Amidine-mediated delivery of CO2 from gas phase to reaction system for highly efficient synthesis of cyclic carbonates from epoxides
Balaka Barkakaty, Kazuhide Morino, Atsushi Sudo and Takeshi Endo*
Molecular Engineering Institute, Kinki University, 11-6 Kayanomori, Iizuka, Fukuoka 820-8555, Japan. E-mail: tendo@mol-eng.fuk.kindai.ac.jp; Fax: +81-948-22-7210; Tel: +81-948-22-7210
First published on 29th October 2009
Abstract
A novel, efficient synthesis of cyclic carbonates from the reaction of epoxides and gaseous CO2 under mild conditions (1 atm, rt to 45 °C) was achieved in reasonable yields (65–83%) by using N-methyltetrahydropyrimidine (MTHP) that facilitated efficient delivery of CO2 from the gas phase to the reaction system.
Introduction
Innovation of novel methods for easy and economical chemical fixation of CO2 and its use as a C1 resource for synthesis of valuable target molecules is the need of this century for the sustenance of life on earth.1 Recently, the synthesis of five-membered cyclic carbonates from epoxides using CO2 has gained much attention, owing to its increased importance as a building block in polymer synthesis, electrolytes, aprotic polar solvents and precursors for pharmaceuticals.2 For this reaction, various catalysts, including metal complexes,3 smectites,4 titanosilicates,5 zeolites,6 metal oxides,7 organic bases,8 alkali metal halides9 and ionic liquids,10 have already been developed and reported. However, high pressure and/or high temperature are important pre-requisites for high efficiency in these catalytic systems, and some of them suffer from the formation of poly(carbonate) as a by-product. Endo and coworkers have reported that alkali metal salts such as lithium bromide catalyzed the reaction at 1 atm pressure to give five-membered cyclic carbonates in high yields and without the formation of poly(carbonate).9 However, in order to achieve a satisfactory rate of reaction, heating the system to 100 °C was required. In other words, catalysis of the reaction at 1 atm and at near ambient temperature is still a highly challenging endeavour. Recently, Endo and coworkers have reported a reversible system for efficient trap-and-release of CO2(g) by N-methyltetrahydropyrimidine (MTHP) and its analogous functionality attached to polymer side chains (Scheme 1).11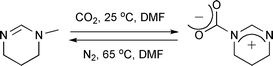 | ||
| Scheme 1 Reversible fixation-release of CO2 by amidine (MTHP). | ||
This intrinsic nature of MTHP prompted us to design a new system for converting epoxides into the corresponding carbonates under milder temperature conditions and at atmospheric pressure, which relies on utilization of MTHP that can naturally capture CO2 molecules from the gas phase and deliver them to the reactive site in a reversible cycle (Scheme 2).
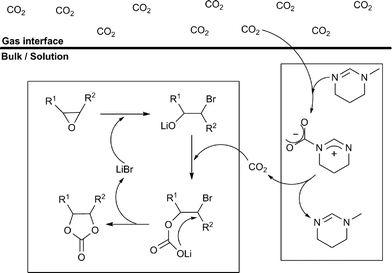 | ||
| Scheme 2 Delivery of CO2 by amidine and its insertion reaction with epoxide. | ||
Herein, we report our achievement of a highly efficient utilization of CO2 at low temperature (<45 °C) and under 1 atm, which was facilitated by participation of MTHP as a “CO2 deliverer”.
Results and discussion
For the present study, two monosubstituted epoxides, n-butyl glycidyl ether (1a), glycidyl phenyl ether (1b), and one sterically hindered disubstituted epoxide, cyclohexene oxide (2), were employed as substrates (Scheme 3). The reactions were carried out at 25 or 45 °C, which were much lower than the previously reported temperature (100 °C)9 for the efficient transformation of epoxide into the corresponding carbonate by using lithium bromide as catalyst under 1 atm CO2. The results are summarized in Table 1.| Entry | Substrate/product | Amount of MTHP/mol% | Time/h | T/°C | Yield/%b |
|---|---|---|---|---|---|
a Conditions: [epoxide]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[MTHP]:[LiBr] = 100:10:25.b Determined by 1H NMR. Isolated yields are shown in parentheses.c Reaction carried out in the absence of LiBr.d Reaction carried out in NMP, the initial concentration of epoxide was 1 M. :[MTHP]:[LiBr] = 100:10:25.b Determined by 1H NMR. Isolated yields are shown in parentheses.c Reaction carried out in the absence of LiBr.d Reaction carried out in NMP, the initial concentration of epoxide was 1 M. | |||||
| 1 | 1a/3a | 0 | 6 | 25 | 48 |
| 2 | 1a/3a | 10 | 6 | 25 | 100 (77) |
| 3c | 1a/3a | 10 | 6 | 25 | 0 |
| 4d | 1b/3b | 0 | 96 | 25 | 45 |
| 5d | 1b/3b | 10 | 24 | 25 | 35 |
| 6d | 1b/3b | 10 | 96 | 25 | 100 (83) |
| 7 | 2/4 | 0 | 72 | 45 | 12 |
| 8 | 2/4 | 10 | 12 | 45 | 25 |
| 9 | 2/4 | 10 | 24 | 45 | 68 |
| 10 | 2/4 | 10 | 48 | 45 | 84 |
| 11 | 2/4 | 10 | 72 | 45 | 95 (65) |
 | ||
| Scheme 3 Amidine catalyzed synthesis of cyclic carbonates from epoxides under mild conditions. | ||
The reaction of 1a was studied first. As a reference experiment, the reaction was performed in the absence of MTHP (entry 1); when a bulk mixture of 1a and LiBr (25 mol%) was stirred at room temperature in a reaction vessel equipped with a 1 atm CO2 balloon, 1a was slowly converted into the corresponding cyclic carbonate 3a, and after 6 h, its yield, determined by 1H NMR analysis of the resulting mixture, had reached 48%. On the other hand, the addition of 10 mol% MTHP under similar conditions remarkably improved the efficiency of the reaction to allow complete conversion of 1a into 3a after 6 h (entry 2). However, use of LiBr was mandatory for the reaction; in its absence, the reaction fails to proceed (entry 3).
This remarkable acceleration was also observed on adding MTHP in the reaction of epoxide 1b (entry 4 vs. entry 5). In this case, use of a solvent was required, because the intrinsically high crystallinity of the corresponding carbonate product 3b prevented efficient progress of the reaction in the bulk state. N-Methylpyrrolidone (NMP) was used as a suitable solvent which allowed the reaction to proceed in the homogeneous system.9 In the absence of MTHP, only 45% yield of the corresponding cyclic carbonate 3b was achieved, even though the reaction time was prolonged to 96 h (entry 4). In contrast, by adding MTHP, the reaction was greatly accelerated to achieve 100% yield of 3b under the same reaction conditions (entry 6).
These successful results at low temperature encouraged us further to apply the MTHP-mediated system for the reaction of cyclohexene oxide 2 (Table 1, entries 8–11). So far, transformation of such 1,2-disubstituted epoxides into the corresponding carbonates by conventional systems has been rather difficult due to steric hindrance.3d,3g,8c,10a,10e For example, when cyclohexene oxide 2 was heated in NMP (at 100 °C, for 24 h) under 1 atm CO2, the reaction was quite slow, leading to only 35% yield of the carbonate 4. This poor efficiency implied the intrinsic low reactivity of 2, and, in fact, its bulk reaction at 45 °C in the absence of MTHP was also very slow, giving the corresponding carbonate 4 in only 12% yield after 72 h (entry 7). In contrast, by adding MTHP, the efficiency of the reaction was improved markedly. The yield of 4 reached 25% after 12 h (entry 8), followed by a smooth progress of the reaction to achieve 95% yield at 72 h (entries 8–11).
However, the catalytic efficiency of the catalysts showed a gradual decrease when recycled (Scheme 4, Table 2). This might be attributed to the gradual decrease of LiBr concentration during the process of product separation by filtration and repeated washing with diethyl ether. Since Li salts are known to have a high affinity for carbonate and ether compounds,12 succesive formation of carbonate products followed by further treatment with diethyl ether probably removes substantial amounts of the required LiBr catalyst from the reaction system, resulting in lower yields of the desired products when the same loadings of catalysts were reused.
 | ||
| Scheme 4 Synthesis of butylglycidyl carbonate from recycled catalysts. | ||
Conclusion
In summary, a highly efficient synthesis of cyclic carbonates from the corresponding epoxides was designed and achieved by utilizing only a catalytic amount of natural CO2 fixer like MTHP along with the conventional LiBr catalyst. This system broadens the scope of such reactions by outlining a key concept to overcome the high activation barrier which is faced by these reactions when using conventional methodologies employing high temperature and high pressure techniques. The reported method can be operated successfully at atmospheric pressure and at 25–45 °C, which allows less energy-demanding and more environmentally friendly chemical fixation of CO2 into valuable compounds. The successful results imply that addition of MTHP as a “CO2 deliverer” might be a versatile strategy to improve the efficiency of various reaction systems which use CO2 as a reactant.Experimental
a) The experimental procedure for the synthesis of 3a
CO2(g) from a gas balloon (purchased from AS ONE, code 9-086-01) was allowed to react with 10 mol% of MTHP (25 mg, 0.25 mmol) at room temperature for 25 min, after which the liquid MTHP turned into white solid. Then, 25 mol% of LiBr (55 mg, 0.64 mmol) followed by 2.55 mmol of n-butyl glycidyl ether (1a, 0.36 ml) were added to the reaction mixture and the reaction was allowed to stir at room temperature under CO2 atmosphere (1 atm) for another 6 h. 1H NMR of the crude reaction mixture in CDCl3 showed 100% conversion of the starting epoxide to the desired carbonate compound after 6 h. Diethyl ether (5 ml) was then added to the reaction mixture and the reaction mixture was allowed to stir for around 10 min in the open air, the liquid part was then separated from the suspension by simple filtration. The residue in the reaction vessel was scratched with a spatula and washed several times with diethyl ether and filtered to ensure removal of any carbonate product trapped in the white solid suspension. Then, the combined filtrate was evaporated to obtain 360 mg of pure n-butyl glycidyl carbonate (3a) in 81% yield.13b) The experimental procedure for the synthesis of 3b
CO2(g) from a gas balloon (purchased from AS ONE, code 9-086-01) was allowed to react with 10 mol% of MTHP (25 mg, 0.25 mmol) in 2.5 mL of NMP at room temperature for 25 min. Then, 25 mol% of LiBr (55 mg, 0.64 mmol) followed by 2.55 mmol of phenyl glycidyl ether (1b, 0.35 ml) were added to the reaction mixture and the reaction was allowed to stir at room temperature under CO2 atmosphere (1 atm) for another 96 h. 1H NMR of the crude reaction mixture in CDCl3 after 96 h showed 100% conversion of the starting epoxide to the desired carbonate compound. The reaction mixture was then poured into 100 mL of water and the precipitate was washed thoroughly with water. The crude cyclic carbonate was then recrystallized from ethanol to obtain 411 mg of pure phenyl glycidyl carbonate (3b) in 83% yield.9ac) The experimental procedure for the synthesis of 4
