A green-by-design biocatalytic process for atorvastatin intermediate
Steven K. Maa, John Grubera, Chris Davisa, Lisa Newmana, David Graya, Alica Wanga, John Gratea, Gjalt W. Huisman*a and Roger A. Sheldon*b
aCodexis, Inc, 200 Penobscot Drive, Redwood City, CA 94603, USA. E-mail: gjalt.huisman@codexis.com
bDelft University of Technology, Department of Biotechnology, Julianalaan 136, 2628, BL, Delft, Netherlands. E-mail: r.a.sheldon@tudelft.nl
First published on 23rd October 2009
Abstract
The development of a green-by-design, two-step, three-enzyme process for the synthesis of a key intermediate in the manufacture of atorvastatin, the active ingredient of the cholesterol lowering drug Lipitor®, is described. The first step involves the biocatalytic reduction of ethyl-4-chloroacetoacetate using a ketoreductase (KRED) in combination with glucose and a NADP-dependent glucose dehydrogenase (GDH) for cofactor regeneration. The (S) ethyl-4-chloro-3-hydroxybutyrate product is obtained in 96% isolated yield and >99.5% e.e. In the second step, a halohydrin dehalogenase (HHDH) is employed to catalyse the replacement of the chloro substituent with cyano by reaction with HCN at neutral pH and ambient temperature. The natural enzymes were highly selective but exhibited productivities that were insufficient for large scale application. Consequently, in vitro enzyme evolution using gene shuffling technologies was employed to optimise their performance according to predefined criteria and process parameters. In the case of the HHDH reaction, this afforded a 2500-fold improvement in the volumetric productivity per biocatalyst loading. This enabled the economical and environmentally attractive production of the key hydroxynitrile intermediate. The overall process has an E factor (kg waste per kg product) of 5.8 when process water is not included, and 18 if included.
Introduction
Atorvastatin calcium is the active ingredient of Lipitor®, the first drug in the world with annual sales to exceed $10 billion. Lipitor is a cholesterol-lowering drug, a member of the statin family of so-called HMG-CoA reductase inhibitors that block the synthesis of cholesterol in the liver.1 The structure of atorvastatin calcium is shown in Fig. 1. In common with other statins, it comprises a chiral 3,5-dihydroxy carboxylate side chain attached to a cyclic nucleus. Atorvastatin is unique in having the side chain attached to nitrogen in the nucleus.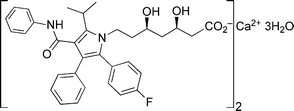 | ||
| Fig. 1 Structure of atorvastatin calcium. | ||
Atorvastatin's high volume demand coupled with the requirement for high chemical and optical purity has led to extensive efforts towards more economic production of its chirality-setting intermediates.1 The key chiral building block in all commercialized syntheses of atorvastatin is ethyl (R)-4-cyano-3-hydroxybutyrate 1, a.k.a. “hydroxynitrile” (see Fig. 2). In the innovator's original commercial process for atorvastatin, the second stereogenic center in atorvastatin (at the 3-hydroxy group) is set by diastereomeric induction, using cryogenic borohydride reduction of a boronate derivative of the 5-hydroxy-3-keto intermediate 2 derived from 1.2
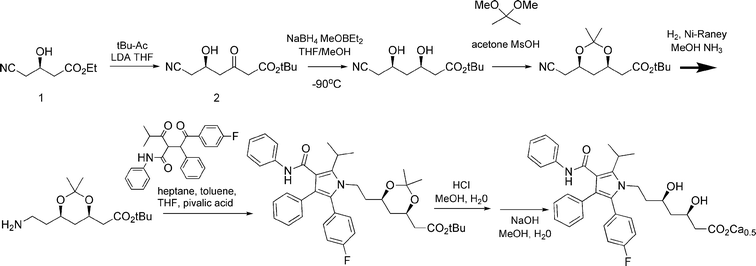 | ||
| Fig. 2 Synthetic route for atorvastatin calcium. | ||
Previous routes that have been used for the industrial production of 1 are depicted in Fig. 3. Early routes involved kinetic resolutions using microbes, and the use of (S)-hydroxy butyrolactone produced from chiral pool raw materials, lactose or malic acid. Later routes have involved asymmetric reduction of ethyl 4-chloroacetoacetate, produced from diketene, using an asymmetric hydrogenation catalyst, microbial cells, or an enzyme. Alternative chemoenzymatic routes have been described, using nitrilases3 or lipases4 but they have not, to our knowledge, been commercialized. These processes tend to require high enzyme loadings,1 making product recovery difficult5 and adding significant costs to the process.
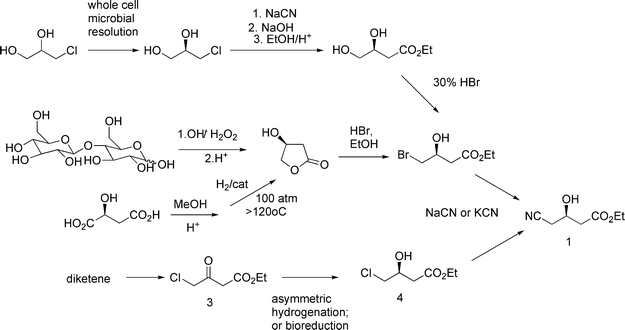 | ||
| Fig. 3 Previous routes to the hydroxynitrile intermediate 1. | ||
The final step to 1 in all of the previous commercial processes involves reaction of an ethyl 3-hydroxy-4-halobutyrate (a.k.a. “halohydrin”) with a cyanide ion in alkaline solution at elevated temperatures to form the hydroxynitrile 1. Alkaline conditions are necessary to form the nucleophilic cyanide anion (the pKa of HCN is about 9). However, the substrate and product are base-sensitive compounds, resulting in extensive by-product formation. In one report, the reaction with the chlorohydrin was conducted at around 80 °C and pH 10 with a reaction yield of ∼85%.6 The product is a high-boiling oil and the by-products include many which are close in boiling point.6 As a result, a troublesome high-vacuum fractional distillation is required to recover product of acceptable quality, which results in still further yield loss and waste.6
Annual demand for 1 is estimated to be in excess of 100 mT, making it highly desirable to reduce the wastes and hazards involved in its manufacture, while reducing its cost and maintaining or, preferably, improving its quality.
It is difficult to imagine a practical process for 1 that does not involve using cyanide, and we are not aware of any that has been proposed. We reasoned that the key to providing a superior process for 1 would be to find and develop a catalyst capable of accomplishing the cyanation reaction under mild conditions at neutral pH, so that by-product formation would be minimized. Enzymes are known that catalyse the enantioselective ring-closing elimination of halohydrins to the corresponding epoxide in a reversible reaction.7 These enzymes are often referred to as halohydrin dehalogenases (HHDHs). It is also known, from the work of Nakamura and co-workers8,9 that these HHDHs accept cyanide as a non-natural nucleophile leading to the irreversible enantioselective formation of β-hydroxynitriles, under neutral, ambient conditions. More recently, this finding was further elaborated and extended to other non-natural nucleophiles by Janssen and co-workers.10–12 The availability of this enzyme led us to propose the two-step, three-enzyme process for the production of 1 shown in Fig. 4. The key step is the HHDH catalysed reaction of the halohydrin 4 with HCN at neutral pH to produce 1.
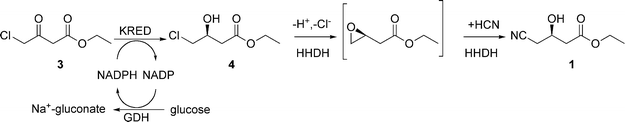 | ||
| Fig. 4 Two-step, three-enzyme process for hydroxynitrile 1. | ||
In addition to being highly efficient catalysts that typically exhibit exquisite chemo-, regio-, and stereoselectivities, enzymes have many benefits from the viewpoint of developing green, sustainable processes.13 They operate under mild conditions (ambient temperature and pressure as well as neutral pH), with water as the preferred reaction medium. They are produced from renewable raw materials and are nontoxic and biodegradable. Enzymes often provide the opportunity to reduce the number of chemical steps in a synthetic process as there is no need for functional group protection and deprotection. Finally, enzymatic reactions are typically performed in standard multi-purpose manufacturing plants. Notwithstanding all these advantages, the use of enzymes as catalysts in the large-scale production of fine chemicals and pharmaceuticals had been hampered by perceived intrinsic limitations of natural enzymes.14,15 Such limitations include insufficient activity towards non-natural substrates, insufficient activity at high substrate loadings due to substrate and/or product inhibition, and low operational stability under such economically viable conditions. While process engineering solutions might alleviate some of these problems,16 the optimal solution is to employ in vitro directed evolution technologies to generate enzymes that operate effectively with the desired substrate and under the desired, more practical process conditions;17 instead of following the traditional path of compromising the process to fit the available catalyst, the catalyst is evolved to fit the desired process. This enables the development of a process that is ‘green by design’.
Herein, we describe a novel, economically viable and environmentally attractive process for the large-scale synthesis of 1 that includes the laboratory evolution of three enzymes to meet pre-defined process parameters. Details of the enzyme evolution aspects of the HHDH enzyme have been reported elsewhere.18
Results and discussion
In the process,19 a ketoreductase (KRED) and glucose dehydrogenase (GDH) are used for the enantioselective reduction of ethyl 4-chloroacetoacetate (3), using glucose as reductant, to ethyl (S)-4-chloro-3-hydroxybutyrate (4). Glucose is oxidized to gluconic acid, which is neutralized by sodium hydroxide via a pH-stat. Subsequent to its recovery, 4 is converted to 1 by reaction with HCN (pKa∼ 9) at neutral pH, catalyzed by a halohydrin dehalogenase (HHDH). This reaction releases the H+ and Cl−, and must also be pH-statted, which is achieved using aqueous NaCN as the base and source of CN−.The activities of natural KRED and GDH for the reduction of 2 to 3 were low (Table 1). With 100 g L−1 of 3, a total of 9 g L−1 natural, recombinantly produced KRED (6 g L−1) and GDH (3 g L−1) were required to complete the reaction in 15 h. However, due to emulsion formation, recovery of 4 was problematic. Although the analytical yield of 4 was >99%, the recovered yield, after an hour to allow for some emulsion separation, was only 85%. To enable a practical large-scale process, the enzyme loadings needed to be drastically reduced.
| Parameter | Process design | Initial process | Final process |
|---|---|---|---|
| Substrate loading/g L−1 | 160 | 80 | 160 |
| Reaction time/h | <10 | 24 | 8 |
| Biocatalyst loading/g L−1 | <1 | 9 | 0.9 |
| Isolated yield/% | >90 | 85 | 95 |
| Chemical purity/% | >98 (GC) | >98 | >98 |
| E.e. of 4/% | >99.5 | >99.5 | >99.9 |
| Phase separation of organic product phase from aqueous phase containing enzyme/min | <10 | >60 | <1 |
| Space–time yield/gproduct L−1 d−1 | >384 | 80 | 480 |
| Catalyst yield (gproduct/gcat) | >160 | 9 | 178 |
DNA shuffling technology20 was used to improve the activity and stability of KRED and GDH while maintaining the nearly perfect enantioselectivity exhibited by the natural KRED. DNA shuffling involves the mutation of a gene encoding the enzyme of interest to generate “libraries” of mutants. These libraries are screened in high throughput, under conditions that approximate the desired manufacturing process, and improved mutants are selected for further evolution. Their genes are recombined in vitro to create the next generation of libraries to be screened for further improved progeny. The gene libraries are transformed and expressed in a host organism, such as Escherichia coli, and screened in high throughput for enzyme variants that exhibit the desired improved properties. DNA shuffling is becoming a proven method to efficiently improve properties of enzymes, such as specific activity, stereoselectivity, optimum pH, organic solvent tolerance, and stability.21
Through several generations of such DNA shuffling, GDH activity was improved by a factor of 13 and KRED activity by a factor of 7. The enantioselectivity of the improved KRED remained >99.5%. With the improved enzymes, the reaction was complete in 8 h with an increased loading of 3 to 160 g L−1, reduced KRED loading to 0.57 g L−1, and lowered GDH loading to 0.38 g L−1 (Table 1). With a 9.5× lowering of the enzyme loading there were no emulsion problems. Phase separation required less than one minute and provided 4 in >95% recovered yield of >99.9% e.e.
The activity improvement for the ketone reduction biocatalysts over the course of evolution compared with the wild type enzyme is shown in Fig. 5.
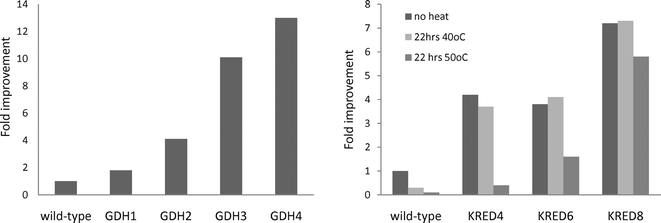 | ||
| Fig. 5 Improvements in the biocatalysts’ performances for the reduction of 3. The numbers identifying the enzymes represent the number of generations of evolution. The KREDs were exposed to thermal challenges before their assay as indicated in the figure. | ||
Similarly, the activity of natural HHDH for cyanation of 4 to 1 (Fig. 6) was extremely low and the enzyme showed poor stability in the presence of the substrate and product. With 20 g L−1 of 4 and 30 g L−1 of recombinantly-produced natural HHDH, the rate of reaction had virtually approached 0 by 72 h. Product recovery was difficult due to work-up challenges in the filtration and phase separation steps caused by the large amount of enzyme. In addition to the extremely low activity and poor stability of the natural HHDH for the cyanation of 4, the enzyme was also strongly inhibited by the product. Since 1 is more water-soluble than the substrate 4, attempts to overcome the inhibition by extraction of product into an organic solvent such as EtOAc, n-BuOAc, or MTBE were unsuccessful. However, after many iterative rounds of DNA shuffling, with screening in the presence of iteratively higher concentrations of product, the inhibition was largely overcome and the HHDH activity was increased >2500-fold compared to the wild-type enzyme.
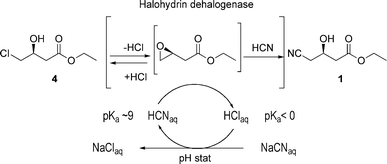 | ||
| Fig. 6 HHDH-catalyzed cyanation of 4. | ||
Fig. 7 shows the progress of cyanation reactions, using HHDH catalysts spanning multiple generations of directed evolution. With the improved HHDH, the reaction of 140 g L−14 using 1.2 g L−1 enzyme was complete in 5 h (Table 2). The product was isolated in 92% yield.
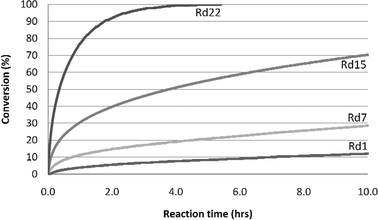 | ||
Fig. 7 Progress of cyanation reactions using HHDH catalysts. Substrate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :biocatalyst = 100:1 (w/w). Rd 1 = expression mutant of wild type enzyme. Rd 22 gave 130 g L−1 product. :biocatalyst = 100:1 (w/w). Rd 1 = expression mutant of wild type enzyme. Rd 22 gave 130 g L−1 product. | ||
The dramatic improvement in the activities of three different enzymes demonstrates that DNA shuffling technologies can enable large-scale enzymatic processes that otherwise would not have been economically viable. Our completely enzymatic process has been scaled-up to 2000 L reactors. In addition to this two-step process, running the reactions as a one-pot process, sequentially or concertedly, was demonstrated on a laboratory scale.19
The green features of the new process
As noted above, previous commercial processes involved kinetic resolution (50% maximum yield), syntheses from chiral pool precursors involving bromine chemistry or asymmetric hydrogenation of 3. All of these processes ultimately involve substitution of halide by cyanide under warm, alkaline conditions (e.g. 80 °C, pH 10), causing extensive by-product formation and the concomitant need for high-vacuum fractional distillation. The process described here is greener than previous processes when assessed according to the twelve principles of green chemistry.22Principle 1: waste prevention. The highly selective biocatalytic reactions afforded a substantial reduction in waste. In the final process, raw material is converted to product with >90% isolated yield affording product that is more than 98% chemically pure with an enantiomeric excess of > 99.9%. Furthermore, the avoidance of alkaline-induced by-products obviates the need for further yield-sacrificing fractional distillation. The highly active evolved biocatalysts are used at such low loadings that countercurrent extraction can be used to minimize solvent volumes. Moreover, the butyl acetate solvent is recycled with an efficiency of 85%. The E factor (kg waste per kg product)23 for the overall process is 5.8 if process water is excluded (2.3 for the reduction and 3.5 for the cyanation). If process water is included, the E factor for the whole process is 18 (6.6 for the reduction and 11.4 for the cyanation). The main contributors to the E Factor, as shown in Table 3, are solvent (EtOAc and BuOAc) losses which constitute 51% of the waste, sodium gluconate (25%), NaCl and Na2SO4 (combined ca. 22%). The three enzymes and the NADP cofactor accounted for < 1% of the waste. The main waste streams are aqueous and directly biodegradable.
| Parameter | Process design | Initial process | Final process |
|---|---|---|---|
| Substrate loading/g L−1 | ≥120 | 20 | 140 |
| Reaction time/h | 8 | 72 | 5 |
| Biocatalyst loading/g L−1 | 1.5 | 30 | 1.2 |
| Isolated yield/% | >90 | 67 | 92 |
| Chemical purity/% | >98 | >98 | >98 |
| E.e. of 1/% | >99.5 | >99.5 | >99.5 |
| Phase separation of organic product phase from aqueous phase containing enzyme/min | <10 | >60 | <1 |
| Space–time yield/gproduct L−1 d−1 | >360 | 7 | 672 |
| Catalyst yield (gproduct/gcat) | 80 | 0.7 | 117 |
| Waste | Quantity (kg per kg HN) | % Contribution to E (excluding water) | % Contribution to E (including water) |
|---|---|---|---|
| ECAA losses (8%) | 0.09 | <2 | <1 |
| Triethanolamine | 0.04 | <1 | <1 |
| NaCl and Na2SO4 | 1.29 | 22 | ca. 7 |
| Na-gluconate | 1.43 | ca. 25 | ca. 9 |
| BuOAc (85% recycle) | 0.46 | ca. 8 | ca.3 |
| EtOAc (85% recycle) | 2.50 | ca. 43 | ca. 14 |
| Enzymes | 0.023 | <1 | <1 |
| NADP | 0.005 | 0.1 | <0.1 |
| Water | 12.250 | — | 67 |
| E factor | 5.8 (18) |
Principle 2: atom economy. The atom economy24 is only 45%. The use of glucose as the reductant for cofactor regeneration is cost effective but not particularly atom efficient. However, glucose is a renewable resource and the gluconate co-product is fully biodegradable.
Principle 3: less hazardous chemical syntheses. The reduction reaction uses starting materials that pose no toxicity to human health or the environment. It avoids the use of potentially hazardous hydrogen and heavy metal catalysts throughout the process thus obviating concern for their removal from waste streams and/or contamination of the product. While cyanide must be used in the second step, as in all practical routes to HN, it is used more efficiently (higher yield) and under less harsh conditions compared to previous processes.
Principle 4: design safer chemicals. This principle is not applicable as the hydroxynitrile product is the commercial starting material for atorvastatin.
Principle 5: safer solvents and auxiliaries. Safe and environmentally acceptable butyl acetate is used, together with water, as the solvent in the biocatalytic reduction reaction and extraction of the hydroxynitrile product; no auxiliaries are used. Solvent use is minimized by employing countercurrent extraction.
Principles 6 and 9: design for energy efficiency, and catalysis. In contrast with previous processes, which employ elevated temperatures for the cyanation step and high pressure hydrogenation for the reduction step, both steps in our process are very efficient biocatalytic transformations. The reactions are run at or close to ambient temperature and pressure and pH 7 and the very high energy demands of high vacuum distillation are dispensed with altogether, resulting in substantial energy savings. The turnover numbers for the different enzymes are >105 for KRED and GDH and >5 × 104 for HHDH. Because of the low enzyme concentration used, immobilization of the biocatalyst to make it recyclable is neither practical, nor economic.
Principles 7 and 10: use of renewable feedstocks, and design for degradation. The enzyme catalysts and the glucose co-substrate are derived from renewable raw materials and are completely biodegradable. The by-products of the reaction are gluconate, NADP (the cofactor that shuttles reducing equivalents from GDH to KRED) and residual glucose, enzyme, and minerals and the waste water is directly suitable for biotreatment.
Principle 8: reduce derivatization. The process avoids derivatization steps, i.e. it is step-economic25 and involves fewer unit operations than earlier processes, most notably by obviating the trouble-prone product distillation or the bisulfite-mediated separation of dehydrated by-products.26
Principles 11 and 12: real-time analysis for pollution prevention, and inherently safer chemistry. The reactions are run in pH-stat mode at neutral pH by computer-controlled addition of base. Gluconic acid generated in the first reaction is neutralized with aq. NaOH and HCl generated in the second step is neutralized with aq. NaCN, regenerating HCN (pKa∼9) in situ. The pH and the cumulative volume of added base are recorded in real time. Feeding NaCN on demand minimizes the overall concentration of HCN affording an inherently safer process.
Conclusions
We have developed and deployed a two-step, three-enzyme, and green-by-design process for the manufacture of the key hydroxynitrile intermediate for atorvastatin. We used directed evolution technologies to optimize the performance of all three enzymes to meet pre-defined process criteria. The overall volumetric productivity per mass catalyst load of the cyanation process was improved ∼2500-fold, comprising a 14-fold reduction in reaction time, a 7-fold increase in substrate loading, a 25-fold reduction in enzyme use, and a 50% improvement in isolated yield. Concomitant significant process simplification resulted in an overall reduction in energy use and waste production. These results demonstrate the power of state-of-the-art enzyme optimization technologies in enabling economically viable, green-by-design biocatalytic processes that would otherwise remain mere laboratory curiosities.Experimental
Materials
Ethyl 4-chloroacetoacetate, ethyl (S)-4-chloro-3-hydroxybutyrate (3), and ethyl 4-(R)-cyano-3-hydroxybutyrate (1) were purchased from Sigma-Aldrich. Ketoreductase (KRED; EC1.1.1.184) glucose dehydrogenase (GDH; EC1.1.1.47) and halohydrin dehalogenase (HHDH; EC3.8.1.5) were produced as semi-purified lyophilisates by fermentation using recombinant E. coli strains carrying the corresponding genes and subsequent work-up as described. The desired enzymes constitute 30–50% of this material by weight.Determination of (S)-ECHB (4) and (R)-HN (1) by chiral GC
:1; temperature program: 80 °C (for 4) or 100 °C (for 1) for 30 min, 15 °C min−1, final T = 180 °C, held for 5 min; detector: 300 °C; 30 mL min−1 H2, 400 mL min−1 air; retention times for enantiomers of 4: S at 24 min, R at 25 min; retention times for enantiomers of 1: R at 22 min, S at 23 min.Preparative enzymatic reduction of ECAA (3) to (S)-ECHB (4)
A 3 L jacketed three-neck flask equipped with a pH electrode connected to a pH stat (Schott system) was charged with 570 ml of 100 mM triethanolamine. The pH was adjusted to 7 using 96% H2SO4 (∼5 ml) and D-glucose (298 g; 1.64 M) was added. The temperature was raised to 25 °C. KRED (854 mg) and GDH (578 mg) were charged as lyophilized powders. Then, Na-NADP (98 mg) was added followed by butyl acetate (370 ml). The reaction was started by the addition of 3 (240 g; 1.46 moles) from an addition funnel while maintaining the pH at 6.9 ± 0.05 using the pH stat with 4 N NaOH feeding. The reaction was completed in ∼8 h at 25 °C at which point 357 ml 4 N NaOH was consumed. A sample was analyzed by GC to check for completion. The reaction mixture was heated to 50 °C for 30 min and 5 g of Celite was added to facilitate filtration. The reaction mixture was cooled to 25 °C and filtered through a Celite pad. The two layers were separated and the aqueous solution was extracted with 350 mL butyl acetate. The two organic extracts were combined and the solvents were removed to obtain 232.61 g (96%) of 4, as a light yellow-colored liquid. The enantiomeric excess of 4 was >99.5% (the R-enantiomer was not detected).Preparative enzymatic cyanation of (S)-ECHB (4) to HN (1)
A 1 L jacketed three-neck round bottom flask equipped with a rubber septum, a pH electrode connected to a pH stat, and a mechanical stirrer was charged with 400 ml 500 mM NaCN. The whole system was airtight since gaseous HCN is generated. The pH of the solution was ∼11.2. The pH electrode was calibrated at room temperature 20–23 °C before use. The vessel was sealed and the pH was adjusted to ∼7.5 using conc. H2SO4 (∼7 mL). HHDH was charged as an aqueous solution (1.05 g in 20 mL de-ionized water) and the reaction mixture was heated to 40 °C. Then, the pH stat control was started and 70 g (0.42 moles) of 4 was added with a syringe over ∼10 min. The pH stat maintained the pH at 7.3 ± 0.05 with the addition of a 25% NaCN solution containing 0.25% NaOH. After ∼18 h, 73 mL of NaCN/NaOH solution was consumed. At that point, the progress of the reaction was checked by GC and no ECHB was detected. The pH of the reaction mixture was adjusted to ∼3 with conc. H2SO4 (∼2.5 mL). For safety reasons, the reaction was cooled to 25 °C. The pH electrode and the rubber septum were removed [caution: HCN gas present]. The flask was equipped with a condenser and nitrogen dip tube extending into the liquid. Vacuum (∼100 mm Hg) was applied from a diaphragm pump (a caustic trap containing ∼4 M NaOH was used to collect the HCN in the off gas). The reaction mixture was heated to 40 °C. A nitrogen bleed was used to facilitate the removal of HCN. After 2.5 h, HCN removal was complete (<5 ppm in the off gas). The mixture was cooled and treated with 3.5 g of Celite and 3.5 mL bleach (containing 6.1% sodium hypochlorite). This mixture was filtered through a Celite pad. The filtrate was extracted four times with 280 mL of ethyl acetate. The combined ethyl acetate phases were concentrated in vacuo to give 61.6 g (93%) of 1. The chemical purity as determined by GC was 99.5% and the chiral purity was >99.5% e.e. for the R-enantiomer (no S-enantiomer was detected).References
- M. Müler, Angew. Chem., Int. Ed., 2005, 44, 362 CrossRef.
- D. E. Butler, T. V. Le, A. Millar and T. N. Nanninga, US5155251 ( 1992).
- G. DeSantis, Z. Zhu, W. A. Greenberg, K. Wong, J. Chaplin, S. R. Hanson, B. Farwell, L. W. Nicholson, C. L. Rand, D. P. Weiner, D. E. Robertson and M. J. Burk, J. Am. Chem. Soc., 2002, 124, 9024 CrossRef CAS; G. DeSantis, K. Wong, B. Farwell, K. Chatman, Z. Zhu, G. Tomlinson, H. Huang, X. Tan, L. Bibbs, P. Chen, K. Kretz and M. J. Burk, J. Am. Chem. Soc., 2003, 125, 11476 CrossRef CAS.
- F. H. Hoff and T. Anthonsen, Tetrahedron: Asymmetry, 1999, 10, 1401 CrossRef CAS.
- D. R. Yazbek, C. A. Martinez, S. Hu and J. Tao, Tetrahedron: Asymmetry, 2004, 15, 2757 CrossRef.
- H. Matsuda, T. Shibata, H. Hashimoto and M. Kitai, U.S. Patent, 5, 908,953, 1999 to Mitsubishi Chemical Corporation.
- M. M. Elenkov, B. Hauer and D. B. Janssen, Adv. Synth. Catal., 2006, 348, 579 CrossRef.
- T. Nakamura, T. Nagasawa, F. Yu, I. Watanabe and H. Yamada, Biochem. Biophys. Res. Commun., 1991, 180, 124 CrossRef CAS.
- T. Nakamura, T. Nagasawa, F. Yu, I. Watanabe and H. Yamada, Tetrahedron, 1994, 50, 11821 CrossRef CAS.
- J. H. Lutje Spelberg, L. Tang, M. van Gelder, R. M. Kellogg and D. B. Janssen, Tetrahedron: Asymmetry, 2002, 13, 1083 CrossRef CAS.
- J. H. Lutje Spelberg, J. E. T. van Hylckama Vlieg, L. Tang, D. B. Janssen and R. M. Kellogg, Org. Lett., 2000, 3, 41.
- G. Hasnaoui, J. H. Lutje Spelberg, E. de Vries, L. Tang, B. Hauer and D. B. Janssen, Tetrahedron: Asymmetry, 2005, 16, 1685 CrossRef CAS.
- R. A. Sheldon, Chirotechnology; Industrial Synthesis of Optically Active Compounds, Marcel Dekker, New York, 1993 Search PubMed; C. H. Wong and G. M. Whitesides, Enzymes in Synthetic Organic Chemistry, Elsevier, Amsterdam, 1994 Search PubMed.
- U. T. Strauss, U. Felfer and K. Faber, Tetrahedron: Asymmetry, 1999, 10, 107 CrossRef CAS.
- A. Bommarius and B. Bommarius-Riebel, Fundamentals of Biocatalysis, Wiley-VCH, Weinheim, 2005 Search PubMed.
- A. Liese, K. Seelbach, and C. Wandrey, Industrial Biotransformations, Wiley-VCH, Weinheim, 2000 Search PubMed; G. J. Lye, P. A. Dalby and J. M. Woodley, Org. Process Res. Dev., 2002, 6, 434 Search PubMed.
- K. A. Powell, S. W. Ramer, S.B. del Cardayre, W. P. C. Stemmer, M. B. Tobin, P. F. Longchamp and G. W. Huisman, Angew. Chem., Int. Ed., 2001, 40, 3948 CrossRef CAS; M. T. Reetz and K.-E. Jaeger, Chem.–Eur. J., 2000, 6, 407 CrossRef CAS; S. B. Rubin-Pitel and H. Zhao, Combinatorial Chemistry & High Throughput Screening, 2006, 9, 247 Search PubMed.
- R. J. Fox, S. C. Davis, E. C. Mundorff, L. M. Newman, V. Gavrilovic, S. K. Ma, L. M. Chung, C. Ching, S. Tam, S. Muley, J. Grate, J. Gruber, J. C. Whitman, R. A. Sheldon and G. W. Huisman, Nat. Biotechnol., 2007, 25, 338 CrossRef CAS.
- US 7125693 and US 7132267 to Codexis.
- W. P. Stemmer, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10747–51 CAS; W. P. Stemmer, Nature, 1994, 370, 389–91 CrossRef CAS; J. E. Ness, M. Welch, L. Giver, M. Bueno, J. R. Cherry, T. V. Borchert, W. P. Stemmer and J. Minshull, Nat. Biotechnol., 1999, 17, 893–6 CrossRef CAS.
- J. F. Chaparro-Riggers, K. M. Polizzi and A. S. Bommarius, Biotechnol. J., 2007, 2, 180 Search PubMed; R. J. Fox and G. H. Huisman, Trends Biotechnol., 2008, 26, 132 CrossRef CAS.
- P. Anastas and J. Warner, Eds., Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- R. A. Sheldon, Chem. Ind. (London, UK), 1992, 903 Search PubMed; R. A. Sheldon, Chem. Commun., 2008, 3352 RSC; R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC.
- B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS; B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS.
- P. A. Wender, M. P. Croatt and B. Witulski, Tetrahedron, 2006, 62, 7505 CrossRef CAS; P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40 CrossRef CAS.
- US 6,140,527 to Kaneka.
| This journal is © The Royal Society of Chemistry 2010 |