Stable carbon and hydrogen isotope analysis of methyl tert-butyl ether and tert-amyl methyl ether by purge and trap-gas chromatography-isotope ratio mass spectrometry: Method evaluation and application†
Dorothea M.
Kujawinski
a,
Manuel
Stephan
a,
Maik A.
Jochmann
a,
Karen
Krajenke
b,
Joe
Haas
c and
Torsten C.
Schmidt
*a
aUniversity Duisburg-Essen, Lotharstr.1, 47048, Duisburg, Germany. E-mail: torsten.schmidt@uni-due.de; Fax: +49 203 379-2108; Tel: +49 203 379-3311
bEnvironmental Assessment and Remediations, 225 Atlantic Avenue, Patchogue, NY 11772, USA. Fax: +1 631 447-6497; Tel: +1 631 447-6400
cNew York State Attorney General, Environmental Protection Bureau, 120 Broadway (26th Floor), New York, NY 10271, USA. Fax: +1 212 416-6007; Tel: +1 212 416-8481
First published on 20th November 2009
Abstract
In order to monitor the behaviour of contaminants in the aqueous environment effective enrichment techniques often have to be employed due to their low concentrations. In this work a robust and sensitive purge and trap-gas chromatography-isotope ratio mass spectrometry method for carbon and hydrogen isotope analysis of fuel oxygenates in water is presented. The method evaluation included the determination of method detection limits, accuracy and reproducibility of δD and δ13C values. Lowest concentrations at which reliable δ13C values could be determined were 5 µg L−1 and 28 µg L−1 for TAME and MTBE, respectively. Stable δD values for MTBE and TAME could be achieved for concentrations as low as 25 and 50 µg L−1. Good long-term reproducibility of δ13C and δD values was obtained for all target compounds. But δD values varying more than 5‰ were observed using different thermal conversion tubes. Thus, a correction of δD values in the analysis of groundwater samples was necessary to guarantee comparability of the results. The applicability of this method was shown by the analysis of groundwater samples from a gasoline contaminated site. By two dimensional isotope analysis two locations within this site were identified at which anaerobic and aerobic degradation of methyl tert-butyl ether occurred.
Environmental impactBiodegradation of methyl tert-butylether (MTBE) at sites contaminated by spillage or leakage of gasoline has been a major topic in environmental research in the last years. The investigation of carbon isotope ratios was employed for this purpose in various lab experiments and in the field. However, non-fractionating processes like dilution can distort the assessment of degradation at spill sites. We therefore applied two-dimensional isotope ratio mass spectrometry for the analysis of stable isotope ratios of carbon and hydrogen in two ethers at a contaminated field site in New York. The contaminant plume could be divided into two zones of different degradation mechanisms. The analysis of hydrogen in addition to carbon has hardly been reported for ethers other than MTBE. |
Introduction
Fuel oxygenates like methyl tert-butyl ether (MTBE), ethyl tert-butyl ether, tert-amyl methyl ether (TAME) and diisopropyl ether are major groundwater pollutants due to their use as octane enhancers in gasoline. Leakage from underground tanks and spills are major sources of fuel oxygenate input into the environment. Due to the high polarity of these compounds sorption to soil is very limited unlike for other gasoline components.1,2 Abiotic degradation of the ethers is considered to be very slow. Therefore, biodegradation is the only relevant sink for this contaminant group.1 Microbial transformation of MTBE and TAME yields tert-butyl alcohol (TBA) and tert-amyl alcohol (TAA), respectively. Nevertheless, the occurrence of these compounds at contaminated sites can only serve as a vague indicator for degradation since they are also used as gasoline additives or exist as production impurities within the associated ether. 1 Hence, the investigation of isotope ratios is a helpful tool in the investigation of transformation processes of fuel oxygenates.While aerobic degradation shows a large hydrogen isotope fractionation, the carbon isotope fractionation is only slightly detectable at early degradation state. Gray et al.3 reported a shift of δ13C value of 8.1‰ at 99.7% biodegradation, whereas shifts of δD were 80 and 156‰ at 90% biodegradation. In contrast, anaerobic degradation of MTBE shows a low shift in δD but a large shift in δ13C values.4
A detailed discussion of the proposed reaction mechanisms and their isotope fractionation is given in Zwank et al.5
The quantification of isotopic enrichment is usually done by a Rayleigh equation:
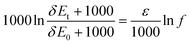 | (1) |
| Compound | Carbon isotope enrichment εC [‰] | Hydrogen isotope enrichment εH [‰] | Conditions | Bacteria | Reference |
|---|---|---|---|---|---|
| MTBE | −2 | −36 | oxic | Pure aerobic strain | 3 |
| −2.4 | |||||
| −1.5 | −66 | oxic | Mixed consortium from a field site | 3 | |
| −1.8 | −29 | ||||
| −1.52 | oxic | Microcosm (aquifer sediment) | 10 | ||
| −1.97 | |||||
| −2.4 | −42 | oxic | Methylibium sp.R8 | 11 | |
| −1.4 | oxic | Microcosm (aquifer sediment) | 6 | ||
| −9.2 | anoxic | Microcosm (aquifer sediment) | 12 | ||
| −14.2 | |||||
| −4.2 | |||||
| −13 | −16 | anoxic | Enrichment culture | 4 | |
| −15.6 | methanogenic | Microcosm | 13 | ||
| −14.4 | methanogenic and sulfate-reducing | Enrichment culture | 14 | ||
| −2.3 | −100 | aerobic | Pseudonocardia K1 | 15 | |
| TAME | −13.7 | methanogenic | Microcosm | 13 | |
| −1.7 | −72 | aerobic | Pseudonocardia K1 | 15 |
Since the sensitivity of gas chromatography-isotope ratio mass spectrometry (GC-IRMS) is limited by low ionization efficiency of the ion source and low abundance of stable isotopes,9 effective enrichment techniques have to be employed to determine in situ enrichment factors of pollutants in the environment.
Purge and Trap (P&T) is a widely-used enrichment method for volatile organic compounds from water and solid samples as it provides very low detection limits and good accuracy. Therefore it has become the standard method for the quantitative analysis of volatile compounds such as MTBE from water by U.S. EPA (United States Environmental Protection Agency). As this method includes many phase-transfer steps, the influence on isotope composition has to be evaluated.
The coupling of P&T and GC-IRMS has already been reported for the analysis of carbon isotope compositions of chlorinated VOCs16–19 from water, benzene, toluene, ethylbenzene, and xylenes (BTEX) from groundwater,20 short chain hydrocarbons in crude oils,21 trihalomethanes from chlorinated humic substances and plant leachates22 and methane from melted ice-cores.23 Few applications of this method for the analysis of fuel oxygenates have already been reported.4,6,12,19,24 Most of these works only report on precision of the measurements, but no information on accuracy is given. Besides chlorinated volatiles Zwank et al.19 investigated P&T-GC-IRMS for MTBE, benzene and toluene. It showed lower detection limits than solid phase microextraction (0.63 µg L−1 for MTBE, 0.25 µg L−1 for toluene), higher reproducibility and less fractionation in carbon isotope compositions (below 0.5‰).
P&T-GC-IRMS methods for hydrogen isotope ratio measurements are rarely reported in literature. Kuder et al.4 measured δD values for MTBE at concentrations as low as 20 µg L−1 and showed MTBE biodegradation at several gasoline spill sites.4 But to our knowledge no detailed method evaluation in respect to accuracy, detection limits and precision has been reported in literature, although these should be investigated thoroughly, especially when measuring environmental samples. Crucial parameters for hydrogen isotope measurements are residence time of the compounds in the reactor and temperature and conditioning of the reactor to achieve a carbon layer. The frequency of conditioning strongly depends on the sample matrix and experience of the operator.25 Incoming water and oxygen might oxidize the carbon coating thus leading to incomplete conversion of the analytes and wrong δD values.25
The aim of this work was to evaluate a P&T method for the analysis of carbon and hydrogen isotope compositions of MTBE, TAME and TBA in respect to method detection limits (MDL), reproducibility and accuracy. Furthermore, this method was used to determine carbon and hydrogen enrichment factors for degradation of TAME and MTBE in gasoline contaminated groundwater.
Experimental
Chemicals
Tert-butyl alcohol (99.8%), methyl tert-butyl ether (99.9%) and isopropyl alcohol (99.9%) were purchased from Fluka (Buchs, Switzerland). Tert-amyl methyl ether (97%) and tert-amyl alcohol (99%) were supplied by Sigma-Aldrich (Steinheim, Germany). Methanol (99.5%) for cleaning purposes and standard solutions was purchased from Sigma-Aldrich (Steinheim, Germany). Triple distilled water from a home made distillation unit was used for the preparation of standard solutions and stock solutions. Helium and nitrogen were purchased from Air Liquide (Düsseldorf, Germany) in a purity of ≥99.999%.Stock and standard solutions
Aqueous stock solutions with concentrations 1 g L−1 were prepared monthly in brown glass vials (20 mL volume) with screw caps with PTFE/rubber septa and stored in a refrigerator at 4 °C. Various standard solutions containing the three investigated compounds were prepared by the dilution of the stock solution in volumetric flasks. For P&T experiments 40 mL glass vials with screw caps and PTFE/silicone septa were used (PAS Technology, Magdala, Germany).Standards for the determination of the long-term stability and accuracy of carbon isotope measurements contained 125 µg L−1 MTBE, 25 µg L−1 TAME and 1 mg L−1 TBA. For hydrogen isotope measurements the concentrations were 150 µg L−1 of each MTBE and TAME and 10 mg L−1 of TBA.
For liquid injections methanolic standard solutions of MTBE and TAME were prepared by mixing aliquots of pure substances with methanol.
Purge and trap
For sample enrichment by P&T an AQUATek 70 autosampler and a Velocity XPT concentrator (both Teledyne Tekmar Company, Mason, OH, USA) were used. A sample volume of 25 mL was transferred to the glass sparger and purged 11 min with N2 at a flow of 40 mL min−1. The analytes were adsorbed on a VOCARB 3000 trap at room temperature. Afterwards the trap was dry purged for 2 min. The compounds were desorbed at 250 °C for 2 min with the carrier gas flow of the GC.After each extraction and desorption the trap was baked out at a temperature of 300 °C with a bake flow of 400 mL min−1 for 30 min. The sparger and the sample loop were rinsed with hot Milli-Q water (Millipore, Schwalbach, Germany) to prevent carry-over effects.
The effluent of the P&T device was cooled in the cryofocusing unit at a temperature of −120 °C. The hold time was 30 s and the heat ramp rate was 15 °C s−1 until a temperature of 160 °C. The device was controlled by the OPTIC3 injector software (GL Sciences Inc., Veldhoven, Netherlands).
Carbon isotope analysis by GC-IRMS
Chromatographic separation was realized by a Trace GC Ultra (Thermo Electron Corporation, Bremen, Germany) equipped with a Restek Stabilwax fused-silica capillary column (60 m × 0.32 mm I.D., film thickness 1 µm, Restek Corp., Bellefonte, PA, US). The temperature program was 1 min at 40 °C; 7 °C min−1 to 110 °C, then 3 °C min−1 to 130 °C and hold for 3 min. Helium was used as carrier gas at a flow rate of 1.5 mL min−1. An OPTIC 3 PTV injector (GL Sciences Inc., Veldhoven, Netherlands) with additional cryotrapping unit was used to introduce the analytes onto the chromatographic column.The detection of carbon isotope ratios was realized by an isotope ratio mass spectrometer MAT 253 (Thermo Electron Corporation) measuring at mass to charge ratios of 44, 45 and 46. The interface between GC and IRMS was a combustion oven GC-Combustion 3 (Thermo Electron Corporation) maintained at a temperature of 960 °C. It was reoxidized after 30–40 measurements. Calibrated CO2 reference gas was used to calculate δ13C values relative to VPDB. The IRMS was tuned on maximum linearity.
The software Isodat Version 2.5 (Thermo Electron Corporation, Bremen, Germany) was used for data processing, acquisition and evaluation.
Hydrogen isotope analysis by GC-IRMS
For the determination of hydrogen isotope ratios the same instruments as for carbon isotope measurements were used. The GC column effluent was converted to hydrogen gas at 1450 °C in a non-porous alumina reactor (32 cm length, 0.5 mm ID). The reactor was conditioned with 1 µL octane prior each set of samples. The backflush mode was activated after 950 s GC run time to protect the pyrolysis reactor from incoming water. Helium was used as a carrier gas at 1.0 mL min−1.The MAT 253 isotope ratio mass spectrometer (Thermo Electron Corporation, Bremen, Germany) was tuned on maximum linearity and detected hydrogen at mass to charge ratios of 2 and 3. All δD values were determined relative to VSMOW. The H3+-factor was determined daily using the standard function of the Isodat Version 2.5 software.
Liquid injections
A methanolic standard solution of TAME and MTBE was measured by direct injection into the GC-IRMS for the correction of δD values of the sample analytes. From this solution 1 µL was injected by a CTC-CombiPAL autosampler (Axel Semrau, Sprockhövel, Germany) into the OPTIC 3 injector run in split mode at a splitflow of 1 mL min−1. To protect the coating of the pyrolysis reactor the incoming methanol was removed by activating the backflush mode. Other GC parameters remained unchanged. The measurement was performed in triplicate.Pure phase isotopic compositions
Determination of pure phase carbon isotope compositions was made in the Helmholtz Center Munich (German Research Center for Environmental Health). Aliquots of the pure liquid standards were introduced into the combustion oven of an elemental analyzer (EA) (NC2500, Thermoquest, San Jose, CA) coupled to an IRMS (Delta XL, Thermo, Bremen). Isotope ratios of the analytes were corrected by measurements of a solid reference material in order to obtain δ13C values relative to VPDB. The reference material was measured with the same instrumental setting and the same internal reference gas. All measurements were performed in triplicate.Determination of method detection limits
MDLs were determined for carbon and hydrogen isotope analysis by P&T-GC-IRMS. Several standard solutions were prepared by dilution of the aqueous stock solutions in volumetric flasks and analyzed by P&T-GC-IRMS methods for carbon and hydrogen isotope analysis.MDLs were calculated by the moving mean procedure described by Jochmann et al.26 In the first step the mean δ13C value from the three highest concentrations was calculated. The δ13C values of these three standards had to lie within an interval ±0.5‰ around this mean value. This interval represents the total analytical error of the measurement and the accuracy of the measurement in respect to the international reference material. In the second step the next mean δ13C value was calculated by implementation of the δ13C value of the next lower concentration. Again, the δ13C values should not deviate more than ±0.5‰ from the moving mean. This procedure was repeated as long as the δ13C value was in this interval and showed a standard deviation of the triplicate measurement below ±0.5‰. The lowest concentration fulfilling these criteria was set as the method detection limit.
For hydrogen isotope analysis the same method was used but the standard deviation should not exceed 5‰. This is the typical precision of hydrogen isotope measurements by GC-IRMS according to the manufacturer and the literature.27
Site description and sampling procedure
Groundwater samples were taken from a dissolved phase petroleum based plume located in New Hyde Park, Long Island, NY (USA). At least two car service stations were suggested as contamination sources releasing gasoline over several years. Approximately 6 ×106 m3 of the aquifer were contaminated with more than 5 t of MTBE resulting in concentrations exceeding 1.3 g L−1. TAME concentrations exceeded 63 mg L−1 in the near-source region. Fig. 1 shows the major contamination sources and the dimensions of the plume. | ||
| Fig. 1 Map of sampling site including MTBE concentrations and carbon isotope compositions. “Aerobic plume section” comprises sampling wells at which dissolved oxygen contents exceeded 1 mg L−1. Black blocks correspond to possible contamination sources. The dark shaded areas mark the spots where aerobic (downstream) and anaerobic (upstream) degradation of MTBE likely occurs. ORC socks have been removed in 2005. | ||
Although there has been a decrease in concentrations of fuel oxygenates (among others due to remediation approaches such as pump & treat, air sparging, vapor extraction and oxygen releasing compound (ORC)), it is still very high. The current remediation action is pump & treat; vapor extraction and air sparging terminated operation in August 2008. The specific locations at which these efforts are made are depicted in Fig. 1.
Table 2 shows concentrations of MTBE, TAME and TBA in the samples determined by EcoTest Laboratories, N. Babylon, N.Y. USA. Additionally the dissolved oxygen concentration (obtained from field screening) and information on the sampling wells are given.
| Sample well | Depth [m] | Distance from source [m] | MTBE | TAME | Dissolved O2 [mg L−1] | ||||
|---|---|---|---|---|---|---|---|---|---|
| [µg L−1] | δ13C [‰] | δD [‰] | [µg L−1] | δ13C [‰] | δD [‰] | ||||
| a n.a. not available. | |||||||||
| MW-16(100) | 30 | 60 | 4600 | −27.78 ± 0.53 | −81 ± 1 | 340 | −27.05 ± 0.64 | −61 ± 7 | n.a. |
| MW-60(110) | 33 | 130 | 7900 | −25.57 ± 0.43 | −72 ± 5 | 450 | −25.49 ± 0.64 | −87 ± 4 | 0.68 |
| MW-20(100) | 30 | 130 | 4800 | −30.09 ± 0.38 | −83 ± 6 | 290 | −28.83 ± 0.75 | −79 ± 4 | 0.88 |
| MW-20(165) | 50 | 130 | 1000 | −27.87 ± 0.47 | −78 ± 5 | 32 | −27.91 ± 0.71 | −75 ± 2 | 2.06 |
| ML-107K | 36 | 130 | 2200 | −26.25 ± 0.41 | −74 ± 3 | 43 | −23.02 ± 0.62 | −73 ± 6 | 0.31 |
| ML-97K | 36 | 140 | 130 | −11.68 ± 0.86 | −59 ± 4 | 3 | n.a | n.a. | 0.28 |
| MW-21(120) | 36 | 140 | 2400 | −30.36 ± 0.70 | −88 ± 3 | 120 | −27.88 ± 0.60 | −75 ± 4 | 0.29 |
| MW-62(130) | 39 | 210 | 37000 | −30.9 ± 0.78 | −84 ± 9 | 1400 | −29.00 ± 0.94 | −81 ± 6 | 0.57 |
| MW-27(115) | 35 | 310 | 13000 | −31.12 ± 0.56 | −82 ± 4 | 200 | −30.01 ± 0.80 | −73 ± 6 | 1.07 |
| MW-44(125) | 38 | 390 | 74 | −29.93 ± 0.47 | −46 ± 3 | 3 | n.a | n.a. | 4.82 |
| MW-37(100) | 30 | 460 | 7700 | −31.13 ± 0.41 | −82 ± 2 | 170 | −29.64 ± 0.73 | −59 ± 8 | 5.24 |
| MW-52(110) | 33 | 540 | 1000 | −32.11 ± 0.47 | −85 ± 3 | 38 | n.a | −80 ± 3 | 1.29 |
| ML-87H | 32 | 620 | 64000 | −31.05 ± 0.41 | −78 ± 2 | 430 | −30.47 ± 0.67 | −67 ± 3 | 0.6 |
The samples were filled into brown 40 mL vials with PTFE/silicone septa without headspace; the vials were pre-preserved with Na3PO4. (approximate pH 10.5) as suggested by the US EPA guideline for compound-specific isotope measurements.28 The samples were stored in a refrigerator at 4 °C until analysis.
Measurement of environmental samples
The target compounds were extracted from the groundwater samples by P&T using the parameters described above and analyzed with both GC-IRMS methods. Although concentrations of the fuel oxygenates were generally very high, P&T was chosen to minimize matrix effects via high dilution of the samples.According to the given concentrations the samples were diluted with triple distilled water to obtain peak amplitudes as high as reference gas peaks (approx. 10 V for carbon isotope measurements and 6 V for hydrogen isotope measurements). Every triplicate measurement of a sample was preceded by the analysis of three standard solutions and followed by the analysis of one standard solution.
Samples containing TAME at concentrations near the MDL for hydrogen isotope measurements (ML107K, MW52(100), MW20(165)) were analyzed with a purge time of 20 min to increase sensitivity. Standards bracketing these samples were analyzed the same way.
All δD values were corrected for errors resulting from pyrolysis and extraction by the following equation similar to Kuder et al.:4
| δDsample = δDmeasured – δDstandard + δDli | (2) |
δ13C values were corrected for fractionation with the difference of pure phase δ13C values and δ13C values of the standards (see Table 3).
| Compound | MTBE | TAME | TBA |
|---|---|---|---|
| a c(MTBE) = 125 µg L−1, c(TAME) = 25 µg L−1, c(TBA) = 1 mg L−1. b (n = 46). c (n = 64). | |||
| MDL for carbon isotope analysis [µg L−1] | 28 | 5 | 375 |
| Amplitude at MDL [mV] | 496 | 1564 | 847 |
| δ13C at MDL concentration [‰] | −28.87 ± 0.48 | −40.10 ± 0.49 | −26.89 ± 0.35 |
| Average δ13C in long-term stability measurements [‰]a | −28.71 ± 0.35 b | −39.58 ± 0.29 c | −27.41 ± 0.32 b |
| EA-IRMS δ13C [‰] | −29.98 ± 0.02 | −40.24 ± 0.35 | −26.06 ± 0.03 |
| Shift of standard δ13C relative to pure phase [‰] | 1.2 ± 0.37 | 0.66 ± 0.54 | −1.35 ± 0.35 |
| MDL for hydrogen isotope analysis [µg L−1] | 25 | 50 | 12500 |
| Amplitude at MDL [mV] | 495 | 1310 | 11332 |
| δD at MDL [‰] | −83 | −73 | −107 |
| δD from liquid injection[‰] | −79 ± 0 | −73 ± 0 | n.d. |
Results and discussion
Water management for hydrogen isotope measurements by P&T-GC-IRMS
Sample enrichment by P&T can lead to a notable amount of water in the system. Water is reduced to H2 and CO in the reactor and thus weakens the carbon layer. Although the trap is dry purged after adsorption of the analytes the period of 2 min recommended by the manufacturer was too short to remove excess water. Higher dry purge times reduced the amount of water entering the reactor but lead to a significant loss of analytes.Kuder et al.4 used a DB-Carbowax pre-column for water separation. But an additional 6-port-valve to switch between column and pre-column and two cryofocusing units had to be installed.
In this work the dry purge time was kept at 1 min and water was separated chromatographically on the polar GC-column. Therefore the backflush mode was activated before the elution of water from the column to protect the reactor.
MDL for carbon and hydrogen isotope measurements by P&T-GC-IRMS
For TAME and MTBE the lowest aqueous concentrations for precise carbon isotope measurements were 5 µg L−1 and 28 µg L−1, respectively. Stable δ13C value for TBA could be achieved down to a concentration of 375 µg L−1.Zwank et al.19 and Kuder et al.4 report smaller MDL for MTBE than determined during this work, 0.63 µg L−1 and 1.85 µg L−1, respectively. Kolhatkar et al.12 report quantitation limits of 5 µg L−1 for MTBE and around 60 µg L−1 for TBA. Higher purge times and purge temperatures used in these works caused lower detection limits. Except for Zwank et al.19 there was no detailed information on the methods used to determine these concentrations.
Stable δD values for MTBE and TAME could be achieved for concentrations as low as 25 and 50 µg L−1, respectively. MDL for TBA was 12.5 mg L−1 due to a high standard deviation and dependency of δD values on concentration. Low extraction efficiency due to a low Henry's law constant and hydrogen exchange reactions with water might be a reason for this, but further research is needed to proof this.
The MDL for hydrogen and carbon isotope measurements are at similar peak-heights for TAME (around 1500 mV) and MTBE (around 500 mV), respectively (see Table 3 and Fig. 2). Also, concentrations of the MDL for carbon isotope measurements of TAME are 10 times lower than for hydrogen isotope measurements. This is in good agreement with the 8 times higher sensitivity of carbon isotope analysis compared to hydrogen isotope analysis reported in literature.9 For yet unknown reasons MDL for MTBE for carbon isotope measurement is as high as for hydrogen. But the MTBE concentrations in the samples are generally much higher than the MDL.
 | ||
| Fig. 2 Calibration line used to determine the MDL for hydrogen isotope analysis by P&T-GC-IRMS of TAME. Squares correspond to δD values whereas diamonds show the peak heights in mV. The dotted lines shows the ±5‰ uncertainty interval (see text). Error bars mark the relative standard deviation from a triplicate measurement. | ||
Long-term stability and accuracy of δ13C values and δD values
The stability of δ13C values was monitored by measuring more than 70 multi-component standards (see Fig. 3). The long-term stability of δ13C values of TBA, MTBE and TAME was very good. The standard deviation of the δ13C values did not exceed ±0.5‰. The variance in peak amplitudes over sequential injections can probably be explained by variations in the temperature of the column cryofocussing unit. During the heating process this has an influence on the initial band width of the analyte on the analytical column. Thus, it would in principle be better to use peak areas for comparison. However, we kept peak amplitudes in this case since this is common practice in isotope ratio mass spectrometry. | ||
| Fig. 3 Long-term stability of δ13C and δD of MTBE. a) Diamonds represent the amplitudes of the peaks, whereas squares show the deviation of the δ13C value of the single standard measurement from the average of all standards b) Diamonds show the peak heights and black squares the obtained δD values. Arrows mark the change of the pyrolysis tube. | ||
Zwank et al.19 found the shift of δ13C values during P&T enrichment of MTBE to be less than 0.5‰ whereas Smallwood et al.29 report a constant fractionation of 0.66‰. The results of these works show that extraction efficiency or purge time, respectively, can be an important parameter for accurate isotope ratio measurements. In this work the extraction efficiency calculated from the slopes of calibration lines obtained from liquid injections and P&T enrichment was about 35% for MTBE and TAME. At a purge time of 11min both ethers showed a shift towards more positive δ13C values whereas TBA showed a more negative δ13C value in comparison to the pure phase (see Table 3). An explanation for this might be insufficient extraction efficiency due to a low Henry`s law constant. But since the fractionation was constant and long-term stability very good, δ13C values of the analytes in the samples were corrected for this.
δD values varied with different pyrolysis tubes (see Fig. 3). In order to garantee comparability of the results, δD values of the sample analytes were corrected based on the procedure outlined in Kuder et al.4 (see Equation 2). Repeated analysis of the same groundwater sample with different pyrolysis tubes demonstrated a very good repeatability of this procedure. The obtained δD values differed only by 1‰.
Measurement of environmental samples
Analysis of the groundwater samples revealed carbon and hydrogen isotope compositions given in Table 2. From these data two-dimensional (2-D) isotope analysis was performed and compared to literature values to identify points at which degradation occurred. Due to dilution and mixing with undegraded substrate, enrichment factors determined from environmental samples are not comparable to those reported in literature.28 In the case of dilution the concentration decrease originates from fractionating biodegradation and non-fractionating dilution with uncontaminated groundwater.6 As a consequence regression curves of logarithmic plots according to the Rayleigh equation are flattened due to high decrease in concentration and low shift in δ values. Derived enrichment factors ε represented by the slopes of these lines are thus lower as well.The comparison of slopes from 2-D isotope analysis diminishes influences of non-fractionating processes as well as mixing with undegraded compounds.8 Here, the resulting lines are derived from a combination of two Rayleigh equations so the slopes show the ratio of both isotope enrichment factors and hence concentrations have no influence. Therefore, εH/εC were compared to literature values during this study.
A 2-D isotope ratio plot in Fig. 4b for MTBE indicates that anaerobic as well as aerobic degradation has occured variably within portions of the MTBE plume at the investigated site. Sample point MW44(125) shows a shift in the δD value of about 40‰ with respect to the lowest δD value obtained at MW21(120), but only a small shift in the δ13C value. This indicates aerobic degradation of MTBE.3 In the nearby well MW 37 sampled at another depth a much higher concentration of MTBE than in MW44 and no isotope shift in δD or δ13C was observed. This shows the heterogeneity of the system and the importance of a careful choice of sampling points, in particular when applying non-integrated, depth-oriented sampling. All groundwater samples downgradient to MW27(115) show dissolved oxygen contents above 1 mg L−1. Aerobic conditions downgradient to the source may result from the former ORC socks. But further geochemical parameters such as Fe2+, Mn2+ and N-species would have been useful to corroborate the assigned classification of aerobic and anaerobic conditions.30 Furthermore, to conclusively evaluate the significance of aerobic degradation in this section of the plume further data points showing a substantial isotope shift would be required.
 | ||
| Fig. 4 a) Double logarithmic plot according to Equation 1 for proposed anaerobic degradation of TAME. Diamonds correspond to δD values and squares to δ13C. b) 2-D plot of δD and δ13C values for MTBE; Squares mark points at which the dissolved O2 concentration was below 1 mg L−1, diamonds show the same for O2 concentrations exceeding 1 mg L−1; the grey shaded area with the dotted lines marks the hypothetical shift in isotopic composition for anaerobic degradation calculated from εC and εH from Kuder et al.4; the lighter shaded area mark the hypothetical shift in isotopic composition for aerobic biodegradation calculated from Grey et al.3 | ||
Sampling point ML97K showed a large shift of about 20‰ in δ13C values but only a medium shift of 18‰ in δD values. The nearby points ML107K and MW60(100) also showed higher δ13C values but rather low shift of δD values indicating an anaerobic degradation mechanism. In Fig. 4b a linear regression was made with all sampling points which had a dissolved oxygen content <1 mg L−1. Measureable dissolved oxygen contents below 1 mg L−1 may be due to diffusion of oxygen through sampling tubes, in particular when taking samples from larger depths.
The obtained line shows a very good fit to the findings of Kuder et al.4 The slope of the regression line was 1.3. Kuder et al. found in an anaerobic batch experiment enrichment factors for carbon to be −13‰ and for hydrogen −16‰ resulting in a slope of 1.2.
In Fig. 4a a double logarithmic plot according to the Rayleigh equation is shown for carbon and hydrogen isotope ratios of TAME during proposed anaerobic degradation. An εC of −1.4‰ and an εH of −3.4‰ was calculated from the slope of a linear regression line resulting in an εH/εC of 2.4. Unfortunately, no εH for anaerobic degradation of TAME in laboratory experiments have been reported in literature so far.
Conclusions
In this work we present a robust and sensitive method to measure carbon and hydrogen isotope compositions of fuel oxygenates and one of their metabolites. For the first time a P&T method evaluation for hydrogen isotope analysis is reported. The applicability was shown by the determination of enrichment factors for anaerobic biodegradation of MTBE and TAME. Also, the need for two-dimensional isotope analysis was emphasized to avoid underestimation of biodegradation as well as to diminish effects of dilution.Notes and references
- T. C. Schmidt, M. Schirmer, H. Weiss and S. B. Haderlein, J. Contam. Hydrol., 2004, 70, 173–203 CrossRef CAS.
- F. Fayolle, J. P. Vandecasteele and F. Monot, Appl. Microbiol. Biotechnol., 2001, 56, 339–349 CrossRef CAS.
- J. R. Gray, G. Lacrampe-Couloume, D. Gandhi, K. M. Scow, R. D. Wilson, D. M. Mackay and B. Sherwood Lollar, Environ. Sci. Technol., 2002, 36, 1931–1938 CrossRef CAS.
- T. Kuder, J. T. Wilson, P. Kaiser, R. Kolhatkar, P. Philp and J. Allen, Environ. Sci. Technol., 2005, 39, 213–220 CAS.
- L. Zwank, M. Berg, M. Elsner, T. C. Schmidt, R. P. Schwarzenbach and S. B. Haderlein, Environ. Sci. Technol., 2005, 39, 1018–1029 CrossRef CAS.
- L. E. Lesser, P. C. Johnson, R. Aravena, G. E. Spinnler, C. L. Bruce and J. P. Salanitro, Environ. Sci. Technol., 2008, 42, 6637–6643 CrossRef CAS.
- M. Elsner, L. Zwank, D. Hunkeler and R. P. Schwarzenbach, Environ. Sci. Technol., 2005, 39, 6896–6916 CrossRef CAS.
- C. Vogt, E. Cyrus, I. Herklotz, D. Schlosser, A. Bahr, S. Herrmann, H. H. Richnow and A. Fischer, Environ. Sci. Technol., 2008, 42, 7793–7800 CrossRef CAS.
- T. C. Schmidt, L. Zwank, M. Elsner, M. Berg, R. U. Meckenstock and S. B. Haderlein, Anal. Bioanal. Chem., 2004, 378, 283–300 CrossRef CAS.
- D. Hunkeler, B. J. Butler, R. Aravena and J. F. Barker, Environ. Sci. Technol., 2001, 35, 676–681 CrossRef CAS.
- M. Rosell, D. Barcelo, T. Rohwerder, U. Breuer, M. Gehre and H. H. Richnow, Environ. Sci. Technol., 2007, 41, 2036–2043 CrossRef CAS.
- R. Kolhatkar, T. Kuder, P. Philp, J. Allen and J. T. Wilson, Environ. Sci. Technol., 2002, 36, 5139–5146 CrossRef CAS.
- P. Somsamak, H. H. Richnow and M. M. Haggblom, Environ. Sci. Technol., 2005, 39, 103–109 CrossRef CAS.
- P. Somsamak, H. H. Richnow and M. M. Haggblom, Appl. Environ. Microbiol., 2006, 72, 1157–1163 CrossRef CAS.
- J. R. McKelvie, M. R. Hyman, M. Elsner, C. Smith, D. M. Aslett, G. Lacrampe-Couloume and B. Sherwood Lollar, Environ. Sci. Technol., 2009, 43, 2793–2799 CrossRef CAS.
- N. R. Auer, B. U. Manzke and D. E. Schulz-Bull, J. Chromatogr., A, 2006, 1131, 24–36 CrossRef CAS.
- D. L. Song, M. E. Conrad, K. S. Sorenson and L. Alvarez-Cohen, Environ. Sci. Technol., 2002, 36, 2262–2268 CrossRef CAS.
- K. M. Beneteau, R. Aravena and S. K. Frape, Org. Geochem., 1999, 30, 739–753 CrossRef CAS.
- L. Zwank, M. Berg, T. C. Schmidt and S. B. Haderlein, Anal. Chem., 2003, 75, 5575–5583 CrossRef CAS.
- C. A. Kelley, B. T. Hammer and R. B. Coffin, Environ. Sci. Technol., 1997, 31, 2469–2472 CrossRef CAS.
- M. J. Whiticar and L. R. Snowdon, Org. Geochem., 1999, 30, 1127–1161 CrossRef CAS.
- B. A. Bergamaschi, M. S. Fram, C. Kendall, S. R. Silva, G. R. Aiken and R. Fujii, Org. Geochem., 1999, 30, 835–842 CrossRef CAS.
- M. Behrens, J. Schmitt, K. U. Richter, M. Bock, U. C. Richter, I. Levin and H. Fischer, Rapid Commun. Mass Spectrom., 2008, 22, 3261–3269 CrossRef CAS.
- J. T. Wilson, R. Kolhatkar, T. Kuder, P. Philp and S. J. Daugherty, Ground Water Monit. Rem., 2005, 25, 108–116 CrossRef CAS.
- A. L. Sessions, J. Sep. Sci., 2006, 29, 1946–1961 CrossRef CAS.
- M. A. Jochmann, M. Blessing, S. B. Haderlein and T. C. Schmidt, Rapid Commun. Mass Spectrom., 2006, 20, 3639–3648 CrossRef CAS.
- J. A. M. Ward, J. M. E. Ahad, G. Lacrampe-Couloume, G. F. Slater, E. A. Edwards and B. Sherwood Lollar, Environ. Sci. Technol., 2000, 34, 4577–4581 CrossRef CAS.
- D. Hunkeler, R. U. Meckenstock, B. Sherwood Lollar, T. C. Schmidt and J. T. Wilson, A Guide for Assessing Biodegradation and Source Identification of Organic Ground Water Contaminants using Compound Specific Isotope Analysis (CSIA), U.S. EPA Office of Research and Development, Report EPA 600/R-08/148, 2008 Search PubMed.
- B. J. Smallwood, R. P. Philp, T. W. Burgoyne and J. D. Allen, Environ. Forensics, 2001, 2, 215–221 CrossRef CAS.
- M. Blessing, T. C. Schmidt, R. Dinkel and S. B. Haderlein, Environ. Sci. Technol., 2009, 43, 2701–2707 CrossRef CAS.
Footnote |
| † Part of a themed issue dealing with water and water related issues. |
| This journal is © The Royal Society of Chemistry 2010 |