CO2(g) from a gas balloon (purchased from AS ONE, code 9-086-01) was allowed to react with 10 mol% of MTHP (200 mg, 2.04 mmol) at room temperature for 25 min, after which the liquid MTHP turned into white solid. Then, 25 mol% of LiBr (443 mg, 5.1 mmol) followed by 20.04 mmol of cyclohexene oxide (2, 2.0 ml) were added to the reaction mixture and the reaction was allowed to stir at 45 °C under CO2 atmosphere for 72 h. After 72 h, the reaction mixture turned into a gel-like precipitate. 1H NMR of the crude reaction mixture in CDCl3 after 72 h showed 95% conversion of the starting epoxide to the desired carbonate compound. Then, the crude reaction mixture was extracted with EtOAc–H2O followed by a further three extractions of the aqueous layer with EtOAc. The combined organic layers were dried over anhydrous MgSO4 and concentrated in vacuo. The residue was purified by column chromatography [hexane–EtOAc (3/2, v/v)] to give 1.85 g of 4 (65% yield).14Acknowledgements
This research was financially supported by the JSR Corporation. We are thankful to Dr. Bungo Ochiai from Yamagata University for providing us with MTHP to carry out our initial investigations.Notes and references
- (a) M. Yoshida, Y. Komatsuzaki and M. Ihara, Org. Lett., 2008, 10, 2083 CrossRef CAS; (b) Y. Sugawara, W. Yamada, S. Yoshida, T. Ikeno and T. Yamada, J. Am. Chem. Soc., 2007, 129, 12902 CrossRef CAS; (c) M. Yoshida, Y. Ohsawa, K. Sugimoto, H. Tokuyama and M. Ihara, Tetrahedron Lett., 2007, 48, 8678 CrossRef CAS; (d) T. Mizuno, M. Mihara, T. Nakai, T. Iwai and T. Ito, Synthesis, 2007, 2524 CrossRef CAS; (e) M. Murakami, N. Ishida and T. Miura, Chem. Commun., 2006, 643 RSC; (f) K. Shimizu, M. Takimoto, Y. Sato and M. Mori, Org. Lett., 2005, 7, 195 CrossRef CAS; (g) G. S. Andrade, J. E. Berkner, C. L. Liotta, C. Eckert, D. A. Schiraldi, A. Andersen and D. M. Collard, Synth. Commun., 2003, 33, 3643 CrossRef CAS; (h) P. Munshi, D. J. Heldebrant, E. P. Mckoon, P. A. Kelly, C.-C. Tai and P. G. Jessop, Tetrahedron Lett., 2003, 44, 2725 CrossRef CAS; (i) E. R. Perez, M. O. da Silva, V. C. Costa, P. R.-F. Ubirajara and D. W. Franco, Tetrahedron Lett., 2002, 43, 4091 CrossRef CAS; (j) T. Mizuno and Y. Ishino, Tetrahedron, 2002, 58, 3155 CrossRef CAS; (k) M. Takimoto and M. Mori, J. Am. Chem. Soc., 2001, 123, 2895 CrossRef CAS; (l) W. McGhee, D. Riley, K. Christ, Y. Pan and B. Parnas, J. Org. Chem., 1995, 60, 2820 CrossRef CAS; (m) H. Hoberg and M. Minato, J. Organomet. Chem., 1991, 406, C25 CrossRef CAS; (n) H. Hoberg, Y. Peres, C. Krueger and Y. H. Tsay, Angew. Chem., 1987, 99, 799.
- (a) A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951 CrossRef CAS; (b) J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663 CrossRef CAS; (c) J. P. Parrish, R. N. Salvatore and K. W. Jung, Tetrahedron, 2000, 56, 8207 CrossRef CAS; (d) J. Bayardon, J. Holz, B. Schaffner, V. Andrushko, S. Verevkin, A. Preetz and A. Borner, Angew. Chem., Int. Ed., 2007, 46, 5971 CrossRef CAS.
- (a) R. L. Paddock and S. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498 CrossRef CAS; (b) H. S. Kim, J. J. Kim, B. G. Lee, O. S. Jung, H. G. Jang and S. O. Kang, Angew. Chem., Int. Ed., 2000, 39, 4096 CrossRef CAS; (c) S.-S. Wu, X.-W. Zhang, W.-L. Dai, S.-F. Yin, W.-S. Li, Y.-Q. Ren and C.-T. Au, Appl. Catal., A, 2008, 341, 106 CrossRef CAS; (d) H. Jing and S. T. Nguyen, J. Mol. Catal. A: Chem., 2007, 261, 12–15 CrossRef CAS; (e) T. Chang, H. Jing, L. Jin and W. Qiu, J. Mol. Catal. A: Chem., 2007, 264, 241 CrossRef CAS; (f) W.-L. Wong, K.-C. Cheung, P.-H. Chan, Z.-Y. Zhou, K.-H. Lee and K.-Y. Wong, Chem. Commun., 2007, 2175 RSC; (g) F. Li, C. Xia, L. Xu, W. Sun and G. Chen, Chem. Commun., 2003, 2042 RSC.
- B. M. Bhanage, S. Fujita, Y. Ikushima, K. Torii and M. Arai, Green Chem., 2003, 5, 71 RSC.
- R. Srivastava, D. Srinivas and P. Ratnaswamy, Catal. Lett., 2003, 91, 133 CrossRef CAS.
- M. Tu and R. J. Davis, J. Catal., 2001, 199, 85 CrossRef CAS.
- (a) K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526 CrossRef CAS; (b) H. Yasuda, L.-N. He and T. Sakakura, J. Catal., 2002, 209, 547 CrossRef CAS.
- (a) H. Kawanami and Y. Ikushima, Chem. Commun., 2000, 2089 RSC; (b) Y.-M. Shen, W.-L. Duan and M. Shi, Adv. Synth. Catal., 2003, 345, 337 CrossRef CAS; (c) A. Barbarini, R. Maggi, A. Mazzacani, G. Mori, G. Sartori and R. Sartorio, Tetrahedron Lett., 2003, 44, 2931 CrossRef CAS.
- (a) N. Kihara, N. Hara and T. Endo, J. Org. Chem., 1993, 58, 6198 CrossRef CAS; (b) N. Kihara and T. Endo, Macromolecules, 1992, 25, 4824 CrossRef CAS.
- (a) J. Sun, S. Zhang, W. Cheng and J. Ren, Tetrahedron Lett., 2008, 49, 3588 CrossRef CAS; (b) V. Calo, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561 CrossRef CAS; (c) Y. J. Kim and R. S. Varma, J. Org. Chem., 2005, 70, 7882 CrossRef CAS; (d) H. Kawanami, A. Sasaki, K. Matsui and Y. Ikushima, Chem. Commun., 2003, 896 RSC; (e) Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255 CrossRef CAS.
- (a) T. Endo, D. Nagai, T. Monma, H. Yamaguchi and B. Ochiai, Macromolecules, 2004, 37, 2007 CrossRef CAS; (b) B. Ochiai, K. Yokota, A. Fujii, D. Nagai and T. Endo, Macromolecules, 2008, 41, 1229 CrossRef CAS; (c) E. R. Perez, R. H. A. Santos, M. T. P. Gambardella, L. G. M. de Macedo, U. P. Rodrigues-Filho, J.-C. Launay and D. W. Franco, J. Org. Chem., 2004, 69, 8005 CrossRef CAS; (d) P. G. Jessop, D. J. Heldebrant, X. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102 CrossRef CAS; (e) Y. Liu, P. G. Jessop, M. Cunningham, C. A. Eckert and C. L. Liotta, Science, 2006, 313, 958 CrossRef CAS; (f) T. Yamada, P. J. Lukac, T. Yu and R. G. Weiss, Chem. Mater., 2007, 19, 4761 CrossRef CAS; (g) T. Yamada, P. J. Lukac, M. George and R. G. Weiss, Chem. Mater., 2007, 19, 967 CrossRef CAS.
- (a) A. M. Belostotskii, E. Markevich and D. Aurbach, J. Coord. Chem., 2004, 57, 1047 CrossRef CAS; (b) W. H. Meyer, Adv. Mater., 1998, 10, 439 CrossRef CAS.
- T. Nishikubo, T. Iizawa, M. Iida and N. Isobe, Tetrahedron Lett., 1986, 27, 3741 CrossRef CAS.
- M. Suzuki and T. Sugai, Bull. Chem. Soc. Jpn., 2004, 77, 1217 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |