Production of p-cymene and hydrogen from a bio-renewable feedstock–1,8-cineole (eucalyptus oil)
Benjamin A. Leita*ab, Andrew C. Wardena, Nick Burkeb, Mike S. O'Sheaa and David Trimmb
aCSIRO Molecular and Health Technologies, Private Bag 10, Clayton South VIC, 3169, Australia. E-mail: benjamin.leita@csiro.au; Fax: +61 3 95458380; Tel: +61 3 95458130
bCSIRO Petroleum Resources, Private Bag 10, Clayton South VIC, 3169, Australia. E-mail: david.trimm@csiro.au; Fax: +61 3 95458380; Tel: +61 2 93854340
First published on 23rd October 2009
Abstract
The catalytic transformation of pure 1,8-cineole was performed in a custom-built down-flow fixed bed pyrolysis rig over various metal-doped alumina pellets controlled at temperatures between 523 K (250 °C) and 873 K (500 °C). Varying amounts of oxygen were added to the feed. Hydrophilic, hydrophobic and gaseous products were analysed separately. The hydrophilic phase was predominantly water, while the composition of the hydrophobic phase varied with catalyst type and contained mainly mixtures of both aromatic and non-aromatic C10 hydrocarbons. The main gases produced were hydrogen, carbon monoxide and carbon dioxide. As the reaction temperature increased, yields of gas phase components increased for all catalysts. The palladium-doped γ-Al2O3 catalyst at ∼250 °C showed excellent yields and selectivity for the continuous production of p-cymene together with hydrogen gas. For the best catalysts and reaction conditions, the process is very atom and carbon efficient, with all ten carbon atoms from the cineole molecule being used in the p-cymene product in an oxygen-free environment. The process uses no solvents and the high yields achieved ensure there is no waste clean-up required.
1. Introduction
The volatility of oil prices and the increasing pressure to find sustainable, bio-derived alternatives to petrochemical feedstocks has encouraged researchers to look into an increasingly diverse range of biological materials for chemical precursors. 1,8-cineole (hereafter referred to as cineole) is the main chemical component of eucalyptus oil (∼ 90%) and is obtained via steam distillation of eucalyptus leaves1 (Fig. 1). Most of the world's eucalyptus oil is produced in China as a by-product of the eucalyptus timber plantation industry. The world market for pharmaceutical and domestic use of eucalyptus oil is about 7000 tonne each year.2 In recent years, the price has been relatively stable at between US $4,500 and US $5,000 per tonne.2 However, the potential production of eucalyptus oil has been estimated to be in the millions of metric tonnes per year, globally, with estimates for current Australian eucalyptus plantings alone reaching the level of 800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes per year.3 With this volume of eucalyptus oil on the market, the prices can be expected to drop well below current values.4
000 tonnes per year.3 With this volume of eucalyptus oil on the market, the prices can be expected to drop well below current values.4 | ||
| Fig. 1 Production of 1,8-cineole. | ||
These production figures and lower costs would make cineole a very attractive feedstock for the production of industrial chemicals from a renewable resource. Thus, it is not surprising that cineole has already been regarded as a potentially attractive renewable feedstock for the production of C10 industrial chemicals and chemical precursors such as menthenols, menthenes, menthadiene and p-cymene.5
p-Cymene (Fig. 2) is of significant interest in that it is already used in the production of p-cresol, fragrances, pharmaceuticals, herbicides and fine chemicals.6 Currently, commercial production of cymenes is from toluene and propylene which are obtained from crude oil. The alkylation and isomerisation steps produce a mixture of the three different isomers of cymene, but primarily yield the m- and p-isomers.7 Further separation to pure p-cymene prior to the p-cresol conversion is currently achieved using the UOP-developed “Cymex” process, where m- and p-cymene are subject to chromatography using adsorbing (molecular sieves) and desorbing (toluene) media.8
 | ||
| Fig. 2 Structure of p-cymene. | ||
Subsequent production of cresols based on the oxidation of cymenes, or cleavage of isopropyl toluenes into cresols and acetone, are direct extensions of the phenol process from benzene.9
There have been investigations into the direct synthesis of pure p-cymene using ZSM-5 catalysts for regioselectivity in the alkylation of toluene10 and some further studies for pure p-cymene production using pinenes as a feedstock.8
These processes rely on non-renewable crude oil feedstocks and processing. Matsuura and Waki11 studied the pyrolysis of bio-derived cineole over calcium oxide and activated alumina, the latter having a catalytic effect on the reaction starting at 220 °C, while the calcium oxide catalysed the system only at 450 °C. The major liquid hydrophobic product from the activated alumina-catalysed reactions between 250 and 350 °C was found to be dipentene, with some isomerisation due to the rearrangement of the double bond. In contrast, Hügel et al. reacted cineole with hydrogen over pumice and silica-supported platinum and palladium catalysts to give mixtures of cis- and trans-p-menthanes at lower temperatures while p-cymene was found to be produced at temperatures greater than 350 °C.5 Buhl et al. investigated the conversion of α-limonene to p-cymene over silica-supported Pd catalysts12 and later from pinenes to p-cymene.8 A more recent study by Martín-Luengo and co-workers reported 100% batch conversion of limonene to p-cymene using mesoporous silica–alumina supports heated by microwave irradiation.13
In this study, we have chosen to use continuous flow fixed bed pyrolysis to perform the catalytic transformation of cineole. This approach is advantageous in that we employ a continuous process that provides more scope for mass production at either commercial scales or in smaller, decentralised plants. In addition, the process is very atom and carbon efficient, with all ten carbon atoms from the cineole molecule being used in the p-cymene product without the use of any solvents while producing no waste stream requiring costly disposal. For the catalysts, we have used high-surface area γ-Al2O3 pellets (200 m2 g−1, Saint-Gobain, USA) initially as a slightly acidic catalyst and then as a support where it was doped, via the wet impregnation technique, with molybdenum, iron, cobalt, chromium and palladium nitrate solutions.14 These catalytic materials were subjected to vapour phase pyrolysis of cineole using a down flow fixed bed tubular reactor at atmospheric pressure with varying amounts of blended oxidant.
2. Experimental
2.1. Catalyst preparation
High surface area γ-Al2O3 pellets (200 m2g−1) were used as a slightly acidic catalyst and as a solid support for molybdenum, iron, cobalt, chromium and palladium metals. The metal-doped γ-Al2O3 catalysts were prepared by a wet impregnation technique using 1 M aqueous solutions of the appropriate metal salts.In a typical procedure, 100 mL of 1 M metal nitrate solution was poured over 70 g of γ-Al2O3 pellets (Saint-Gobain NorPro, USA) that had been heated in a vacuum oven at 90 °C overnight. The mixture was stirred briefly with a spatula and left to stand at room temperature overnight. The resultant metal-impregnated γ-Al2O3 pellets were collected, washed three times with deionised water and dried in a vacuum oven at 90 °C overnight. The coated pellets were then transferred to a crucible and calcined in air at 350 °C for 12 h. As controls, undoped γ-Al2O3 pellets were subjected to the same treatment as the metal doped samples before use, and glass beads were used as a blank reaction surface.
2.2. Catalyst characterization
Prior to surface analysis, samples were degassed under vacuum at 300 °C overnight using a VacPrep 061 Degasser. The BET surface area was determined by N2 adsorption at 77 K using a Micromeritics Tristar 3000. X-Ray diffraction (XRD) measurements were carried out using a Phillips DW 1130 machine with Cu Kα (1.542 Å) radiation (40 kV, 25 mA) over the range 5–80° 2θ at a scan rate of 1° min−1 with 0.1° step size.2.3. Catalytic activity measurements
The vapour phase catalytic conversion of cineole was performed using an electrically heated tubular down-flow reactor (13.5 mm internal diameter, 300 mm length) with the catalyst held as a fixed bed at atmospheric pressure. A K-type thermocouple was used to monitor the temperature of the bed. All thermocouples, furnaces, heating bands and mass flow controllers (MFC) were controlled and data were logged using specially designed software.15The liquid product was collected at 40 °C in a stainless steel trap. The gaseous products were sent through a second trap at 0 °C to an online gas chromatograph (GC).
In a typical variable temperature experiment, 3 g of catalyst was loaded into a stainless steel mesh basket, which was placed inside the tubular reactor. The furnace was set to an initial temperature of 250 °C and the temperature was allowed to equilibrate for one hour. Cineole was injected upstream of the pre-heater at a rate of 0.5 mL min−1 with an ISCO 500D syringe pump. The carrier gas was a blended mixture of the amount of oxygen required in argon containing a 5.1% helium internal standard; this was fed at a constant rate of 150 mL min−1. Once at equilibrium, gas samples were taken and liquid products collected. The furnace temperature was then raised by 50 °C and the procedure was repeated until the final reaction temperature of 500 °C was reached.
2.4. Analysis of liquid products
The liquid product obtained for the majority of the samples consisted of an oily, hydrophobic phase and an aqueous phase. The analysis of liquid products was performed using a GCMS fitted with an auto sampler. For analysis, 10 μL of the hydrophobic phase was dissolved in 1.5 mL of acetonitrile (Aldrich) that had been doped with 0.1% mesitylene (Aldrich) as an internal standard. Chromatographic standards of 1,8-cineole, p-cymene and dipentene were run using the same sample preparation method. Major products were also characterised by 1H and 13C NMR. The yield of p-cymene was defined as the percentage of p-cymene in the whole hydrophobic phase. Selectivity for p-cymene is defined as the percentage of p-cymene in the non-cineole fraction of the hydrophobic phase. Unless otherwise noted, the non-cineole fraction of the hydrophobic phase was comprised of the products shown in Scheme 1. The hydrophilic phase was found to be mainly water.
2.5. Analysis of gaseous products
Analysis of the reagent and product gases was performed with an online Shimadzu GC 8A fitted with a Valco sampling valve with a 1 mL sample loop. The GC was fitted with a 12 m HAYESEP Q column and a thermal conductivity detector (TCD). The GC was calibrated with a calibration gas mixture containing known concentrations of hydrogen (5.02%), helium (4.92%), carbon monoxide (4.97%) and carbon dioxide (10.20%) (BOC) before each run. Argon was used as the reference gas for the TCD in the GC analyses, allowing for maximum sensitivity to hydrogen. An internal standard gas mixture containing 5.1% helium in argon was used as the carrier gas for the liquid cineole feedstock. Online gas analysis was conducted at the time of collection of liquid samples. Results for the gas phase analysis were accurate to within ±3%.3. Results and discussion
3.1 Catalyst characterization
The XRD patterns of the metal doped γ-Al2O3 catalysts prepared by the wet impregnation technique are shown in Fig. 3. The pure γ-Al2O3 showed some crystalline nature while the molybdenum-doped catalyst clearly shows a more crystalline nature with intense peaks at 2θ = 12.9, 23.4, 25.8 and 27.4° corresponding to α-MoO3 in the orthorhombic phase.16 The other metal-doped γ-Al2O3 samples display XRD patterns associated with the support material. There is some evidence of added crystallinity in all of the metal-doped samples. | ||
| Fig. 3 The XRD patterns of the metal-doped γ-Al2O3 catalysts. | ||
The BET surface areas for all as-prepared catalysts and γ-Al2O3 are given in Table 1. The surface area of the γ-Al2O3 was the highest while all other catalysts were lower in surface area. Metal doping of the γ-Al2O3 using the wet impregnation technique has shown a minimal amount of surface area loss with the exception of the Mo-doped catalyst.
3.2 Catalytic activity
Products of the pyrolysis process were separated into three phases: hydrophobic and hydrophilic liquid, and gaseous (Scheme 1). Table 2 shows the compositions of the hydrophobic phases produced during the variable temperature experiments using the five metal-doped catalysts and the undoped γ-Al2O3 catalyst. Gaseous phase composition is shown in Fig. 7. In all experiments, the only detectable hydrophilic phase component was water, while the hydrophobic phase generally contained a mixture of both aromatic and non-aromatic C10 hydrocarbons with dipentene and p-cymene as the major products. The hydrophilic liquids were analysed by NMR and GCMS. The only gases detected were hydrogen, carbon monoxide and carbon dioxide. The gas phase composition was dependent on gas feed composition and reaction temperature.| 0% O2 | 7.3% O2 | 14.6% O2 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst | Furnace T/°C | Bed T/°C | p-Cymene/% | Dipentene/% | Cineole/% | Bed T/°C | p-Cymene/% | Dipentene/% | Cineole/% | Bed T/°C | p-Cymene/% | Dipentene/% | Cineole/% |
| γ-Al2O3 | 250 | 214 | 6 | 0 | 93 | 199 | 0 | 0 | 95 | 195 | 6 | 0 | 94 |
| 300 | 212 | 23 | 0 | 76 | 210 | 46 | 0 | 45 | 216 | 18 | 0 | 78 | |
| 350 | 262 | 31 | 0 | 65 | 248 | 49 | 0 | 46 | 252 | 42 | 0 | 54 | |
| 400 | 285 | 47 | 0 | 35 | 283 | 58 | 0 | 34 | 312 | 51 | 0 | 41 | |
| 450 | 357 | 39 | 0 | 20 | 343 | 60 | 0 | 21 | 360 | 51 | 0 | 34 | |
| 500 | 406 | 26 | 0 | 6 | 393 | 50 | 0 | 10 | 397 | 47 | 0 | 24 | |
| Mo-γ-Al2O3 | 250 | 199 | 9 | 0 | 51 | 205 | 9 | 0 | 52 | 228 | 32 | 0 | 19 |
| 300 | 184 | 16 | 0 | 51 | 250 | 54 | 0 | 18 | 266 | 52 | 0 | 25 | |
| 350 | 207 | 14 | 0 | 48 | 279 | 52 | 0 | 17 | 292 | 41 | 0 | 41 | |
| 400 | 274 | 8 | 0 | 34 | 318 | 52 | 0 | 18 | 325 | 38 | 0 | 48 | |
| 450 | 335 | 33 | 0 | 24 | 356 | 54 | 0 | 22 | 358 | 34 | 0 | 53 | |
| 500 | 378 | 56 | 0 | 15 | 393 | 56 | 0 | 25 | 393 | 38 | 0 | 50 | |
| Cr-γ-Al2O3 | 250 | 205 | 11 | 0 | 88 | 200 | 23 | 0 | 42 | 224 | 14 | 0 | 83 |
| 300 | 220 | 18 | 0 | 81 | 225 | 9 | 0 | 91 | 248 | 11 | 0 | 89 | |
| 350 | 250 | 2 | 0 | 83 | 265 | 14 | 0 | 85 | 302 | 23 | 0 | 73 | |
| 400 | 293 | 18 | 0 | 40 | 307 | 25 | 0 | 71 | 346 | 36 | 0 | 49 | |
| 450 | 344 | 23 | 0 | 24 | 349 | 39 | 0 | 45 | 387 | 46 | 0 | 30 | |
| 500 | 394 | 35 | 0 | 13 | 392 | 47 | 0 | 33 | 427 | 55 | 0 | 17 | |
| Fe-γ-Al2O3 | 250 | 224 | 0 | 12 | 87 | 227 | 0 | 61 | 19 | 224 | 0 | 4 | 95 |
| 300 | 228 | 0 | 23 | 76 | 249 | 0 | 41 | 53 | 261 | 0 | 50 | 42 | |
| 350 | 250 | 0 | 56 | 42 | 282 | 0 | 42 | 54 | 286 | 0 | 48 | 44 | |
| 400 | 279 | 0 | 76 | 15 | 321 | 0 | 45 | 46 | 318 | 0 | 51 | 37 | |
| 450 | 325 | 0 | 64 | 4 | 363 | 0 | 48 | 40 | 363 | 0 | 52 | 32 | |
| 500 | 379 | 0 | 0 | 40 | 408 | 0 | 53 | 35 | 406 | 0 | 58 | 25 | |
| Co-γ-Al2O3 | 250 | 209 | 0 | 0 | 100 | 233 | 0 | 3 | 97 | 228 | 0 | 34 | 63 |
| 300 | 229 | 0 | 55 | 42 | 252 | 0 | 4 | 96 | 248 | 0 | 26 | 74 | |
| 350 | 267 | 0 | 66 | 26 | 289 | 0 | 10 | 90 | 297 | 0 | 26 | 74 | |
| 400 | 315 | 0 | 71 | 15 | 323 | 0 | 26 | 71 | 344 | 0 | 33 | 63 | |
| 450 | 361 | 0 | 64 | 8 | 365 | 0 | 44 | 48 | 411 | 0 | 47 | 45 | |
| 500 | 411 | 0 | 57 | 7 | 419 | 0 | 55 | 23 | 459 | 0 | 58 | 22 | |
| Pd-γ-Al2O3 | 250 | 214 | 57 | 0 | 37 | 234 | 89 | 0 | 10 | 293 | 93 | 0 | 5 |
| 300 | 231 | 56 | 0 | 42 | 252 | 91 | 0 | 8 | 301 | 94 | 0 | 5 | |
| 350 | 269 | 67 | 0 | 29 | 293 | 95 | 0 | 5 | 317 | 89 | 0 | 5 | |
| 400 | 305 | 77 | 0 | 18 | 328 | 94 | 0 | 3 | 350 | 81 | 0 | 4 | |
| 450 | 347 | 85 | 0 | 11 | 374 | 68 | 0 | 1 | 393 | 50 | 0 | 2 | |
| 500 | 385 | 88 | 0 | 6 | 425 | 50 | 0 | 2 | 440 | 52 | 0 | 1 | |
Fig. 4 shows p-cymene yield as a function of catalyst bed temperature for all catalysts. Cineole remained relatively unchanged for all experiments using the glass beads. The amount of oxygen blended into the carrier gas had only a mild effect on the hydrophobic phase products. A moderate level of introduced oxygen (7.3%) generally increased the yields of p-cymene, while higher levels (14.6%) decreased p-cymene yields and increased the formation of CO and CO2.
 | ||
| Fig. 4 Yield of p-cymene versus bed temperature with 7.3% oxygen. | ||
Experiments performed with the undoped γ-Al2O3 pellets showed some formation of p-cymene with yields of 47, 60 and 50%, for 0, 7.3 and 14.6% oxygen, respectively at around 400 °C. The selectivity of the undoped γ-Al2O3 pellets at this temperature was approximately 70% for p-cymene. The major product formed for both the iron- and cobalt-doped γ-Al2O3 catalysts was dipentene, with no apparent production of p-cymene.
The chromium-doped γ-Al2O3 catalyst catalysed the formation of p-cymene with a maximum yield of 55% using 14.6% oxygen at ∼400 °C. At this bed temperature, the conversion of cineole ranged from 67–87% depending on the amount of oxygen used. The selectivity of this catalyst to p-cymene was initially quite high (>95%) but with very little conversion of the cineole (9–15%). At 400 °C, selectivity was again reasonable, being approximately 70% (Fig. 5).
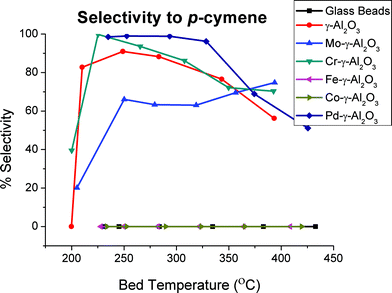 | ||
| Fig. 5 Selectivity for p-cymene versus bed temperature with 7.3% oxygen. | ||
The molybdenum-doped γ-Al2O3 catalyst also produced p-cymene as the major product with a maximum yield of 56% at 0 and 7.3% oxygen and ∼390 °C. At lower temperatures, the selectivity of this catalyst was quite low (<20%) but reached 66% at 378 °C. p-Cymene was the major product produced with high selectivity for the palladium-doped γ-Al2O3 catalyst. This catalyst gave excellent conversion efficiencies to p-cymene at much lower bed temperatures compared to the other catalysts tested. The experiments with this catalyst containing some oxygen in the carrier gas showed almost complete conversion of the cineole while conversion efficiencies to p-cymene were 95% at a reaction bed temperature of approximately 250 °C. The selectivity of this catalyst for p-cymene was also very high, being >98% at this bed temperature.
Catalysts such as the undoped alumina and the chromium-based catalyst displayed very high selectivities for p-cymene (96 and 100%, respectively) at very low bed temperatures (∼220 °C) but had poor overall conversion of cineole (24 and 9%, respectively). The molybdenum catalyst displayed both poor selectivity and overall production of p-cymene, whereas the iron and cobalt catalysts produced no p-cymene under any of the reaction conditions tested.
There was a general trend towards decreasing selectivity with increasing bed temperatures greater than ∼250 °C for most of the catalysts, with the notable exception of the palladium catalyst, which maintained very high selectivity for p-cymene in a zero-oxygen environment. Conversion increased from 37% at 214 °C to nearly complete cineole consumption (94%) at 385 °C. Addition of oxygen at moderate levels increased the selectivity to p-cymene to 99–100% at low bed temperatures while exhibiting excellent conversion of cineole (>90%).
Interestingly, the undoped alumina, molybdenum, chromium and palladium catalysts produced no dipentene in any of the runs, yet selectivity for dipentene was very high under mild conditions for the iron and cobalt catalysts. Conversion of cineole was poor for cobalt (<10%) but slightly better for iron, reaching a maximum of 58% at 250 °C, with 97% selectivity. As was the case for p-cymene, selectivity towards dipentene dropped off past ∼250 °C for both the iron and cobalt catalysts. The addition of a moderate level of oxygen during runs with the iron catalyst dramatically improved the conversion level of cineole from 13 to 81% at low bed temperatures However, additional oxygen past 7.3% reduced cineole conversion to 5%. Selectivity for dipentene was only marginally affected by the varying oxygen levels. The best result for the cobalt catalyst was 74% cineole conversion with 89% selectivity for dipentene at 267 °C with no oxygen.
3.3 Analysis of gas products
The yields of hydrogen for the different catalysts with 7.3% oxygen in the carrier gas are shown in Fig. 6.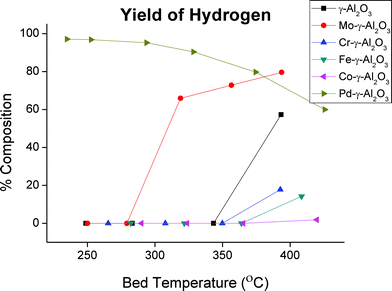 | ||
| Fig. 6 Yield of hydrogen for experiments with 7.3% oxygen. | ||
The only gas product observed during oxygen-free experiments was hydrogen. Across all catalysts tested, increasing the amount of oxygen in the carrier gas increased the production of CO and CO2.
The effect of oxygen in the carrier gas stream on the composition of the gas phase produced by the palladium doped γ-Al2O3 catalyst is shown in Fig. 7. When oxygen was used in the reaction, the amount of hydrogen produced decreased, while the amount of CO2 increased. This effect was more pronounced at higher temperatures. The CO production was negligible for all the palladium-doped γ-Al2O3 catalyst experiments. Almost total conversion of oxygen was observed during experiments containing oxygen.
 | ||
| Fig. 7 Gaseous products formed with the palladium doped γ-Al2O3 catalyst. | ||
3.4 Reaction mechanism
Based on the observed products, a possible reaction mechanism for the conversion of cineole into p-cymene over γ-Al2O3 supported catalysts is shown in Scheme 2. The reaction consists of a dehydration step followed by a dehydrogenation step. According to the thermophysical and theoretical study undertaken by Leal et al.,17 the weakest bond in the cineole molecule is the oxygen to carbon bond with only one methyl group attached. This bond is broken first, and the dehydration of cineole takes place very rapidly, producing a mixture of doubly unsaturated isomers. In the second step, dehydrogenation and rearrangement of the double bonds occurs to produce p-cymene.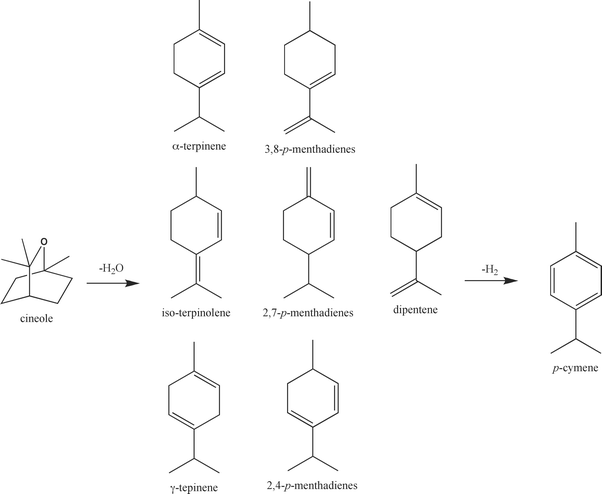
The undoped γ-Al2O3 catalyst was active for both the dehydration and dehydrogenation reactions. The dehydration reaction yielded a mixture of non-aromatic intermediates. This was followed up by the dehydrogenation of these intermediates into the fully aromatic p-cymene (Scheme 2). Similar products were also observed for the chromium- and molybdenum-doped γ-Al2O3 catalyst. For the cobalt- and iron-doped γ-Al2O3 catalyst, there was no observation of p-cymene in the products indicating that these catalysts are not able to affect the dehydrogenation step. The major products for both the cobalt- and iron-doped γ-Al2O3 catalysts suggest that these catalysts were only capable of performing the dehydration of the cineole while also giving some selectivity towards dipentene. They were not able to perform the dehydrogenation step.
For the palladium-doped γ-Al2O3 catalyst, a very high selectivity towards the p-cymene was observed. This catalyst is capable of performing both the dehydration of the cineole followed by a rapid dehydrogenation into p-cymene. Due to the absence of dehydration products observed when using the palladium-doped γ-Al2O3 catalyst, the palladium plays a vital role in the dehydrogenation step. This is not surprising as palladium is a known hydrogenation/dehydrogenation catalyst. The high selectivity of the dehydrogenation step to p-cymene over the Pd catalyst suggests this step proceeds at a greater rate than the cineole dehydration step.
4. Conclusions
This study has shown that a continuous synthetic route exists for the conversion of cineole to valuable products. The high temperature catalytic transformation of the bio-renewable feedstock, 1,8-cineole, over a number of metal-doped γ-Al2O3 catalysts has shown p-cymene to be the major hydrophobic liquid product and hydrogen the major gaseous product. From an industrial perspective, the separation of the liquid products for runs with high selectivity is trivial owing to the in situ separation of the aqueous and hydrophobic phases. The results exemplify dual-function catalytic activity (dehydration and dehydrogenation) on a bio-renewable feedstock to produce alkylbenzenes. While cobalt- and iron-doped γ-Al2O3 catalysts were only able to perform the dehydration step, the palladium-doped γ-Al2O3 pellets showed the best activity, yielding >90% p-cymene while producing large amounts of hydrogen in the exit gas at a bed temperature of ∼250 °C. Future work is focusing on studying the palladium catalyst system in more detail including: optimizing the reaction conditions to maximize both conversion of cineole and selectivity to p-cymene, and studying catalyst lifetime.Acknowledgements
The authors wish to thank the CSIRO OCE PDF funding scheme for financial support for this project.References
- J. L. Simonsen, and L. N. Owen, ‘The Terpenes’ 2nd edn, vol. 1, Cambridge University Press, Cambridge, 1973, p. 423 Search PubMed.
- Deeptha RajkumarEucalyptus oil units for dumping duty on Chinese importsThe Hindu Business Line, Thursday, Feb 12, 2004 Search PubMed.
- J. Bartle, G. Oslen, D. Cooper and T. Hobbs, Int. J. Global, Energy Issues, 2007, 27(2), 115–137 CrossRef.
- G. N. Pain, Eucalyptus Oil Report, Verve Energy, 2006 Search PubMed.
- H. M. Hügel, W. R. Jackson, C. D. Kachel and I. D. Rae, Aust. J. Chem., 1977, 30, 1287–1292 CAS.
- A. K. Mukhopadhyay, Industrial Chemical Cresols and Downstream Derivatives, Marcel Dekker, New York, 2005 Search PubMed.
- D. M. Roberge, D. Buhl, J. P. M. Niederer and W. F. Hölderich, Appl. Catal., A, 2001, 215, 111–124 CrossRef CAS.
- R. W. Neuzil, D. H. Rosback, R. H. Jensen, J. R. Teague and A. J. deRosset, Chemtech, 1980, 8, 498.
- H. Fiege, in: Ullmann's Encyclopedia of Industrial Chemistry, Vol. A8, VCH, Weinheim, 1985, p. 25 Search PubMed.
- J. S. Beck, W. O. Haag, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger and J. Weitkamp, vol. 4, Wiley-VCH, Weinheim, 1996, p. 2123 Search PubMed.
- T. Matsuura and T. Waki, J. Sci. Hiroshima Univ., 1957, 20(3), 177–186 Search PubMed.
- D. Buhl, D. M. Roberge and W. F. Hölderich, Appl. Catal., A, 1999, 188, 287 CrossRef CAS.
- M. A. Martín-Luengo, M. Yates, M. J. Martínez Domingo, B. Casal, M. Iglesias, M. Esteban and E. Ruiz-Hitzky, Appl. Catal., B, 2008, 81, 218–224 CrossRef CAS.
- B. Leita, P. Gray, N. Burke, D. Trimm, provisional patent filed 16th July 2009, Australian Provisional Patent Application No. 2009903333.
- ECE Fast, Development of the data logging software ‘RAMPX’.
- A. Kido, H. Iwamoto, N. Azuma and A. Ueno, Catal. Sury. Jpn., 2002, 6, 45 Search PubMed.
- S. Aparicio, R. Alcalde, M. J. Da'vila, B. Garcıa and J. M. Leal, J. Phys. Chem. B, 2007, 111, 3167–3177 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |