Study of Na+/H+ exchange-mediated pHi regulations in neuronal soma and neurites in compartmentalized microfluidic devices†
Lucas
Vitzthum‡
ad,
Xinzhi
Chen‡
ab,
Douglas B.
Kintner
a,
Yu
Huang
d,
Shing-Yan
Chiu
c,
Justin
Williams
d and
Dandan
Sun
*ab
aDept. of Neurological Surgery, University of Wisconsin Medical School, T513 Waisman Center, 1500 Highland Ave., Madison, WI 53705, USA. E-mail: sun@neurosurg.wisc.edu; Fax: +1 (608) 263-1409; Tel: +1 (608) 263-4060
bDept. of Neuroscience Training Program, Madison, WI 53705, USA
cDept. of Physiology, School of Medicine and Public Health, Waisman Center, Madison, WI 53705, USA
dDept. of Biomedical Engineering, Univ. of Wisconsin, Madison, WI 53705, USA
First published on 14th December 2009
Abstract
Regulation of intracellular pH (pHi) in neurons is crucial to maintain their physiological function. In the current study, newly-developed polydimethylsiloxane (PDMS) microfluidic devices were used to independently investigate pHi regulation in neuronal soma and neurites. Embryonic cortical neurons were cultured in PDMS microfluidic devices with soma growing in one chamber (seeded) and neurites extending through a set of perpendicular microchannels into the opposite parallel chamber (non-seeded). Neurons in the microchambers were characterized by the vital dye calcein-red, polarized mitochondria, and expression of neuronal specific β-tubulin (type-III), axonal Tau-1 protein, dendritic microtubule associated protein (MAP-2), and Na+/H+ exchanger isoform 1 (NHE-1). Neurites exhibited higher resting pHi than soma (7.16 ± 0.09 vs. 6.90 ± 0.15). The neurites had a proton extrusion rate 3.7-fold faster than in soma following NH4Cl prepulse-mediated acidification (p < 0.05). The difference in the pHi regulation rates between neurites and soma can be accounted for by the larger surface area to volume ratio in the neurites. Interestingly, pharmacological inhibition of NHE-1 activity blocked the pHi regulation in soma and in neurites by ∼70% (p < 0.05). Taken together, our study demonstrated that the microfluidic devices provide a useful tool to study neuronal pHi regulation in soma and their neurites. We conclude that NHE-1 plays an important role in regulation of pHi in both compartments.
Insight, innovation, integrationIntracellular pH (pHi) in neurons can rapidly fluctuate in response to neuronal activity or pathological insults. The tight regulation of intracellular pH homeostasis is required for neuronal function in the central nervous system. Changes in pHi affect the functions of multiple enzymes, ion channels, and other macromolecules, thus affecting synaptic transmission, neuronal excitability, and gap junction conductance. In the current study, we developed an enclosed, compartmentalized microfluidic platform to examine the differential pHi regulation in neuronal soma and neurites. Our study demonstrated that the microfluidic devices provide a useful tool to study neuronal pHi regulation in soma and their processes. We conclude that Na+/H+ exchanger isoform 1 plays an important role in regulation of pHi in both compartments. |
Introduction
Intracellular pH (pHi) can rapidly fluctuate in response to neuronal activity or pathological insults.1 The tight regulation of intracellular and extracellular pH (pHi and pHo) homeostasis is required for neuronal function in the central nervous system (CNS). Changes in pHi (even in a small range) affect the functions of multiple enzymes, ion channels, and other macromolecules, thus affecting synaptic transmission, neuronal excitability, and gap junction conductance.2–4 pHi may function as an important variable in regulating neurotransmitter release.5,6 The Na+/H+ exchangers (NHEs) are acid extrusion proteins which catalyze the electrochemically neutral exchange of Na+ into the cell and H+ out of the cell at a ratio of 1 Na+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H+.7 NHEs are extremely sensitive to pHi and characterized by the fact that they are allosterically activated by cytosolic H+, thereby promoting the rapid extrusion of acid when pHi drops below a threshold level.7,8 The Na+/H+ exchanger 1 (NHE-1) is the most ubiquitous isoform in the CNS9 and plays an important role in regulation of pHi in neurons and astrocytes.8 NHE1 activity is stimulated by growth factors, peptide hormones,7 and pathological stimuli such as ischemic-hypoxia.10,11
:1 H+.7 NHEs are extremely sensitive to pHi and characterized by the fact that they are allosterically activated by cytosolic H+, thereby promoting the rapid extrusion of acid when pHi drops below a threshold level.7,8 The Na+/H+ exchanger 1 (NHE-1) is the most ubiquitous isoform in the CNS9 and plays an important role in regulation of pHi in neurons and astrocytes.8 NHE1 activity is stimulated by growth factors, peptide hormones,7 and pathological stimuli such as ischemic-hypoxia.10,11
We recently found that pHi regulation rates in the dendrites of cultured cortical neurons grown in culture dishes were 330% higher than in the soma primarily because of the higher surface area to volume rates in dendrites.12 The traditional in vitro neuronal culture models using culture dishes can not be used to independently investigate differential pHi regulation between soma and neurites. Thus, the in vitro compartmentalized neuronal culture models make this type of study possible. Campenot first introduced a compartmentalized fluidic neuronal culture using a device consisting of a Teflon® divider attached to a collagen coated Petri dish with silicon grease.13 These devices required great skill to assemble and were difficult for imaging and prone to leakage.13 Recently, compartmentalized microfluidic devices have been made of biologically inert polydimethylsiloxane (PDMS) that allows neurites to grow into isolated fluidic environments.14 Such devices have been applied in studying of axonal injury and regeneration,14 axon myelination,15 and drug-induced axonal degeneration.16 Moreover, the enclosed microfluidic system provides a more physiologically relevant micro-environment (larger cell surface area to culture medium ratios) than culture dishes.17
In the current study, we developed an enclosed, compartmentalized microfluidic platform to examine the differential pHi regulation in neuronal soma and neurites. Cultured cortical neurons were characterized with vital dyes (calcein and Mitotracker) and immunocytochemistry staining. Resting pHi levels and pHi recovery rates were measured in neurites and soma independently. Our report suggests that NHE-1 plays an important role in neuronal pHi regulation.
Methods
Materials
Hanks balanced salt solution (HBSS) was from Mediatech Cellgro (Manassas, VA). Neurobasal medium, B-27 supplement, MitoTracker green, calcein-AM, SYTO 60, and BCECF-AM were from Invitrogen (Carlsbad, CA). HOE 642 was a kind gift from Aventis Pharma (Frankfurt, Germany). Nigericin and antibody for MAP-2 were purchased from Sigma (St. Louis, MO). Antibody for β-Tubulin (type-III) was obtained from Promega (Madison, WI). Tau-1 antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). SU-8 2 and SU-8 2001 were purchased from Microchem (Newton, MA). Polydimethylsiloxane (PDMS) was purchased from Dow Corning (Sylgard 184, Midland, MI). G116 antibody was a kind gift from Dr Leong L. Ng (Leicester, UK).Fabrication of microfluidic device
The microfluidic devices were fabricated in PDMS using rapid prototyping and soft lithography.18,19 The layout of the microfluidic devices is shown in Fig. 1A, which consisted of two parallel chambers (seeded and non-seeded, 0.4 mm wide × 16 mm long × 0.25 mm tall) and bridging microchannels (30 μm wide × 200–300 μm long × 3 μm tall). Two corresponding transparency masks were printed from an EPS file using a 6000 dpi high-resolution printer (Imagesetter Inc, Madison, WI). A dual-layered negative master was fabricated by patterning dual layers of the epoxy based photoresist SU-8 2 (one for the microchannels, one for the two chambers). The photoresist was spun on a silicon wafer at a spin speed of 1500 rpm for 40 s to obtain an approximate height of 3 μm for the bridging microchannels. The photoresist SU-8 2100 was subsequently spun on the silicon wafer (1100 rpm for 40 s, 0.25 mm in height) to create the non-seeded and seeded chambers. Both mask layers were carefully aligned with a technique (error < 15 μm) as described recently.20 Microfluidic devices were fabricated by molding PDMS against this negative master. After degassing, PDMS (10:1 ratio base to catalyst) and the wafer were heated in a hot plate at 120 °C for 30 min. Inlets and outlets for fluid exchange in each chamber (3 mm in diameter) were made in the resulting PDMS microdevice (1.5 mm thick). PDMS microdevices were placed on poly-D-lysine coated glass coverslips (22 × 22 mm) and sterilized by autoclave.
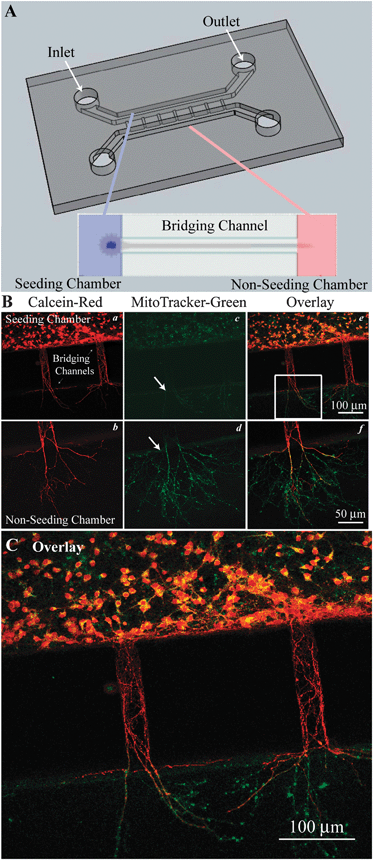 | ||
| Fig. 1 Cortical neuron cultures in PDMS microfluidic devices A. Microfluidic devices were adhered to a poly-d-lysine (PDL) coated coverslips to create a water tight, gas permeable chamber. The devices had two larger parallel chambers (0.4 mm wide, 0.25 mm tall) labeled as the seeded and non-seeded chambers. Inlet and outlet ports through the PDMS were created for cell loading and media exchange. These chambers were connected by a series of 200–300 μm long micro-bridging channels (30 μm wide, 3–5 μm tall) that exclude neuronal soma, but through which neurites can extend. Expanded view shows neuronal soma in the seeded channel with processed extending through the bridging channel into the non-seeded channel. B. Neurons cultured in the microfluidic device were imaged with calcein and MitoTracker dyes to access cell viability (a–f). C. An enlarged view of panel B (e) provides detailed information. | ||
Primary neuron culture in microfluidic devices
Pure cortical neurons from mouse embryonic day 14–16 fetuses (E14–16) were prepared as described before.21 In brief, the cortices were removed from E14–16 fetuses and treated with 0.5 mg ml−1 trypsin at 37 °C for 25 min. The cells were centrifuged at 300 g for 4 min at 4 °C. The cell pellet was diluted in B-27 supplemented Neurobasal medium (2%) containing 0.5 mM L-glutamine and penicillin/streptomycin (100 units ml−1 and 0.1 mg ml−1, respectively). As shown in Fig. 1A, the cells were seeded at a density of 500 cells mm−2 in the seeded chamber by passive pumping.22 Briefly, the seeded chamber was rinsed with Neurobasal medium by adding a drop (∼20 μL) into the inlet and aspirating from the outlet (Fig. 1A). The non-seeded chamber was similarly rinsed with the fresh medium. Cells were seeded via passive pumping by adding ∼10 μL of a 2 × 106 ml−1 cell suspension to the inlet which then flowed into the seeded chamber due to a low surface tension created by the large drop of medium placed on the outlet. The microfluidic devices were placed in an incubator at 37 °C (model 3130, Thermo Forma, Waltham, MA) with 5% CO2 and atmospheric air. A blank PDMS piece (∼0.5 mm thick) was placed over the chamber ports on day 2 in vitro (DIV 2) to prevent medium evaporation. DIV 8–11 cultures were used in the study.Measurement of pHi after NH4+/NH3 pre-pulse
Neuronal cultures in the microfluidic device were incubated with 10 μM BCECF-AM in HCO3−-free HEPES buffered medium at 37 °C for 30 min. Microfluidic devices were then mounted on a temperature controlled (37 °C) imaging chamber (Warner Instruments, Hamden, CT). The chamber was placed on the stage of the TE 300 inverted epifluorescence microscope and visualized with a 40× oil-immersion objective. The cells were excited every 1–20 s at 490 and 440 nm, and the emission fluorescence at 535 nm recorded. Images were collected using a Princeton Instruments MicroMax CCD camera. Regions of interest were drawn either in the cytosol of 20 cells (seeded side) or on neurites (non-seeded side) and analyzed with MetaFluor image-processing software. At the end of each experiment, the ratio of the background-corrected fluorescence emissions (F490/F440) was calibrated using the high K+/nigericin technique.23 In this study, the inherent error in determining pHi with the BCECF method is ∼1.5% (supplemental Fig. 1, ESI†). Microchamber fluid replacement in these experiments was accomplished by placing a drop of solution with a 25 μl Hamilton syringe on the inlet of one microchamber and quickly drawing the fluid through the outlet with the corner of a Kimwipe®.For the pre-pulse treatment, either soma (the seeded-chamber) or neurites (non-seeded chambers) were subjected to an acid load by transient application (5 min) of 30 mM NH4+/NH3 solution.24 NH4+/NH3 solutions were prepared by replacing 30 mM NaCl in the HCO3−-free HEPES-buffered solution with an equimolar concentration of NH4Cl. Application of NH4+/NH3 solution caused pHi to rise as NH3 gas diffused quickly into the cell and combined with H+ to form NH4+. After 5 min, the NH4+/NH3 solution was replaced with HCO3−-free HEPES-buffered solution, causing NH3 to leave the cell and trapping H+ in the cell. This is the classic method of placing an artificial acid load on cells to characterize pHi regulation.25 Interestingly, we observed that the pre-pulse treatment applied to one microchamber (soma or neurites) did not cause either alkalization or acidification in the opposite chamber (data not shown). This suggests that we are able to investigate pHi regulation in these compartments (i.e. between soma and neurites), separately. Before returning cells to normal HCO3−-free HEPES-buffered solution to acidify the cells, the image acquisition interval rates was increased to 1 HZ in order to monitor pHi recovery. pHi recovery rate was determined from the slope of a fitted linear regression fit to the initial linear portion of the trace after NH4+/NH3 application (5–30 s).24
Resting pHi was averaged from the first ∼2 min incubation of cells with HCO3−-free HEPES-buffered normal solution before the pre-pulse was performed. In the experiments with specific NHE-1 inhibitor HOE 642, either soma or neurites were incubated with HOE 642 (1 μM) for 10 min prior to and then throughout the NH4+/NH3 pre-pulse.
Live cell imaging
To determine surface area to volume ratios in pure neuron cultures growing in microchamber devices, neurons were loaded with 0.5 μM calcein-AM green (a vital dye) and 5 μM SYTO 60 (nucleus dye) for 30 min at 37 °C. The coverslips were then placed in the perfusion chamber on the stage of a Leica DMIRE2 confocal microscope and visualized with a 100× 1.4 NA apochrome oil-immersion objective. An ∼15 μm (neurites) to 30 μm (somata) thick image stack (150–250 slices at 512 by 512 pixels) was collected sequentially (ex. 488 nm argon laser line, em. 500–545 nm and ex. 543 HeNe laser, em 544–677) and imported into ImageJ (Version 1.41, NIH). A cellular region (somata, nucleus, or neurite) was defined and the surface area was calculated by summing the product of the region perimeter by the distance between each image section (0.12 μm). The volume of the region was calculated with the region area and intra-section distance. The somata volume was corrected by subtracting the calculated volume for the nucleus volume. In other experiments, neurons in microchambers were further characterized by duel-labeling with 0.5 μM calcein-AM red and 0.2 μM MitoTracker green, a mitochondrial-selective fluorescent dye, for 30 min. Calcien-AM and Mitotracker images were sequentially captured with a Leica DMIRE2 confocal microscope using a 20× objective and 543 nm HeNe and a 488 nm argon lasers, respectively.Immunofluorescence staining
PDMS microfluidic devices were removed from the glass coverslips and the cells on the coverslips were gently rinsed with PBS (pH 7.4) and fixed with 4% paraformaldehyde in PBS.24 After rinsing, cells were incubated with blocking solution (10% normal goat serum, 0.4% Triton X-100, and 1% bovine serum albumin in PBS) for 1 h at room temperature. Cells were incubated for 1 h at 37 °C with polyclonal G116 NHE-1 antibody (1:50), mouse monoclonal MAP-2 antibody (1:200), monoclonal β-Tubulin (type-III) antibody (1:200) or rabbit polyclonal Tau-1 antibody (1:100). After rinsing in PBS, cells were incubated with goat anti-mouse Alexa 488-conjugated IgG (1:100; Invitrogen), goat anti-rabbit Alexa 546 conjugated IgG (1:100; Invitrogen, for confocal microscopy) or goat anti-rabbit Alexa 594 conjugated IgG (1:100; Invitrogen, for epifluorescence microscopy) for 1 h at 37 °C. 1 μg mL−1 To-pro-3 iodide was used to stain nuclei for confocal image collection. Fluorescence images were captured with a Nikon TE 300 inverted epifluorescence microscope (20×) using a Princeton Instruments MicroMax CCD camera and MetaMorph image-processing software. Negative controls were incubated with secondary antibodies only. Identical digital imaging acquisition parameters were used in both negative control and experimental images. Images of Tau-1 (20×) were captured with a Leica DMIRE2 confocal microscope using an 543 nm HeNe laser for Alexa Fluor 546, and a 640 nm GreNe laser for To-pro-3 iodide signals.
Statistics
Paired Student’s t-test was used for comparisons between groups and analysis of variance with the Bonferroni post-test for multiple comparisons (SigmaStat, Systat Software, Point Richmond, CA). When comparison groups did not have normal distribution or equal variance, a Mann–Whitney Rank Sum Test was used. A P-value of <0.05 was considered as significantly different.Results
Neuronal growth and differentiation in the microfluidic system
Neurons were grown in the PDMS microfluidic system as described in Methods. DIV 8–11 cultures were loaded with the vital dye calcein red and MitoTracker green (Fig. 1B). Both soma and neurites retained calcein-red, indicating intact plasma membrane integrity of neurons in the culture (Fig. 1B, a). Fig. 1B(a, b) also illustrate that the neurites grew across the bridging channels and protruded into the non-seeded chamber. Concurrently, neuronal cultures were stained with the mitochondrial dye MitoTracker-green, which reacts with thiol groups of proteins and forms conjugates in the mitochondria, thus labeling mitochondria independent of their membrane potential. As shown in Fig. 1B (c, d, arrows), punctate MitoTracker-green signals were detected in soma and neurites. Interestingly, a high level of mitochondria was distributed in neurites in the non-seeded chamber (punctuate, rod-shape individual mitochondria in d, f). In contrast, Mitotracker-green signals were less evident in the neurites in the bridging channels, suggesting that MitoTracker green does not diffuse quickly within the intracellular space or through the bridging channels (Fig. 1B, c). An overlay of the calcein red and MitoTracker-green images revealed that the distal neurites expressed less calcein red (Fig. 1C). Taken together, these data illustrate that cortical neurons (in the seeded chamber) were able to grow and extend their neurites through the micro-bridging channels into the non-seeded chamber. Mitochondria appear to be enriched in the distal neurites in a similar manner as in vivo.Characterization of neurons in microfluidic devices
Neurons cultured in the PDMS microfluidic devices were further characterized with immunofluorescence staining assays. First, expression of three neuronal marker proteins [β-Tubulin (type-III), Tau-1 and MAP-2] in the cultures was examined. As shown in Fig. 2A, β-Tubulin was abundantly expressed in neuronal soma and throughout the neurites. Moreover, dendritic marker protein MAP-2 was expressed in soma and dendritic neurites in the bridging channels and non-seeded chamber (Fig. 2B). On one occasion, small immature neurons migrated through the bridging channel and grew in the non-seeded chamber (arrow, Fig. 2B). In Fig. 2C, D, the axonal marker protein Tau-1 was expressed in the neurites growing into the non-seeded chamber, but with less immunointensity in neuronal soma (in the seeded-chamber) or in the neurites of the bridging channels. Negative control studies for these proteins were conducted by omitting the primary antibodies and illustrated no fluorescence signals (insets of Fig. 2A–C). These findings demonstrate that cortical neurons grow in the microfluidic devices with neurites (both axons and dendrites) extruding into the non-seeded chambers.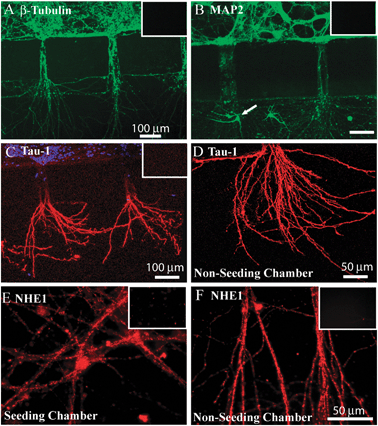 | ||
| Fig. 2 Characterization of neurons cultured in microfluidic devices A. Neurons cultured in microfluidic device at DIV 8–11 stained for β-Tubulin. B. Neurons stained for MAP-2. C, D. Neurons stained for Tau-1 in microfluidic device (C) or in the non-seeded chamber (D). E, F. Neurons stained for NHE-1 protein in the seeded-(E) and non-seeded chambers (F). Inset, negative controls by omitting primary antibodies for β-Tubulin, MAP-2, Tau-1 or NHE-1 antibody. Scale bars = 100 μm in A, B, C. and = 50 μm in D, E, F. | ||
Next, we evaluated expression of an important pHi regulatory protein NHE-1. As shown in Fig. 2E, F, NHE-1 was present in soma of the seeded-chamber and neurites in the non-seeded chambers. Negative control studies were shown in insets of Fig. 2E, F. No or low fluorescence signals in these controls were detected under the same data acquisition settings.
Regulation of pHi in soma and neurites
We then independently determined pHi regulatory responses in soma of the seeded chambers and neurites in the non-seeded chambers. pHi recovery rate was measured following the NH3/NH4+ prepulse-induced acidification. In the absence of HCO3−, pHi regulation largely reflects NHE-mediated H+ efflux.26 As shown in Fig. 3A, when neuronal somata in the seeded chamber was exposed to 30 mM NH3/NH4+, pHi rose rapidly as NH3 diffused into the cell and combined with H+ to form NH4+ (a–b) and then declined slowly (b–c). Removal of the NH3/NH4+Cl solution from the seeded chamber caused somata pHi to decrease because the newly formed NH4 rapidly left cells as NH3, trapping H+ inside of cells (c–d). Neurons were able to quickly restore somata pHi at a rate of 1.25 ± 0.29 pH units min−1 in < 3 min and returned to their basal levels (6.90 ± 0.15 pH). In some cases, there was an alkaline overshoot (Fig. 3A). In the presence of the potent NHE-1 inhibitor HOE 642 (1 μM), somata pHi recovery rate was significantly reduced (0.35 ± 0.13 pH units min−1, p < 0.05, Fig. 3A and C) and the alkaline overshoot was blocked.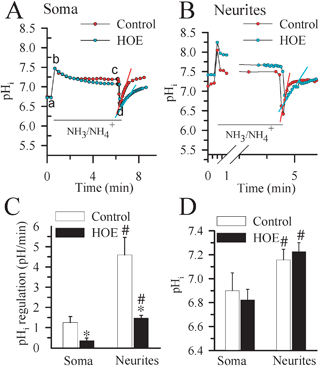 | ||
| Fig. 3 pHi regulation in soma and neurites. pHi recovery rates were determined in cortical neurons cultured in microfluidic devices at DIV 8–11. Representative tracings showing the pHi changes in neuronal soma (A.) and neurites (B.) subjected to a prepulse acidification by exposure to 30 mM NH4Cl for 5 min. Each point is the average of 20 regions of interest. pHi recovery rates were determined by fitting a slope to the pH values over the linear portion of recovery following acidification. Red and blue slope lines are shown to illustrate the quantified rate of recovery. In some studies, 1 μM HOE 642 was present 10 min before and throughout the remaining NH3/NH4+ prepulse experiments. C. Summary data of calculated pHi recovery rates D. Summary data of resting pHi values for neuronal soma and neurites. Data are mean ± SEM. n = 5–6. * p < 0.05 vs. control; # p < 0.05 vs. soma. | ||
After NH3/NH4+ prepulse-induced acidification, the neurites exhibited much faster pHi recovery kinetics (4.59 ± 0.86 pH units ,min−1, Fig. 3B, C). Inhibition of NHE-1 with HOE 642 reduced the recovery rate to 1.47 ± 0.14 pH units min−1 in neurites (p < 0.05, Fig. 3B, C). Moreover, a more alkaline basal pHi (7.16 ± 0.09) was detected in the neurites than in soma (Fig. 3D).
Determination of surface area and volume (A/V) ratio in soma and neurites
The apparent higher pHi recovery rates in the neurites could result from significantly larger A/V ratios in the neurites. A/V ratios were determined in somata (arrow) and neurites (arrow-head) as illustrated in Fig. 4A and B. Fig. 4C shows that the A/V ratio in the neurites is 2.8 times higher than that of soma. Interestingly, the A/V ratios of soma and neurites were higher in the microfluidic devices than in cells cultured on coverslips in traditional plastic 35 mm culture dishes (Fig. 4C).12 Although digital 3D reconstruction from 2D data sets will tend to underestimate surface areas by smoothing the images, this estimate of A/V ratios served as approximation for correcting pHi regulation rates.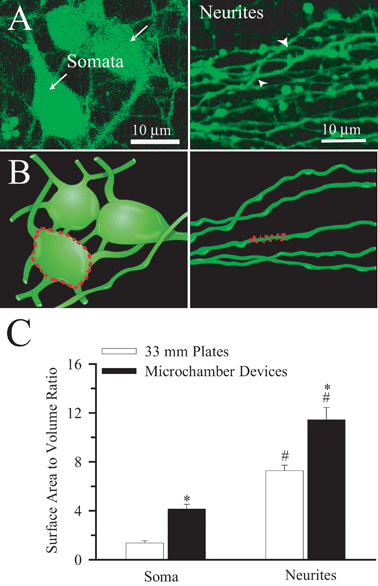 | ||
| Fig. 4 Topography and surface area to volume ratio of cultured neurons A. A single 2D confocal image of neurons growing in the seeded-chamber loaded with 0.5 μM calcein-AM from a stack (30 μm) of 250 images (Left Panel). A single 2D confocal image of neurites as they emerged into the non-seeded chamber from a stack (15 μm) of 150 images (Right Panel). Arrows: somata. Arrowheads: neurites B. Rendition of a 3D image produced from A to illustrate the surface area to volume ratio determination in soma and dendrites (dotted lines). C. Summary data of surface area to volume (A/V) ratios calculated in the soma of the seeded-chamber and neurites that extended into the non-seeded chamber. The A/V ratios in microdevices were further compared to ones collected in neurons grown on 35 mm plastic plates.12 Data are mean ± SEM. n = 8–14 cells. * p < 0.05 vs. 33 mm plastic plates. # p < 0.05 vs. soma. | ||
Consequently, we corrected the pHi recovery rate for the A/V ratios in soma and neurites (Table 1). After the correction, the differences in H+ extrusion rates in the two regions were abolished. Once the soma pHi recovery rates were corrected for A/V ratio they were comparable to the corrected rates that were observed in neurons in 35 mm culture dishes (0.30 ± 0.07 vs. 0.30 ± 0.01 pH units min−1, respectively).12 However, the A/V-corrected pHi recovery rates in neurites grown in microfluidic devices (0.40 ± 0.08 pH units min−1) remained significantly higher than in neurites grown in the culture dishes (0.19 ± 0.04, p < 0.05).12
Discussion
Characterization of cultured neurons in microfluidic devices
Our results showed viable neuronal growth in the seeded-chamber with neurites expanding through the bridging channels and into the non-seeded chamber. In most cases, the calcein signals were colocalized with the MitoTracker signals, indicating the intact integrity of plasma membrane and mitochondria. Damaged neurons would not retain either dye. Taken together, these results demonstrate that we have developed a viable compartmentalized microfluidic system for culturing neurons.Microfluidic devices with micro-bridging channels lengths greater than 200 μm exhibit almost exclusively axonal growth and a few dendrites in the non-seeded chambers at DIV 14.14 In contrast, both dendrites and axons extend through 150 μm long bridging channels at DIV 14.14 In our study, we used 200–300 μm long bridging channels. We observed abundant expression of neuronal specific β-III tubulin and dendritic protein MAP-2 in both soma and neurites, while the neurites in the non-seeded chambers of DIV 8–11 neuron cultures exhibited abundant expression of the axonal marker protein Tau-1. Because both axons and dendrites were present in the non-seeded chambers in the current study, we are unable to differentiate between the two with regard to pHi measurement in the non-seeded chamber. Therefore, in the non-seeded channels we referred to them as neurites.
There was a noticeable lack of MitoTracker-labeled mitochondria within the micro-bridging channels. This most likely resulted from restricted access of the MitoTracker dye to the mitochondria within the micro-bridging channels. To reach mitochondria within the micro-bridging channel, a dye must either enter the bridging channel and then be taken up by the cell or enter the cell and then be transported through the cytoplasm to reach mitochondria in the neurites of the bridging channel. Movement of the dye depends on molecular diffusion which is driven by concentration gradients of the dye. Upon entering the cell, MitoTracker is concentrated in nearby mitochondria where it covalently binds to proteins and would be unavailable to reach mitochondria located further away. However, we can not rule out that the low density of mitochondria in the micro-bridging channel determined with MitoTracker staining could be the result of its shallow height (3 μm) which would limit mitochondrial congregation.
Differential regulation of pHi in soma vs. neurites
The high fluidic resistance of the micro-bridging channels produces a small but sustained flow between the seeded and non-seeded chambers that counteracts diffusion. The fluidic isolation between the two chambers can last for 20 h owing to the high fluidic resistance of the micro-bridging channels.14 Thus, the fluidic resistance allows us to measure pHi regulation in soma and neurites independently. We found that neurites exhibited more alkaline resting pHi than in soma (7.16 ± 0.09 vs. 6.90 ± 0.15). The neurites were able to change [H+] 3.7-fold faster than soma following NH4Cl prepulse-mediated acidification (p < 0.05). Interestingly, pharmacological inhibition of NHE-1 activity blocked the pHi regulation in soma and neurites by ∼70% (p < 0.05). Moreover, the presence of NHE-1 isoform protein was detected in both neuronal soma and neurites, supporting its role in the pHi regulation. Investigating the mechanisms involved in pHi regulation in soma and dendrites is important to understand how the cell counteracts the robust intracellular acidification that occurs during synchronous neural activity.27 In addition, we have recently reported that the selective vulnerability of dendrites to ischemic insults may involve overstimulation of NHE-1 activity.12The more alkaline basal pHi observed in neurites suggests that increased NHE1 function could have significant metabolic consequences under physiological and pathological conditions. For example, NHEs are major intraterminal pHi regulators on GABAergic presynaptic nerve terminals and play an important role in the neuronal excitability by increasing inhibitory tones.1 It is recently reported that NHE1 accumulates at the leading edge of neurites where it alkalizes growth cones and regulates membrane protrusion and directional motility.28 On the other hand, NADPH oxidase is inhibited by low pHi, thus, increased NHE1 activity could increase formation of reactive oxygen species and potentiate reactive oxidative damage in neurites.29
We found that the A/V ratio in the neurites is 2.8 times higher than that of soma grown in microfluidic devices. In addition, the A/V ratios of soma and neurites were higher in the microfluidic devices than in the traditional plastic plates. The reason for the difference in A/V ratios between neurons grown in culture dishes vs. microfluidic devices is not clear. It could be because of the difference in “effective culture volume” (ECV) between the two.30 The ECV is a measure of the cell’s control over its local environments and a function of the magnitude of mass transport along each axis, the effects of diffusion and convection, and the degree of protein adsorption to surfaces.30In vivo, extracellular molecules such as secreted growth factors have a crucial role in determining cell shape.31 These signals would be lost through dilution in culture dishes. We speculate that the neuronal cell shape and A/V ratios in microscale cultures result from extracellular microenvironments which are not present in culture dishes.
In addition, the significantly higher NHE-1 activity in the neurites in microfluidic devices than in the culture dishes could also result from the density of NHE-1 expression or post-translational regulation (phosphorylation state of the protein). Further investigation is needed to define the influence of the culture microenvironment on pHi regulation through NHE-1.
In summary, we reported here that dissociated pure cortical neurons were cultured in the microfluidic devices with neurites grown across a 200–300 μm bridging channels. The microfluidic devices allowed us to successfully examined pHi in the two compartments independently. We detected that neurites exhibited more alkaline resting pHi than in soma. The neurites had a pHi regulation rate 3.7-fold faster than in soma following acidification. These findings suggest that the NHE1’s role in maintaining alkaline resting pHi in neurites may regulate function of NMDA receptor activity, voltage-gated Ca2+ channel, and synaptic vesicle-mediated exocytosis.
Acknowledgements
This work was supported in part by NIH grants R01NS38118 and R01NS048216 (D. Sun), and an AHA Established-Investigator award grant N0540154 (D. Sun); MS RG-4054-A-8 (S.Y. Chiu).References
- I. S. Jang, M. S. Brodwick, Z. M. Wang, H. J. Jeong, B. J. Choi and N. Akaike, J. Neurochem., 2006, 99, 1224–1236 CrossRef CAS.
- G. C. Tombaugh, J. Neurophysiol., 1998, 80, 1702–1712 CAS.
- L. E. Trudeau, V. Parpura and P. G. Haydon, J. Neurophysiol., 1999, 81, 2627–2635 CAS.
- G. G. Somjen and G. C. Tombaugh, pH modulation of neuronal excitabiliity and central nervous system functions, in pH and Brain Function, ed. K. Kaila and B. R. Ransom, Wiley-Liss, New York, 1998, pp. 373–394 Search PubMed.
- M. Chesler and K. Kaila, Trends Neurosci., 1992, 15, 396–402 CrossRef CAS.
- M. Chesler, Physiol Rev., 2003, 83, 1183–1221 CAS.
- J. Orlowski and S. Grinstein, Pfluegers Arch., 2004, 447, 549–565 CrossRef CAS.
- D. B. Kintner, Y. Wang and D. Sun, Front. Biosci., 2007, 12, 762–770 CrossRef CAS.
- E. Ma and G. G. Haddad, Neuroscience, 1997, 79, 591–603 CrossRef CAS.
- J. Luo, H. Chen, D. B. Kintner, G. E. Shull and D. Sun, J. Neurosci., 2005, 25, 11256–11268 CrossRef CAS.
- D. B. Kintner, A. Look, G. E. Shull and D. Sun, Am. J. Physiol.: Cell Physiol., 2005, 289, C934–C945 CrossRef CAS.
- D. B. Kintner, X. Chen, S. Y. Chiu, A. Baba, T. Matsuda, M. S. Cohen, J. Orlowski, J. Taunton and D. Sun, 2009 SFN abstract, program 301.7.
- R. B. Campenot, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 4516–4519 CrossRef CAS.
- A. M. Taylor, M. Blurton-Jones, S. W. Rhee, D. H. Cribbs, C. W. Cotman and N. L. Jeon, Nat. Methods, 2005, 2, 599–605 CrossRef CAS.
- J. Park, H. Koito, J. Li and A. Han, Biomed. Microdevices., 2009 Search PubMed.
- I. H. Yang, R. Siddique, S. Hosmane, N. Thakor and A. Hoke, Exp. Neurol., 2009, 218, 124–128 CrossRef CAS.
- T. M. Pearce and J. C. Williams, Lab Chip, 2007, 7, 30–40 RSC.
- G. M. Whitesides, E. Ostuni, S. Takayama, X. Jiang and D. E. Ingber, Annu. Rev. Biomed. Eng., 2001, 3, 335–373 CrossRef CAS.
- A. O’Neill, J. Soo Hoo and G. M. Walker, Lab Chip: Chips and Tips, 2006 Search PubMed.
- H. Yu, R. Pope and J. Williams, Conf. Proc. IEEE Eng Med. Biol. Soc., 2009 Search PubMed.
- X. Chen, D. B. Kintner, J. Luo, A. Baba, T. Matsuda and D. Sun, J. Neurochem., 2008, 106, 1563–1576 CrossRef CAS.
- G. M. Walker and D. J. Beebe, Lab Chip, 2002, 2, 131–134 RSC.
- G. Boyarsky, B. Ransom, W. R. Schlue, M. B. Davis and W. F. Boron, Glia, 1993, 8, 241–248 CrossRef CAS.
- D. B. Kintner, G. Su, B. Lenart, A. J. Ballard, J. W. Meyer, L. L. Ng, G. E. Shull and D. Sun, Am. J. Physiol.: Cell Physiol., 2004, 287, C12–C21 CrossRef CAS.
- W. F. Boron and P. De Weer, J. Gen. Physiol., 1976, 67, 91–112 CrossRef CAS.
- J. Luo, D. B. Kintner, G. E. Shull and D. Sun, J. Biol. Chem., 2007, 282, 28274–28284 CrossRef CAS.
- S. Makani and M. Chesler, J. Neurosci., 2007, 27, 7438–7446 CrossRef CAS.
- W. C. Sin, D. M. Moniz, M. A. Ozog, J. E. Tyler, M. Numata and J. Church, J. Neurosci., 2009, 29, 8946–8959 CrossRef CAS.
- A. M. Brennan, S. W. Suh, S. J. Won, P. Narasimhan, T. M. Kauppinen, H. Lee, Y. Edling, P. H. Chan and R. A. Swanson, Nat. Neurosci., 2009, 12, 857–863 CrossRef CAS.
- G. M. Walker, H. C. Zeringue and D. J. Beebe, Lab Chip, 2004, 4, 91–97 RSC.
- M. Bibel and Y. A. Barde, Genes Dev., 2000, 14, 2919–2937 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplemental Fig. 1 to show the inherent error in determining pHi with the BCECF method. See DOI: 10.1039/b918440f |
| ‡ These Authors Contributed Equally. |
| This journal is © The Royal Society of Chemistry 2010 |