative data were calibrated to CH− (m/z 13.008), OH− (m/z 17.003), C2H− (m/z 25.008), and C11H19O2− (m/z 183.138, [dpm]−), in the low mass region. A number of peaks of increasing mass were assigned and added to the calibration set for an accurate mass calibration. Mass accuracy was better than 10 ppm. The mass resolution (m/Δm) was up to 8000 depending on the sample. Also for these experiments a portable glove bag was used to transfer the sample to the UHV chamber.3 Results and discussion
STM measurements
In the case of monolayers of Fe4C9SAc, STM studies were rather difficult due to the presence of a compact layer of molecules, which could be only partly resolved. A representative image of Fe4C9SAc-exDCM samples is shown in Fig. 2a. The topography image clearly shows a dense coverage throughout the gold surface. Image analysis reveals that the layer is made up of round objects whose average diameter is 2.5 ± 0.2 nm (see statistics in Fig. 2b), in good agreement with the size estimated for Fe4 from the crystal structure (1.8 × 1.8 × 3.4 nm).20 The best imaging conditions were found to correspond to an average tunneling interaction regime, with bias voltages around 0.3–0.4 V and tunneling currents of the order of 10 pA; no significant dependence was found on the tunneling conditions for what concerns the ability of the tip to displace Fe4 units from the surface. Molecular resolution, however, could be achieved only with interactions not stronger than those indicated above. | ||
| Fig. 2 STM topography images of typical Fe4C9SAc-exDCM (a), -exHex (c) and -exTol (d) monolayers (tunneling conditions: U = 0.35 V, I = 9 pA). Diameter statistics extracted from four images of Fe4C9SAc-exDCM samples are shown in (b). | ||
This is probably due to the fact that the tip, while not being able to actually remove the molecules from the surface, as seen instead on Mn12 adsorbates from DCM,29 scans through them producing noisy and unresolved images.
The absence of any long-range ordering in the adsorbate is an expected consequence of the bulky cluster core, which prevents lateral interactions between aliphatic chains and thus disfavours the closely-packed arrangement typical of alkanethiol SAMs.35 Very similar morphologies, i.e. a densely-packed layer of molecules, were found in samples Fe4C9SAc-exHex and -exTol (see Fig. 2c and d); on the other hand, STM imaging of Fe4C9SAc-exDiox samples failed to detect molecular adsorbates at the surface.
STM imaging is an important tool to detect monolayer formation and to optimize deposition protocols, although topographic information alone can be deceptive and can lead to erroneous conclusions. A clearer view on the compositional and structural properties of the adsorbates was provided by XPS and ToF-SIMS data, which are presented in the following sections.
XPS measurements
In order to have dependable reference data to study monolayers, we carried out a preliminary XPS characterisation on a massive sample of Fe4C9SAc obtained by the drop-cast method on gold. The survey scan (reported in ESI†) shows no strong signals from the underlying gold substrate, thus confirming that the technique is sampling bulk Fe4C9SAc. Detailed spectra of the S2p and C1s peaks are shown in Fig. 3a and b, respectively (Fe2p and O1s peaks are reported in ESI†). Atomic percentages are gathered in Table 1 and compared with calculated values, which agree very closely with the observed composition.| Fe4C9SAc samples | Fe | S | C | O |
|---|---|---|---|---|
| Calcd for Fe4O20C96S2 | 3.3 | 1.6 | 78.7 | 16.4 |
| Drop cast | 2.9 | 1.5 | 78.5 | 17.1 |
| BE | 711.1 | 163.9 | 284.8 | 531.2 |
| -exDCM Monolayer | 3.7 | 2.0 | 71.5 | 22.8 |
| BE | 711.2 | 162.7 | 284.8 | 532.2 |
| -exHex Monolayer | 4.6 | 2.4 | 69.5 | 23.5 |
| BE | 710.9 | 162.1, 163.6 | 284.8 | 531.9 |
| -exTol Monolayer | 3.7 | 2.3 | 73.3 | 20.7 |
| BE | 711.2 | 162.2 | 284.8 | 531.9 |
 | ||
| Fig. 3 Detailed XPS spectra of S2p (a) and C1s (b) peaks carried out on a drop cast and monolayer samples of Fe4C9SAc (labelling scheme in the legend of panel b). In panel a) SI, SII, and SIII indicate the BE of surface-bound, surface-free, and oxidized sulphur components, respectively. In panels (c) and (d) we show the deconvolution for the C1s peaks of the drop cast and the -exDCM absorbate, respectively. In (e), the grey scale coding on the ligands describes the various components used to deconvolute the C1s signal. Fe2p and O1s peaks are reported in ESI.† | ||
A deeper analysis of the C1s signal was attempted by peak deconvolution, and three components were found (see Fig. 3c): a dominant one at 284.8 eV that can be ascribed to aliphatic and adventitious carbon; a second component at higher BE (286.3 eV) that is attributed to oxygen-bound carbon in the trimethyolol group and in dpm ligands (see Fig. 3e); a very weak component at even higher BE (288.4 eV) that can be coherently assigned to the electron-depleted thioacetyl carbon. Relative integral values of the three components, which are reported in Table 2 along with their BEs and FWHMs, are in close agreement with the expected values. The O1s peak could not be resolved into the three components originating from trimethylol, dpm and acetyl groups. Indeed, a limited peak shift is expected for these types of oxygen atoms.32,36 The sulfur S2p photopeak is centered at 163.9 eV, i.e. close to the BE exhibited by unbound thioacetyls in acetyl-protected dithiols27 or in derivatives of calix[4]arene37 on gold (163.4–163.6 eV for S2p(3/2)).
XPS studies on monolayer samples Fe4C9SAc-exDCM, -exHex and -exTol required very long acquisition times to improve the signal-to-noise ratio. A survey scan, taken in the energy range of 0–1300 eV (see ESI†), is dominated by the strong peaks originating from gold photoelectrons and shows additional signals from iron, sulfur, carbon and oxygen. Detailed scans of the S2p and C1s peaks are shown in Fig. 3a and b respectively, while Fe2p and O1s peaks are reported in ESI.† The spectral features are very similar to those observed in the drop-cast sample, all photopeaks showing relatively low broadening and the expected BEs. No significant charging effects are evident, indicating that the neutrality of the single layer of molecules is easily restored through the conducting gold surface.
The atomic percentages obtained by peak integration (Table 1) are in reasonable agreement with the calculated ones. In particular, the Fe:S ratio is in the range 1.6–1.9 (vs. 1.9 in the drop-cast sample and an expected value of 2.0), suggesting that the clusters are largely intact at the gold surface. On the other hand, carbon and oxygen integrals do not perfectly agree with the expected values, the former being less than expected, and the latter in slight excess.
The analysis of the S2p photopeaks provides important information on the arrangement of the Fe4 units on the gold surface. According to the molecular structure revealed by X-ray diffraction, the alkyl chains are long enough to allow the simultaneous binding of the two sulfur-based “alligator clips” to the gold surface, giving rise to a surface-parallel (or lying down) phase, using classical SAM terminology.27
Alternatively, Fe4 units could interact asymmetrically with the surface via only one sulfur atom, affording an upright (or standing up) phase as schematized in Fig. 4.
 | ||
| Fig. 4 Schematic view of the standing-up and lying-down grafting modes of the Fe4C9SAc SMM, with the relative orientation of the easy axis of magnetization indicated by the black arrow. The sulfur atom not bound to gold of the standing-up configuration is depicted as a dark sphere. | ||
Indeed, the S2p peak in Fe4C9SAc-exDCM and Fe4C9SAc-exTol samples is observed at 162.7–162.2 eV vs. 163.9 eV in the bulk reference (Fig. 3). The shift toward lower BE is taken as evidence of an interaction with gold27,36–38 (a similar shift is observed in the S2s photopeaks reported in ESI†). Since the peak is slightly broader as compared with bulk Fe4C9SAc, we cannot completely rule out the presence of a fraction of surface-free S atoms in these samples. The contribution from unbound sulfur is clearly detectable in the Fe4C9SAc-exHex sample, whose S2p photopeak can be fitted with two components centered at 162.1 (surface-bound sulfur, component SI in Fig. 3a) and 163.6 eV (surface-free sulfur, component SII). From the integrated areas, the two sulfur species occur in comparable amounts in the -exHex sample (see ESI†). We attribute these surface-free sulfur atoms to molecules wired to the surface via a single alligator clip or, less likely, forming an overlayer. The simultaneous presence of both surface-parallel and upright configurations of the SMM, with a prevalence of the first, would be consistent with the powder-like magnetic response of the Fe4C9SAc-exDCM sample observed within the resolution of XMCD measurements.22 In fact, the lying-down and standing-up molecules are expected to have their easy axes of magnetization in the plane of the substrate and roughly orthogonal to it, respectively (see Fig. 4). A 2:1 proportion of surface-perpendicular and parallel configurations would closely mimic a random distribution, while the ratio between surface-bound and surface-free sulfur atoms (5:1) would still be consistent with XPS data.
Deconvolution of the C1s signal39 (Fig. 3d) afforded the results reported in Table 2 for the Fe4C9SAc-exDCM sample. Similar results were obtained for Fe4C9SAc-exHex and Fe4C9SAc-exTol, the C1s spectra being superimposable. The excellent agreement with the calculated percentages provides a strong indication that acetyl groups are still present in the monolayers.40 From XPS data alone, however, it is not possible to ascertain whether acetyl groups are still bound to sulfur or remain quantitatively entrapped in the monolayer.28 On the other hand, Fe4C9SAc-exDiox samples showed peculiar features, with no detectable XPS signal from sulfur, a little amount of carbon with a shoulder at higher BE and anomalous amounts of iron and oxygen (see Fig. 3a,b and ESI† for details). This finding is coherent with the results of STM imaging (see previous section), that was not able to evidence morphological features comparable to those observed in the other samples.
ToF-SIMS measurements
Secondary ion mass spectrometry proved to be an extremely powerful tool for the inspection of monolayers of Fe4 on gold. Its soft ionisation mechanism is particularly suited for the desorption of fragile molecules, such as the metal ion polynuclear systems under study. Moreover, the technique is completely surface sensitive, since only the first molecular layers of the sample are involved in the ionic bombardment and subsequent desorption.First, positive and negative ToF-SIMS spectra of Fe4C9SAc drop cast on Si were acquired as a bulk reference (Fig. 5).
 | ||
| Fig. 5 Positive (a) and negative (b) ToF-SIMS spectra of Fe4C9SAc drop cast on Si (* signals from silicon grease contamination). | ||
The characteristic peaks detected and their assignments are listed in Table 3. In the high mass region (m/z 800–2200) of the positive ion spectrum (Fig. 5a), peaks are detected which are assigned to ion fragments generated by loss of one, two, and three dpm moieties from the molecular ion. Similar ion fragments have been previously observed on thin films of non-functionalized Fe4 derivatives obtained by thermal evaporation.41
| Drop cast | Monolayers on Au | Fragment | |||
|---|---|---|---|---|---|
| -exDCM | -exHex | -exTol | -exDiox | ||
| Positive ions | |||||
| 56 | 56 | Fe+ | |||
| 57 | 57 | 57Fe+/FeH+ | |||
| — | 197 | Au+ | |||
| 239 | 239 | [Fe + dpm]+ | |||
| 422 | 422 | [Fe + 2dpm]+ | |||
| 733 | 733 | [3Fe + 3dpm + O]+ | |||
| 1036 | 1036 | [3Fe + 3dpm + L + O]+ | |||
| 1380 | very weak (vw) | [M − 3dpm]+ | |||
| — | 1488 | [M − 2dpm − SAc]+ | |||
| — | 1520 | vw | 1520 | — | [M − 2dpm − Ac]+ |
| 1563 | 1563 | vw | 1563 | — | [M − 2dpm]+ |
| — | — | — | — | 1568 | [M* − dpm]+ |
| weak | 1671 | [M − dpm − SAc]+ | |||
| — | — | — | 1686 | — | [M − dpm − SAc − H + O]+ |
| — | 1702 | 1702 | 1702 | — | [M − dpm − Ac]+ |
| 1746 | 1746 | [M − dpm]+ | |||
| — | 1900 | [M − dpm − Ac + Au]+ | |||
| — | 2051 | vw | 2051 | — | [M − SAc + Au]+ |
| — | — | — | 2066 | — | [M − SAc − H + O + Au]+ |
| — | 2083 | vw | 2083 | — | [M − Ac + Au]+ |
| — | 2126 | [M + Au]+ | |||
| Negative ions | |||||
| 32 | 32 | S− | |||
| 75 | 75 | [SAc]− | |||
| 183 | 183 | [dpm]− | |||
| — | 197 | Au− | |||
| 902 | — | [2Fe + dpm + 2L]− | |||
| 1085 | — | [2Fe + 2dpm + 2L]− | |||
| 1507 | — | [M − Fe − 2dpm]− | |||
| 1929 | — | 1929 | — | 1929 | [M]− |
| — | 1934 | [M*]− ≡ [M − Ac + 3O]− | |||
| — | vw | vw | vw | 2131 | [M* + Au]− |
The intense peak at m/z 1036 is attributed to the ion fragment with formula [3Fe + 3dpm + L + O]+ originated by rearrangement of the Fe4 complex after loss of one Fe atom, three dpm groups and one tripodal ligand. In the low mass region (m/z 0–800, Fig. 5a), Fe+ and other characteristic ion peaks are observed. The high mass region of the negative ion spectrum (m/z 800–2200, Fig. 5b) is characterised by the molecular ion peak at m/z 1929 ([M]−), and other signals assigned to molecular ion fragments generated by loss of Fe atoms and/or dpm moieties.
Positive and negative ToF-SIMS spectra were then recorded on Fe4C9SAc monolayers prepared by dissolving the Fe4 complex in the four solvents. The recorded spectra present a rich variety of molecular secondary ions and show peaks assigned to molecular ions ([M]−), to key molecular ion clusters ([M + Au]+) and fragments (e.g. [M − dpm]+, [M − 2dpm]+, [M − 3dpm]+), demonstrating the presence of Fe4C9SAc complexes on Au.
In Fig. 6, we gather the high mass regions (m/z 800–2200) of the positive ion spectra recorded on the four samples. Besides the distinctive peaks of the Fe4C9SAc complex detected in the drop-cast sample, new characteristic signals are observed in the spectra of monolayers (Table 3). The Fe4C9SAc-exDCM sample (Fig. 6a) shows peaks at m/z 1702 ([M − dpm − Ac]+) and m/z 1520 ([M − 2dpm − Ac]+) assigned to molecular ion fragments generated by loss of the acetyl group, as well as ion–Au and molecular fragments–Au clusters at m/z 1900 ([M − dpm − Ac + Au]+), m/z 2083 ([M − Ac + Au]+), and m/z 2126 ([M + Au]+). These last peaks are not observed in the drop-cast sample, thus excluding that they are originated by the combination with the Au+ of the beam. The positive ion spectra acquired on Fe4C9SAc-exHex, -exTol, and -exDiox show some differences and similarities in the fragmentation patterns.
 | ||
| Fig. 6 Positive ToF-SIMS spectra of Fe4C9SAc-exDCM (a) and (b), -exHex (c), -exTol (d), and -exDiox (e) SAMs on Au. The mass regions m/z 800–2200 (a) and m/z 1500–2200 (b–e) are shown. | ||
The -exDCM and -exHex (Fig. 6c) spectra are identical, except for differences in the peak relative intensities, whereas the -exTol (Fig. 6d) spectrum shows additional features. Finally, the -exDiox spectrum (Fig. 6e) differs from the previous three spectra by the absence of the peaks at m/z 1900, 1702, and 1520 and the presence of a new peak at m/z 1568 tentatively attributed to an oxidized molecular ion fragment [M* − dpm]+ (Table 3), as discussed later.
The high mass regions (m/z 800–2200) of the negative ion spectra are characterised by very intense signals from the Au substrate (Fig. 7). All monolayer spectra show an intense peak at m/z 1934, slightly above the molecular ion. A more detailed investigation of the isotopic distribution (see ESI†) allows us to assign it to a species [M*]− with formula [Fe4(LL*)(dpm)6]−, where L* is an oxidized tripodal ligand containing a terminal sulfonate group (R–SO3−). Such a partially-oxidized species is absent in the drop-cast sample and is accompanied by the molecular ion peak in -exHex and -exDiox samples.
 | ||
| Fig. 7 Negative ToF-SIMS spectra of Fe4C9SAc drop cast (a) -exDCM (b), -exHex (c), -exTol (d), and -exDiox (e) SAMs on Au. | ||
The oxidation of sulfur to sulfonate has been previously observed in non-tightly packed layers of thiolated molecules upon exposure to atmospheric oxygen.42–44 The extent of oxidation was closely related to the surface coverage and to the layer thickness, which determine the permeability of the layer to atmospheric oxidants.45
A similar situation is expected to hold in the monolayers under study, where a tight-packing arrangement of alkylsulfanyl chains cannot be achieved due to the bulky cluster core. Notice that a hint of a S2p component around 168–169 eV (SIII component in Fig. 3a), attributable to oxidised sulfur,27,37 is only visible in the XPS spectra of the -exHex and -exDiox samples. The failure to clearly resolve such oxidized sulfur species in XPS spectra may be related to the different sensitivities and detection mechanisms of the two techniques. In fact, whereas XPS provides a quantitative detection of all S-containing species, the ToF-SIMS spectra are influenced by the interaction strength of the analyte with the substrate, as well as by ionization yields. In this respect, sulfonates are expected to be very easily detected by the ToF-SIMS technique because of their weak interaction with the surface and their negative charge. As a confirmation of our hypothesis, the ozone-induced oxidation of alkanethiol monolayers on gold was reported to become detectable at a much earlier stage by ToF-SIMS than by XPS.46
The absence of the [M*]− peak in the bulk reference as well as other peculiarities observed in the fragmentation patterns of -exDCM, -exHex and -exTol monolayers, like the extensive clustering with Au, support the occurrence in these samples of a large fraction of intact Fe4 complexes interacting with the Au surface via their terminal alligator clips. In addition, fragments are detected in all monolayer samples which still contain one ([M − 2dpm − SAc]+, [M − dpm − SAc]+, [M − dpm − Ac]+, [M − dpm − Ac + Au]+, [M*]−, [M* + Au]−) or two ([M + Au]+, [M − dpm]+, [M − 3dpm]+) acetyl groups. Combined with the XPS signature of an extensive S–Au interaction (especially in sample Fe4C9SAc-exDCM), the ToF-SIMS data suggest that absorption can be largely (though not exclusively) promoted by unprotected thioacetyls.
4 Conclusions
We have studied the morphology, composition and structure of a tetrairon(III) SMM functionalized with thioacetyl alligator clips and deposited on gold from four different solvents. Using DCM and toluene, the features of the S2p peak observed in XPS spectra at around 162 eV established an extensive S–Au interaction, thus ruling out the presence of a dominant fraction of aggregated or multilayered material on the surface. ToF-SIMS analysis confirmed the predominantly monolayer nature of the adsorbate by showing the presence of characteristic ion patterns and of extensive clustering with Au. In particular, we detected a partially-oxidized molecular ion featuring one terminal sulfonate group, as previously observed in alkanethiol monolayers deposited on gold. According to STM imaging, the adsorbates comprise disordered but densely-packed arrays of molecules.Deposition from n-hexane afforded similar results, though with a broader S2p photopeak, taken as evidence of comparable amounts of surface-bound (162.1 eV) and surface-free (163.6 eV) sulfur. By contrast, use of 1,4-dioxane failed to give adsorbates showing the expected morphology and XPS signatures.
The work presented here not only addressed the questions concerning the specific system under study, but also suggests some general methodological guidelines for the study and characterisation of monolayers of complex systems. The deposition of intact SMM layers is directly proved by their XMCD-detected magnetism.22 However, laboratory-scale instrumentation can be employed with success to individuate the most effective deposition protocols, provided that several techniques are used in parallel. In this respect, combined STM, XPS and ToF-SIMS studies contributed to gain considerable insight into the nature of SMM adsorbates and must be regarded as a valuable tool for the investigation of nanostructured molecular materials.
Acknowledgements
This work was supported by the EC projects: FP6-NMP3-CT-2005-515767 NE-MAGMANet, FP7-ICT-2007-211284 MolSpinQIP, ERANET “NanoSci-ERA: NanoScience in the European Research Area” (SMMTRANS – Three terminal transport through Single Molecule Magnets). Financial support by Italian projects: CNR-INSTM Promo, MIUR FIRB RBNE033KMA, MSE Industria 2015, ALADIN, and CARIPARO 2006 (Multi-layer optical devices based on inorganic and hybrid materials by innovative synthetic strategies) is also acknowledged.Notes and references
- C. Joachim, J. Gimzewski and A. Aviram, Nature, 2000, 408, 541 CrossRef CAS.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS.
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, UK, 2006 Search PubMed.
- D. Gatteschi, A. Cornia, M. Mannini and R. Sessoli, Inorg. Chem., 2009, 48, 3408 CrossRef CAS.
- A. Cornia, A. C. Fabretti, M. Pacchioni, L. Zobbi, D. Bonacchi, A. Caneschi, D. Gatteschi, R. Biagi, U. Del Pennino, V. De Renzi, L. Gurevich and H. S. J. Van Der Zant, Angew. Chem., Int. Ed., 2003, 42, 1645 CrossRef CAS.
- L. Zobbi, M. Mannini, M. Pacchioni, G. Chastanet, D. Bonacchi, C. Zanardi, R. Biagi, U. Del Pennino, D. Gatteschi, A. Cornia and R. Sessoli, Chem. Commun., 2005, 1640 RSC.
- A. Naitabdi, J. P. Bucher, P. Gerbier, P. Rabu and M. Drillon, Adv. Mater., 2005, 17, 1612 CrossRef CAS.
- G. G. Condorelli, A. Motta, I. L. Fragala, F. Giannazzo, V. Raineri, A. Caneschi and D. Gatteschi, Angew. Chem., Int. Ed., 2004, 43, 4081 CrossRef CAS.
- B. Fleury, L. Catala, V. Huc, C. David, W. Z. Zhong, P. Jegou, L. Baraton, S. Palacin, P. A. Albouy and T. Mallah, Chem. Commun., 2005, 2020 RSC.
- R. V. Martinez, F. Garcia, R. Garcia, E. Coronado, A. Forment-Aliaga, F. M. Romero and S. Tatay, Adv. Mater., 2007, 19, 291 CrossRef CAS.
- L. Bogani, L. Cavigli, M. Gurioli, R. L. Novak, M. Mannini, A. Caneschi, F. Pineider, R. Sessoli, M. Clemente-Leon, E. Coronado, A. Cornia and D. Gatteschi, Adv. Mater., 2007, 19, 3906 CrossRef CAS.
- M. Mannini, Ph. Sainctavit, R. Sessoli, Ch. Cartier dit Moulin, F. Pineider, M. A. Arrio, A. Cornia and D. Gatteschi, Chem.–Eur. J., 2008, 14, 7530 CrossRef CAS.
- S. Voss, M. Burgert, M. Fonin, U. Groth and U. Rudiger, Dalton Trans., 2008, 499 RSC.
- S. Voss, M. Fonin, U. Rudiger, M. Burgert, U. Groth and Y. S. Dedkov, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 045102 CrossRef.
- M. Mannini, F. Pineider, Ph. Sainctavit, L. Joly, A. Fraile-Rodriguez, M. A. Arrio, Ch. Cartier dit Moulin, W. Wernsdorfer, A. Cornia, D. Gatteschi and R. Sessoli, Adv. Mater., 2009, 21, 167 CrossRef.
- A. L. Barra, A. Caneschi, A. Cornia, F. F. De Biani, D. Gatteschi, C. Sangregorio, R. Sessoli and L. Sorace, J. Am. Chem. Soc., 1999, 121, 5302 CrossRef CAS.
- S. Accorsi, A. L. Barra, A. Caneschi, G. Chastanet, A. Cornia, A. C. Fabretti, D. Gatteschi, C. Mortalo, E. Olivieri, F. Parenti, P. Rosa, R. Sessoli, L. Sorace, W. Wernsdorfer and L. Zobbi, J. Am. Chem. Soc., 2006, 128, 4742 CrossRef CAS.
- R. W. Saalfrank, H. Glaser, B. Demleitner, F. Hampel, M. M. Chowdhry, V. Schunemann, A. X. Trautwein, G. B. M. Vaughan, R. Yeh, A. V. Davis and K. N. Raymond, Chem.–Eur. J., 2002, 8, 493 CrossRef CAS.
- L. Gregoli, C. Danieli, A. L. Barra, P. Neugebauer, G. Pellegrino, G. Poneti, R. Sessoli and A. Cornia, Chem.–Eur. J., 2009, 15, 6456 CrossRef CAS.
- A. L. Barra, F. Bianchi, A. Caneschi, A. Cornia, D. Gatteschi, L. Gorini, L. Gregoli, M. Maffini, F. Parenti, R. Sessoli, L. Sorace and A. M. Talarico, Eur. J. Inorg. Chem., 2007, 4145 CrossRef CAS.
- M. Mannini, F. Pineider, Ph. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M. A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Nat. Mater., 2009, 8, 194 CrossRef CAS.
- G. Pellegrino, A. Motta, A. Cornia, I. Spitaleri, I. Fragalà and G. Condorelli, Polyhedron, 2009, 28, 1758 CrossRef CAS.
- L. Bogani, C. Danieli, E. Biavardi, N. Bendiab, A. L. Barra, E. Dalcanale, W. Wernsdorfer and A. Cornia, Angew. Chem., Int. Ed., 2009, 48, 746 CrossRef CAS.
- C. Danieli, A. Cornia, C. Cecchelli, R. Sessoli, A. L. Barra, G. Ponterini and B. Zanfrognini, Polyhedron, 2009, 28, 2029 CrossRef CAS.
- J. M. Tour, L. Jones, D. L. Pearson, J. J. S. Lamba, T. P. Burgin, G. M. Whitesides, D. L. Allara, A. N. Parikh and S. Atre, J. Am. Chem. Soc., 1995, 117, 9529 CrossRef CAS.
- K. Lau, C. Huang, N. Yakovlev, Z. Chen and S. O'Shea, Langmuir, 2006, 22, 2968 CrossRef CAS.
- M. Béthencourt, L. Srisombat, P. Chinwangso and T. Lee, Langmuir, 2009, 25, 1265 CrossRef CAS.
- F. Pineider, M. Mannini, R. Sessoli, A. Caneschi, D. Barreca, L. Armelao, A. Cornia, E. Tondello and D. Gatteschi, Langmuir, 2007, 23, 11836 CrossRef CAS.
- T. Ishida, W. Mizutani, H. Azehara, F. Sato, N. Choi, U. Akiba, M. Fujihira and H. Tokumoto, Langmuir, 2001, 17, 7459 CrossRef CAS.
- J. Kang, S. Liao, R. Jordan and A. Ulman, J. Am. Chem. Soc., 1998, 120, 9662 CrossRef CAS.
- D. Briggs and M. Seah, Practical Surface Analysis 2nd Edn., Vol. 1, Auger and X-Ray Photoelectron Spectroscopy, Wiley, Chichester, 1990 Search PubMed.
- D. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709 CrossRef.
- N. Ghonaim, M. Nieradko, L. Xi, H. Nie, J. Francis, O. Grizzi, K. Yeung and L. Lau, Appl. Surf. Sci., 2008, 255, 1029 CrossRef CAS.
- A. Ulman, Chem. Rev., 1996, 96, 1533 CrossRef CAS.
- S. Watcharinyanon, C. Puglia, E. Gothelid, J. Backvall, E. Moons and L. Johansson, Surf. Sci., 2009, 603, 1026 CrossRef CAS.
- B. Genorio, T. He, A. Meden, S. Polanc, J. Jamnik and J. M. Tour, Langmuir, 2008, 24, 11523 CrossRef CAS.
- U. Gelius, P. Heden, J. Hedman, B. Lindberg, R. Manne, R. Nordberg, C. Nordling and K. Siegbahn, Phys. Scr., 1970, 2, 70 CAS.
- M. Beerbom, R. Gargagliano and R. Schlaf, Langmuir, 2005, 21, 3551 CrossRef CAS.
- Gold adsorbates of the ligand H3L (DCM solvent, 0.5 mg/mL, 40 h immersion) show a single S2p peak at 162.3 eV and a pronounced, carbonyl-type C1s component at 288.8 eV (see ESI†). The latter is absent in monolayers of the free thiol (DCM solvent, 0.3 mg/mL, 45 h immersion).
- L. Margheriti, M. Mannini, L. Sorace, L. Gorini, D. Gatteschi, A. Caneschi, D. Chiappe, R. Moroni, F. Buatier de Mongeot, A. Cornia, F. M. Piras, A. Magnani and R. Sessoli, Small, 2009, 5, 1460 CrossRef CAS.
- S. Sohn, M. Schroder, D. Lipinsky and H. Arlinghaus, Surf. Interface Anal., 2004, 36, 1222 CrossRef CAS.
- N. Crivillers, M. Mas-Torrent, J. Vidal-Gancedo, J. Veciana and C. Rovira, J. Am. Chem. Soc., 2008, 130, 5499 CrossRef CAS.
- Y. Li, J. Huang, R. T. McIver and J. C. Hemminger, J. Am. Chem. Soc., 1992, 114, 2428 CrossRef CAS.
- K. Wolf, D. Cole and S. Bernasek, Anal. Chem., 2002, 74, 5009 CrossRef CAS.
- L. Sun and J. Gardella, Langmuir, 2002, 18, 9289 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of ligand and samples preparation, 2H-NMR stability experiments, XPS and ToF-SIMS additional spectra. See DOI: 10.1039/b916895h |
| This journal is © The Royal Society of Chemistry 2010 |