Mechanisms underlying the cholesterol-lowering properties of soluble dietary fibre polysaccharides
Purnima
Gunness
and
Michael John
Gidley
*
Centre for Nutrition and Food Sciences, The University of Queensland, Hartley Teakle Building, St Lucia, Queensland 4072, Australia. E-mail: m.gidley@uq.edu.au; Fax: +61 7 3365 1177; Tel: +61 7 3365 2145
First published on 30th September 2010
Abstract
A number of studies have shown a positive relationship between diets rich in soluble dietary fibres (SDF) such as β-glucan, pectin, guar gum and psyllium, and reduced serum cholesterol and thus a decreased risk of cardiovascular disease (CVD). Three major biological mechanisms have been proposed to explain the cholesterol-reducing effects of SDF: prevention of bile salt (BS) re-absorption from the small intestine leading to an excess faecal BS excretion; reduced glycemic response leading to lower insulin stimulation of hepatic cholesterol synthesis; and physiological effects of fermentation products of SDF, mainly propionate. Evidence for the latter mechanism is inconclusive, whereas in vivo, ex vivo and in vitro experiments suggest that BS micelles “bind” to SDF preventing their re-absorption. Whereas, glycemic responses to SDF have been studied extensively, the nature of interactions between bile salt micelles and SDF that lead to incomplete BS re-absorption are poorly defined. Three potential physicochemical mechanisms are proposed together with suggestions for in vitro experiments to test them.
1. Introduction
Over the past 50 years there has been a growing interest in the cholesterol-reducing effects of soluble dietary fibres (SDF). The United States Food and Drugs Administration (FDA) has accepted health claims for food containing oat products (oats, oat bran and oat flour), barley and psyllium husks1 for their cholesterol reducing properties. It is well established that cholesterol is a major risk factor for cardiovascular disease (CVD).2 Many studies have shown a positive relationship between diets rich in soluble dietary fibres such as β-glucan (βG) from oats and barley, pectin from fruits, guar gum and psyllium, and reduced serum total cholesterol (TC) and low density lipoprotein cholesterol (LDL-C). The FDA has therefore recommended incorporation of 3 g/d of βG or 7 g/d of soluble dietary fibre from psyllium seed husks to diets low in cholesterol and saturated fat in order to help reduce TC and LDL-C1,3 and thus lower the risk of CVD. Arabinoxylan (AX), the major SDF in wheat and rye, is believed to have similar cholesterol-reducing health benefits to βG,4,5 but has been studied less extensively.The human body obtains cholesterol from two sources: de novo synthesis mainly in the liver accounting for approximately 700–900 mg/d, and directly from the diet accounting for up to 300–500 mg/d.6 Cholesterol is eliminated from the body by excretion from the gastrointestinal tract (approximately 600 mg/d), conversion to bile acids/salts (BA/BS) (approximately 400 mg/d), loss of dead skin cells (approximately 100 mg/d), synthesis of steroid hormones (approximately 50 mg/d), and by integration into cell membranes of actively dividing cells.7 The liver maintains cholesterol homeostasis. Hepatocyte cholesterol is catabolised into bile acids (BA) and steroid hormones, secreted as is in bile, used in cell membranes or stored as cholesteryl esters. The major route by which hepatic cholesterol is eliminated is via BA synthesis through a cascade of 14 enzymatic reactions with cholesterol 7α-hydroxylase being the rate limiting enzyme.8 Cholesterol homeostasis is also maintained by modulation of 3-hydroxy 3-methylglutaryl co-enzyme A reductase (HMG-Co AR), a rate limiting enzyme in cholesterol biosynthesis and by cholesterol-carrying low density lipoprotein (LDL) receptors.7
In vivo studies have shown that ingestion or administration of some viscous SDF results in a 35 to 65% excess excretion of BA/BS in the faeces9,10 leading to a substantial reduction in total plasma cholesterol (TC) and low density lipoprotein cholesterol (LDL-C) by diversion to synthesis of BA/BS, but with no effect on high density lipoprotein-cholesterol (HDL-C), the ‘good’ cholesterol.11–13 It is believed that the excess BS arises because SDF hinders BS reabsorption into the enterohepatic circulation.
SDF may also lower TC and LDL-C levels through effects on postprandial glycaemia. Viscous SDF reduces the rate of intestinal absorption of glucose with subsequent decrease in insulin production by the pancreas.14 As insulin is an activator of HMG-Co AR, it is possible that reduced insulin levels could lead to a decrease in cholesterol synthesis.15 A further potential role for viscosity is interference in efficient emulsion formation, particularly the ability to form the very small mixed micelles (ca 10 nm) associated with efficient absorption from the digestive tract.16
In addition, other studies have shown that SDF causes an increase in short chain fatty acids (SCFA) such as acetate, propionate and butyrate, products of their bacterial colonic fermentation. These SCFA, particularly propionate, may also indirectly cause a decrease in blood cholesterol through inhibition of hepatic cholesterol synthesis.17,18
Therefore, the properties that could contribute to the cholesterol-reducing effect of SDF in animals and humans are the prevention of BS re-absorption (with subsequent excess faecal excretion), and decreased hepatic cholesterol synthesis modulated by insulin and/or by fermentation products such as propionate.19
2. Proposed biological mechanisms
2.1 Excess faecal BS excretion
In humans, primary bile acids such as cholic acid (CA, 3α,7α,12α-trihydroxy-5β-cholanic acid) and chenodeoxy cholic acid (CDCA, 3α,7α-dihydroxy-5β-cholanic acid) are synthesised de novo in the liver from cholesterol as their sole precursor and are conjugated by N-acyl linkage with glycine or taurine before being temporarily stored in the gall bladder. As the conjugated BA are stronger acids than the un-conjugated forms, they remain ionised at the pH prevailing in the biliary tract and the small intestine; they are therefore termed “bile salts” (BS).20 In the gall bladder, the concentration of conjugated BS is high (300 mM) compared with their critical micelle concentrations (8.86 mM for glycocholate,21 2.5 mM for glycochenodeoxycholate22 in 0.15 M NaCl) causing aggregation of these amphipathic molecules and the formation of mixed micelles with biliary cholesterol and phosphatidylcholine.23 The liberation of gastric chyme and stimulation by cholecystokinin cause the flow of bile from the gall bladder into the duodenum and proximal jejunum, where BS micelles help in the solubilisation of food components such as cholesterol, fat soluble vitamins, and other lipids.20 Diffusion of micelles then delivers solubilised components as well as both biliary and dietary cholesterol across the unstirred water layer covering the luminal side of the enterocytes, thus facilitating uptake of cholesterol and other lipophilic components by the enterocytes.24 Once this role is fulfilled, the BS micelles transit the remainder of the small and large intestine where they are progressively reabsorbed into the enterohepatic circulation by the hepatic portal vein. In the terminal ileum the conjugated BS are reabsorbed from the intestinal lumen into the enterocytes by apical sodium-dependent bile acid transporters (ASBT). The BS are then passed into the hepatic portal vein via heterodimer protein transporters, organic solute transporter α/organic solute transporter β (OSTα/OSTβ) present on the basolateral membrane of the enterocytes.25 Bacteria present in the lumen of the ileum cause the deconjugation of some of the conjugated BS resulting in the passive absorption of free BS starting from the jejunum and throughout the small intestine.26,27 Therefore, in the terminal ileum there is a mixture of conjugated and deconjugated BS.28 The BS which escape intestinal absorption undergo bacterial enzymatic transformation (e.g. dehydroxylation at C-7 and epimerisation) in the large intestine to form secondary BS.29 Thus, the primary BS chenodeoxycholic acid is converted to lithocholic acid (LCA, 3α,12α-dihydroxy-5β-cholan-24-oic acid) and cholic acid to deoxycholic acid (DCA, 3α-hydroxy-5β-cholan-24-oic acid). DCA is passively absorbed across the colonic epithelium into the enterohepatic circulation while most of the LCA is excreted in the faeces.30 The enterohepatic circulation is very efficient at reabsorbing and recycling approximately 95% of the BS at each passage through the intestine and back to the liver. Small amounts, 400 to 600 mg/d are lost by faecal excretion but are compensated by hepatic synthesis from cholesterol25 thus maintaining cholesterol homeostasis.31 The pool of BS in bile is therefore composed of both primary (e.g. CDCA, CA) and secondary BS (e.g. DCA and trace of LCA) and this pool size is maintained at a constant level as the daily BS losses are replenished by biosynthesis.20The presence of viscous SDF in the small intestine has been shown to prevent at least some BS from being re-absorbed into the enterohepatic circulation, resulting in excess excretion of BS in faeces.32,33 This causes a depletion of BA in the liver and consequently cholesterol is rapidly catabolised in the hepatocyte to replenish the BA pool via activation of cholesterol 7α-hydroxylase. Further, cholesteryl esters are also metabolised and production of LDL-C surface membrane receptors are increased to enhance the uptake of LDL-C from the blood stream, thus lowering blood cholesterol concentration.34
2.2 Low glycemic response
Insulin is known to have an influence on carbohydrate metabolism, protein synthesis and lipogenesis. In addition, this hormone also activates HMG-Co AR which promotes the synthesis of hepatic cholesterol.35 Viscous SDF such as oat βG,36 guar gum and pectin37 slow down the digestion of macronutrients by delaying gastric emptying, slowing down the transport/mixing of digestive enzymes, and increasing the resistance of the unstirred water layer lining the mucosa to intestinal absorption.38,39 Consequently, glucose and other macronutrient absorption is reduced.36,40 This decreased postprandial glucose level is accompanied by a decline in insulin level,41 potentially resulting in the inhibition of HMG-Co AR and a subsequent reduction in hepatic cholesterol synthesis.14,42 The hormonal action on cholesterol biosynthesis is complex. In an ileostomy study Lundin et al. (2004)42 reported that a diet high in rye taken as isocaloric meals seven times per day as opposed to meals taken three times per day, decreases blood glucose and insulin levels and lowers the excretion of urinary C-peptide leading to a reduction in cholesterol. The authors argued that the bile salt excretion mechanism is not sufficient as in another study rye did not show any cholesterol-lowering effect even though an excess BS was excreted in the ileostomy bags.43 Further, Jones et al.44 also found that cholesterol synthesis was dependent on meal frequency and correlated with insulin levels. Lundin et al.42 explained their results as the combined action of reduced insulin and enhanced BS excretion leading to a decrease in plasma cholesterol. As insulin plays a central role in so many diet-induced responses, it is difficult to design experiments to determine its exact role in controlling cholesterol levels. Furthermore, the same types of rheological effects of SDF in digesta are likely to cause both a reduced insulin response and restriction of bile salt re-absorption, making the two mechanisms difficult to disentangle.2.3 Fermentation products of SDF
As soluble dietary fibres are neither digested nor absorbed in the small intestine, they undergo anaerobic bacterial fermentation in the caecum and colon, to produce short chain fatty acids (SCFA) such as acetate, propionate, and butyrate. Butyrate is utilised by the colonocytes as their primary source of energy45 whereas acetate and propionate are at least partially absorbed into the hepatic portal vein.17,18 It has been proposed that the SCFA, particularly propionate, deplete plasma cholesterol by inhibiting hepatic cholesterol metabolism via one or more of several mechanisms including reduction of HMG-Co AR activity46,47 and inhibition of acetyl-Co A reductase which catalyses the synthesis of acetyl-Co A from acetate.18 These proposed mechanisms are in line with the fact that propionate intervenes in glycolysis, gluconeogenosis and ketogenosis in the liver.48 The role of propionate in blood cholesterol reduction has been studied in vitro18,48 and in vivo by supplementing the diet with propionate49,50 and by intracaecal infusion of propionate.51,52 The in vitro studies were conducted on isolated rat liver cells incubated in propionate at different concentration levels from 1.0 to 2.5 mM18 and it was found that the inhibition of cholesterol biosynthesis increased with propionate concentration. These concentrations are lower than previously obtained in similar experiments (18 mM) and are closer to the concentration of propionate in the hepatic portal vein (0.8 mM).49 However in the latter report, no change in hepatic cholesterol synthesis was observed in the whole animal experiment in which rats were fed propionate even though there was a decrease in blood cholesterol. Similarly, with the intracaecal infusion experiments no cholesterol reducing role could be attributed to propionate. Moreover, only a few studies on the effects of SCFA on hepatic cholesterol metabolism in humans have been reported.53,54 Results obtained from these experiments are inconsistent, and further human studies are needed to underpin the role of propionate in blood cholesterol reduction. If the effect of propionate on hepatic cholesterol metabolism is involved in the cholesterol-lowering properties of SDF, it is not likely to be the dominant mechanism. This is because SDF fermentation in the colon occurs irrespective of the molecular size of the polysaccharides, whereas cholesterol-lowering effects are associated with viscosity i.e. only polymeric SDF.55From this overview of the three potential mechanisms, it is concluded that excess bile salt secretion can play a role in reducing plasma cholesterol and has the benefit of being a specific, measurable, and testable hypothesis. A central role for insulin cannot be ruled out but is very difficult to test directly, whereas a role for propionate and other fermentation products is not currently established. The remainder of this review will discuss potential physicochemical mechanisms leading to bile salt excretion. It is noted that there is a close parallel with mechanisms that attenuate macronutrient digestion and which could therefore contribute to the insulin-based mechanism as well.
3. Physicochemical mechanisms
3.1 In vivo and ex vivo results
Ileostomy studies are very useful in understanding the relationships between viscosity, ileal BS content and consequently blood cholesterol, as samples collected contain only BS with little or no SDF microbial fermentation. Studies comparing native viscous βG with less viscous, hydrolysed low molecular weight βG, showed an increase in BS excretion and synthesis in patients fed native βG in their breakfast cereal.33 The increase in BS synthesis as measured by an increase in serum concentration of 7α-hydroxy-4-cholesten-3-one (a marker for BA synthesis56) suggests a decrease in blood cholesterol in humans57 and in pigs.4Viscous but non-fermentable substances such as hydroxypropyl methyl cellulose (HPMC) have been used to show that increased intestinal viscosity is positively related to a decrease in plasma cholesterol in hamsters.55 Using the same animal model, Carr et al.58 showed that HPMC (Methocel, K100M: 22% methyl substitution and 8.1% hydroxypropyl substitution) at different viscosity grades (14-1698 cP59) caused an increase in lumen viscosity compared with cellulose which is very slowly fermentable but not viscous. The cholesterol reducing effect of the viscous HPMC suggests that viscosity and not fermentability is primarily responsible for the cholesterol reducing behaviour of SDF as HPMC is poorly fermented by colonic bacteria.60 In a recent human study, Reppas et al.61 have confirmed and extended these findings by showing that HPMC (particularly high viscosity grades) reduces plasma cholesterol.
An effect of βG molecular weight (and therefore viscosity) on plasma cholesterol has recently been demonstrated in a human intervention trial.62 This study showed that oat βG provided as a component of breakfast cereal reduced plasma cholesterol to similar extents if it had a high (ca 2 MDa) or intermediate (ca 500 kDa) molecular weight, but that the effect was halved for a lower molecular weight βG of ca 200 kDa.62 Studies on interactions between viscous soluble polymers and BA/BS have been undertaken in vitro,63in vivo64 and ex vivo using in vivo samples.55 Most of these studies have been interpreted to suggest that the SDF interact with BA in one or both of two different ways, either they are bound to the BA/BS molecules65,66 or the bile salt micelles are entrapped in a viscous or gelatinous network formed by the polymer.9,33,67 However, the nature of this entrapment and the differences between ‘viscous’ networks (which are temporary and caused by polymer entanglement or particle packing) and ‘gelatinous’ networks (which are essentially permanent and based on physical cross-links)68 have not been investigated. This is a difficult area to investigate and the rheology of digesta in vivo is poorly understood due to its complex multi-phase nature68 and limited access to samples. In ileostomy studies,33 the increase in BS ileal content and hepatic BA synthesis were attributed to the entrapment of BA by SDF in the small intestine. The investigators supported the idea that high viscosity oat-bran βG has an effect on the BS-cholesterol mixed micelles in the intestinal lumen. They argued that, as the relative amounts of BS and cholesterol ileal excretion were similar to the ratio of BS to cholesterol (1 mol to 0.08 mol) in mixed micelles found in other ileostomy studies,9,57 oat-bran βG entraps the entire micelles. Other reports have speculated that SDF forms a barrier which prevents the formation of BA micelles and increases the unstirred water layer lining the intestinal mucosal surface.69–71
There are therefore three hypotheses (Fig. 1–3) that have been proposed for explaining the incomplete re-absorption of BS and their subsequent (partial) excretion:
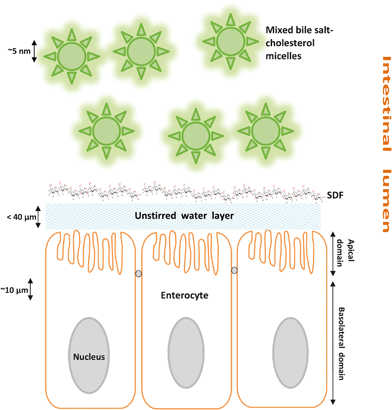 | ||
| Fig. 1 Illustration of the potential barrier property of SDF in the small intestine. | ||
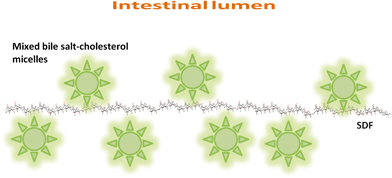 | ||
| Fig. 2 Schematic representation of molecular interactions between SDF and mixed bile salt-cholesterol micelles in the intestinal lumen. | ||
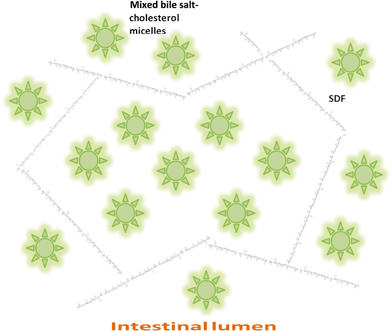 | ||
| Fig. 3 Entrapment of mixed bile salt-cholesterol micelles in a SDF network. | ||
Hypothesis 1: SDF increases the barrier properties of the unstirred layer between micelles and intestinal absorptive cells (Fig. 1)
Hypothesis 2: SDF and BS are associated/complexed at a molecular level (Fig. 2)
Hypothesis 3: SDF forms a local matrix that entraps BS micelles (Fig. 3)
3.2 In vitro studies
Many investigations of SDF interactions with BA have been undertaken in vitro, mostly through binding type experiments. Although there are a number of different types of approach, a common in vitro binding experiment involves mixing BA with the SDF source to be tested, incubating, centrifuging,63,65,72 filtering using a dialysis membrane73–75 and measuring the unbound BA. A major drawback of most of these studies is the use of bile salt concentrations (typically 1 mM) lower than the critical micellar concentration. The majority of studies allow a certain time for interactions to occur and then determine the unbound fraction. A few studies73,75 have reported the time course of BS micelle passage across membranes in the presence and absence of SDF. This is not only a closer approximation to the re-absorption thought to occur in vivo but has also been carried out at sufficiently high BS concentrations to ensure micelle formation. There has been very little data reported on the nature of any molecular interactions between BS micelles and SDF. One of the few examples is the report of Bowles et al.76 who investigated specific interactions between barley βG and glycocholic acid using solid state 13C nuclear magnetic resonance (NMR) spectroscopy. However, no evidence for specific molecular interactions was found. The methods, results and limitations of the reported techniques to analyse interactions between SDF and BS/BA are summarised in Table 1.| Reference | SDF types studied | Methods used to analyse fibre-BA interactions | Main findings | Limitations |
|---|---|---|---|---|
| 63, 72 and references cited therein, 74 and references cited therein | • Wide range of polymers and particulate suspensions | In vitro simulation of gastro-intestinal conditions, typically involving treatment of fibre sample with acid and pepsin followed by neutralisation and addition of bile salts. | Many soluble polymers and hydrated particulate fibre preparations ‘bind’ bile salts to significant extent. | Centrifugation step not representative of in vivo conditions |
| • βG (barley, oat flours & brans) | ||||
| • Arabinoxylan (rye, wheat flours & brans) | ‘Bound’ bile salts separated by centrifugation | Non-hydrated particulate fibres show low (e.g. resistant starch) or very low (e.g. cellulose or lignin) binding | ‘Binding’ may be reversible under in vivo conditions of reabsorption | |
| • Pectin (fruit & vegetable cell wall materials) | Analysis of unbound BA by HPLC after derivatisation and fluorescence detection | Bile salt concentrations typically 1 mM or lower, less than both critical micelle and typical in vivo concentrations (5–20 mM in small intestine) | ||
| 73 and references cited therein | • Soybean fibre | Taurocholic acid (16 mM) mixed with fibre in pH 7 buffer and dialysed (molecular weight cut off (MWCO) 1200)) against same buffer. Time course of dialysed BA compared with and without fibre | Fibre-containing samples delayed but did not completely prevent transport of taurocholic acid compared with control. | Single BA used |
| • Barley flour | Transport across dialysis membrane slow even in absence of fibre (e.g. 33% after 18 h73) | |||
| • Barley βG | ||||
| 75 | • Barley meal and extrudates | Bile salts (1 mM) mixed with 250 mg fibre in 10 mL buffer for 2 h at 37 °C. Unbound bile sample (1 mL) separated by ultrafiltration at 3 Bar with MWCO 3000 and analysed by HPLC | Extrusion or autoclaving increase bile binding. | Bile salts below critical micelle concentration |
| Dihydroxy bile acid more bound than trihydroxy. | Small sample of unbound bile recovered after ultrafiltration | |||
| • Oat/barley βG products | Bile salts (10 mM) mixed with fibres in pH 7 buffer. Dialysed (MWCO 6–8 kDa for proteins) with agitation. Equilibrated dialysate concentration used to infer binding. | Equilibration complete within 5 h. | Structural basis of ‘binding’ not clarified - many factors inferred to contribute | |
| Many phenolic compounds also bind | ||||
| 76 | • Processed barley βG | 13C NMR of hydrated (25% water) mixtures of βG and either glycocholic acid or Congo Red dye. | No measurable change in βG chemical shifts and no rigidification of BA in presence of βG. | Limited sensitivity and broad NMR signals |
| Both rigid (CP/MAS) and mobile (SP/MAS) segments probed. | Congo Red immobilised by βG. | Does not rule out dynamic interactions |
4. Future prospects
Despite the interest in the topic, very few experiments have been performed that could be used to identify the nature of the molecular interactions between SDF and BA/BS molecules and/or micelles even though numerous reports exist on the in vitro binding of SDF and BA as summarised in Table 1. However, methods and approaches can be proposed to test each of the hypotheses represented in Fig. 1–3. Thus, to study whether the SDF forms a barrier preventing mixed BA-cholesterol micelles from reaching the intestinal absorptive cells (Fig. 1), the simplest model is to use dialysis membranes or other semi-permeable barriers that allow passage of BS molecules but not polymeric SDF. The first reports of this type of experiment have started to appear,73,75 but there is a lot of scope for further studies, potentially extending to monolayers of epithelial cells as a more realistic model.The nature of molecular associations i.e. whether the SDF is associated/complexed on a molecular level with BA/BS, and/or forms a local network that entraps the BS micelles can be studied by a variety of structural techniques. Examples include high resolution NMR spectroscopy, small angle X-ray and neutron scattering, together with microcalorimetry to define the energetics of any interactions and binding stoichiometry as a function of SDF and BS type and concentration.
Solution state 13C NMR spectroscopy is a suitable technique for characterising SDF/BS interactions, at least in model systems, as both components are sufficiently mobile in solution to give narrow resonances. First results using this methodology77 show clear evidence for (i) close molecular interactions (Fig. 2) between BS micelles and a barley β-glucan (systematic chemical shift changes in BS resonances as a function of β-glucan concentration) and (ii) the dynamic nature of the interaction (no measurable change in line widths for either BS or SDF resonances). In contrast, a wheat arabinoxylan was found to cause increased line widths but no systematic chemical shift changes for BS micelles,77 consistent with local aggregates of arabinoxylan causing entrapment and reduced mobility of micelles (Fig. 3). The presence of aggregates within ‘solutions’ of arabinoxylan has also been shown to result in reduced diffusion of a probe molecule of the same size range as BS micelles.78
Small angle scattering (X-ray and/or neutron) gives information on the size, shape and surface characteristics of entities with sizes in the range 1–100 nm. As both micelles and polymers are in this size range, they can be investigated both separately and in combination. If there are specific molecular interactions between micelles and the polymer (Fig. 2), then scattering patterns of mixtures of the two would be predicted to be non-additive, with the expectation79 that association of BS micelles would result in a shrinking of the SDF polymer chain. Small angle neutron scattering has the advantage80 that selective contrast matching should be able to be used to examine the structure of BS micelles or SDF in the presence of the other component.
A combination of structural studies with stoichiometric and calorimetric analyses can confidently be predicted to shed further light on the interactions of pure bile salts and SDF. The challenge will then be to extend results from model systems to biological bile and whole foods. This will be difficult given the greater heterogeneity and the presence of many other components, but is necessary in order to connect in vitro data to in vivo data. If successful, this will allow mechanism-based prediction of potential in-body BS binding of SDF from different origins and in different food formulations.
References
- FDA, Food labelling: Health claims; soluble fibre from certain foods and coronoary heart disease, Report 0020–6563, Federal Register 73(37), USA, 2008 Search PubMed.
- T. A. Pearson, S. N. Blair, S. R. Daniels, R. H. Eckel, J. M. Fair, S. P. Fortmann, B. A. Franklin, L. B. Goldstein, P. Greenland, S. M. Grundy, Y. L. Hong, N. H. Miller, R. M. Lauer, I. S. Ockene, R. L. Sacco, J. F. Sallis, S. C. Smith, N. J. Stone and K. A. Taubert, Circulation, 2002, 106, 388–391 CrossRef.
- D. J. A. Jenkins, C. W. C. Kendall, V. Vuksan, E. Vidgen, T. Parker, D. Faulkner, C. C. Mehling, M. Garsetti, G. Testolin, S. C. Cunnane, M. A. Ryan and P. N. Corey, Am. J. Clin. Nutr., 2002, 75, 834–839 CAS.
- H. N. Laerke, C. Pedersen, M. A. Mortensen, P. K. Theil, T. Larsen and K. E. B. Knudsen, J. Sci. Food Agric., 2008, 88, 1385–1393 CrossRef CAS.
- H. W. Lopez, M.-A. Levrat, C. Guy, A. Messager, C. Demigné and C. Rémésy, J. Nutr. Biochem., 1999, 10, 500–509 CrossRef CAS.
- J. M. Dietschy, Klin. Wochenschr., 1984, 62, 338–345 Search PubMed.
- D. W. Russell, Cardiovasc. Drugs Ther., 1992, 6, 103–110 CrossRef CAS.
- D. W. Russell and K. D. R. Setchell, Biochemistry, 1992, 31, 4737–4749 CrossRef CAS.
- A. Lia, G. Hallmans, A. S. Sandberg, B. Sundberg, P. Aman and H. Andersson, Am. J. Clin. Nutr., 1995, 62, 1245–1251 CAS.
- Z. Madar and A. Stark, Agro Food Industry Hi-Tech, 1995, 6, 40–42 Search PubMed.
- B. H. Arjmandi and D. Reevesm Robert, J. Nutr., 1992, 122, 246 CAS.
- C. Moundras, S. R. Behr, C. Remesy and C. Demigne, J. Nutr., 1997, 127, 1068–1076 CAS.
- A. S. Truswell, Eur. J. Clin. Nutr., 2002, 56, 1–14 CrossRef CAS.
- A. T. Erkkila and A. H. Lichtenstein, Journal of Cardiovascular Nursing, 2006, 21, 3–8 Search PubMed.
- K. S. Juntunen, D. E. Laaksonen, K. Autio, L. K. Niskanen, J. J. Holst, K. E. Savolainen, K.-H. Liukkonen, K. S. Poutanen and H. M. Mykkanen, Am. J. Clin. Nutr., 2003, 78, 957–964 CAS.
- Z. Haikal, B. Play, J.-F. Landrier, A. Giraud, O. Ghiringhelli, D. Lairon and D. Jourdheuil-Rahmani, Lipids, 2008, 43, 401–408 CrossRef CAS.
- S. Bridges, J. Anderson, D. Deakins, D. Dillon and C. Wood, Am. J. Clin. Nutr., 1992, 56, 455–459 CAS.
- R. S. Wright, J. W. Anderson and S. R. Bridges, Proc. Soc. Exp. Biol. Med., 1990, 195, 26–29 Search PubMed.
- Z. Y. Chen, R. Jiao and K. Y. Ma, J. Agric. Food Chem., 2008, 56, 8761–8773 CrossRef CAS.
- R. Z. Vlahcevic, M. D. Heuman and B. P. Hylemon, in Hepatology- A textbook of liver disease, ed. D. Zakim and D. T. Boyer, W.B. Sauders Company, Philadelphia, 1996, vol. 1, ch. 13, pp. 376–417 Search PubMed.
- S. Reis, C. G. Moutinho, C. Matos, B. de Castro, P. Gameiro and J. L. F. C. Lima, Anal. Biochem., 2004, 334, 117–126 CrossRef CAS.
- K. Matsuoka, M. Suzuki, C. Honda, K. Endo and Y. Moroi, Chem. Phys. Lipids, 2006, 139, 1–10 CrossRef CAS.
- A. F. Hofmann, News Physiol. Sci., 1999, 14, 24–29 Search PubMed.
- D. Q. H. Wang, Annu. Rev. Physiol., 2007, 69, 221–248 CrossRef CAS.
- P. A. Dawson, T. Lan and A. Rao, J. Lipid Res., 2009, 50, 2340–2357 CAS.
- E. Krag and S. F. Phillips, J. Clin. Invest., 1974, 53, 1686–1694 CrossRef CAS.
- E. R. Schiff, N. C. Small and J. M. Dietschy, J. Clin. Invest., 1972, 51, 1351–1362 CrossRef CAS.
- A. F. Hofmann, Front. Biosci., 2009, 2584–2598 CrossRef CAS.
- J. P. Hamilton, G. Xie, J.-P. Raufman, S. Hogan, T. L. Griffin, C. A. Packard, D. A. Chatfield, L. R. Hagey, J. H. Steinbach and A. F. Hofmann, Am. J. Physiol.: Gastrointest. Liver Physiol., 2007, 293, G256–263 CrossRef CAS.
- J. M. Ridlon, D.-J. Kang and P. B. Hylemon, J. Lipid Res., 2005, 47, 241–259.
- J. Y. L. Chiang, J. Lipid Res., 2009, 50, 1955–1966 CAS.
- J. A. Marlett and M. H. Fischer, J. Nutr., 2002, 132, 2638–2643 CAS.
- L. Ellegard and H. Andersson, Eur. J. Clin. Nutr., 2007, 61, 938–945 CrossRef CAS.
- M. S. Brown and J. L. Goldstein, Angew. Chem., Int. Ed. Engl., 1986, 25, 583–602 CrossRef.
- M. R. Lakshmanan, C. M. Nepokroeff, G. C. Ness, R. E. Dugan and J. W. Porter, Biochem. Biophys. Res. Commun., 1973, 50, 704–710 CrossRef CAS.
- K. Queenan, M. Stewart, K. Smith, W. Thomas, R. G. Fulcher and J. Slavin, Nutr. J., 2007, 6, 6 CrossRef.
- D. J. A. Jenkins, A. R. Leeds, T. M. S. Wolever, D. V. Goff, K. Alberti, M. A. Gassull and T. D. R. Hockaday, Lancet, 1976, 308, 172–174 CrossRef.
- J. Salas-Salvado, M. Bullo, A. Perez-Heras and E. Ros, Br. J. Nutr., 2006, 96, S45–51 CrossRef CAS.
- D. Lairon, B. Play and D. Jourdheuil-Rahmani, J. Nutr. Biochem., 2007, 18, 217–227 CrossRef CAS.
- I. Bourdon, W. Yokoyama, P. Davis, C. Hudson, R. Backus, D. Richter, B. Knuckles and B. O. Schneeman, Am. J. Clin. Nutr., 1999, 69, 55–63 CAS.
- J. G. Potter, K. P. Coffman, R. L. Reid, J. M. Krall and M. J. Albrink, Am. J. Clin. Nutr., 1981, 34, 328–334 CAS.
- E. A. Lundin, J. X. Zhang, D. Lairon, P. Tidehag, P. Aman, H. Adlercreutz and G. Hallmans, Eur. J. Clin. Nutr., 2004, 58, 1410–1419 CrossRef CAS.
- J. X. Zhang, E. Lundin, G. Hallmans, H. Adlercreutz, H. Andersson, I. Bosaeus, P. Aman, R. Stenling and S. Dahlgren, Am. J. Clin. Nutr., 1994, 59, 389–394 CAS.
- P. J. Jones, C. A. Leitch and R. A. Pederson, Am. J. Clin. Nutr., 1993, 57, 868–874 CAS.
- S. A. L. W. Vanhoutvin, F. J. Troost, H. M. Hamer, P. J. Lindsey, G. H. Koek, D. M. A. E. Jonkers, A. Kodde, K. Venema and R. J. M. Brummer, PLoS One, 2009, 4, e6759 CrossRef Article No.: e6759.
- W. J. L. Chen, J. W. Anderson and D. Jennings, Proc. Soc. Exp. Biol. Med., 1984, 175, 215–218 Search PubMed.
- M. A. Levrat, M. L. Favier, C. Moundras, C. Remesy, C. Demigne and C. Morand, J. Nutr., 1994, 124, 531–538 CAS.
- P. M. Nishina and R. A. Freedland, J. Nutr., 1990, 120, 668–673 CAS.
- R. J. Illman, D. L. Topping, G. H. McIntosh, R. P. Trimble, G. B. Storer, M. N. Taylor and B. Q. Cheng, Ann. Nutr. Metab., 1988, 32, 97–107 CrossRef CAS.
- C. S. Venter, H. H. Vorster and D. G. Van der Nest, J. Nutr., 1990, 120, 1046–1053 CAS.
- K. E. Beaulieu and M. I. McBurney, J. Nutr., 1992, 122, 241–245 CAS.
- T. M. S. Wolever, F. Brighenti, D. Royall, A. L. Jenkins and D. J. A. Jenkins, Am. J. Gastroenterol., 1989, 84, 1027–1033 CAS.
- T. Todesco, A. Rao, O. Bosello and D. Jenkins, Am. J. Clin. Nutr., 1991, 54, 860–865 CAS.
- T. Wolever, P. Spadafora and H. Eshuis, Am. J. Clin. Nutr., 1991, 53, 681–687 CAS.
- D. D. Gallaher, C. A. Hassel, K. J. Lee and C. M. Gallaher, J. Nutr., 1993, 123, 244–252 CAS.
- G. Sauter, F. Berr, U. Beuers, S. Fischer and G. Paumgartner, Hepatology, 1996, 24, 123–126 CAS.
- J. Zhang, G. Hallmans, H. Andersson, I. Bosaeus, P. Aman, P. Tidehag, R. Stenling, E. Lundin and S. Dahlgren, Am. J. Clin. Nutr., 1992, 56, 99–105 CAS.
- T. P. Carr and D. D. Gallaher, J. Nutr., 1996, 126, 1463–1469 CAS.
- D. D. Gallaher, C. A. Hassel and K. J. Lee, J. Nutr., 1993, 123, 1732–1738 CAS.
- M. J. Ferguson and G. P. Jones, J. Sci. Food Agric., 2000, 80, 166–170 CrossRef CAS.
- C. Reppas, S. Z. Swidan, S. W. Tobey, M. Turowski and J. B. Dressman, Eur. J. Clin. Nutr., 2009, 63, 71–77 CrossRef CAS.
- T. M. Wolever, S. M. Tosh, A. L. Gibbs, J. Brand-Miller, A. M. Duncan, V. Hart, B. Lamarche, B. A. Thomson, R. Duss and P. J. Wood, Am. J. Clin. Nutr., 2010 DOI:10.3945/ajcn.2010.2917.
- T. S. Kahlon, M. M. Chiu and M. H. Chapman, Cereal Chem., 2009, 86, 329–332 CrossRef CAS.
- G. Dongowski, M. Huth, E. Gebhardt and W. Flamme, J. Nutr., 2002, 132, 3704–3714 CAS.
- G. Dongowski, Food Chem., 2007, 104, 390–397 CrossRef CAS.
- S. Sayar, J. L. Jannink and P. J. White, J. Agric. Food Chem., 2006, 54, 5142–5148 CrossRef CAS.
- S. S. Cho and C. Clark, in Handbook of dietary fiber, ed. S. S. Cho and M. L. Dreher, Marcel Dekker, Inc., New York, 2001, pp. 473–495 Search PubMed.
- M. A. Eastwood and E. R. Morris, Am. J. Clin. Nutr., 1992, 55, 436–442 CAS.
- E. Theuwissen and R. P. Mensink, Physiol. Behav., 2008, 94, 285–292 CrossRef CAS.
- J. A. Marlett, in Dietary fiber in health and disease, ed. D. Kritchevsky and C. Bonfield, Plenum Press, New York, 1997, vol. 427, pp. 109–121 Search PubMed.
- C. Edwards, Adv. Exp. Med. Biol., 1990, 270, 95–104 CAS.
- H. J. Kim and P. J. White, J. Agric. Food Chem., 2010, 58, 628–634 CrossRef CAS.
- S. H. Han, S. W. Lee and C. Rhee, Nutr. Res. Pract., 2009, 3, 149–155 Search PubMed.
- M. Huth, G. Dongowski, E. Gebhardt and W. Flamme, J. Cereal Sci., 2000, 32, 115–128 CrossRef CAS.
- H. T. Simonsen, M. S. Nielsen, N. J. Christensen, U. Christensen, T. V.l. Cour, M. S. Motawia, B. P. M. Jespersen, S. B. Engelsen and B. L. Mller, J. Agric. Food Chem., 2009, 57, 2056–2064 CrossRef CAS.
- R. K. Bowles, K. R. Morgan, R. H. Furneaux and G. D. Coles, Carbohydr. Polym., 1996, 29, 7–10 CrossRef CAS.
- P. Gunness, B. M. Flanagan and M. J. Gidley, J. Cereal Sci., 2010, 10.1016/j.jcs.2010.07.009 Search PubMed.
- K. J. Shelat, F. Vilaplanaa, T. M. Nicholson, H. K. Wong, M. J. Gidley and R. G. Gilbert, Carbohydr. Polym., 2010, 82, 46–53 CrossRef CAS.
- J. Mata, J. Patel, N. Jain, G. Ghosh and P. Bahadur, J. Colloid Interface Sci., 2006, 297, 797–804 CrossRef CAS.
- A. Lopez-Rubio and E. P. Gilbert, Trends Food Sci. Technol., 2009, 20, 576–586 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |