Molecular tracers for the PET and SPECT imaging of disease
Sally L.
Pimlott
*a and
Andrew
Sutherland
*b
aWest of Scotland Radionuclide Dispensary, NHS Greater Glasgow and Clyde, Glasgow G11 6NT, UK
bWestChem, Department of Chemistry, The Joseph Black Building, University of Glasgow, Glasgow, UK G12 8QQ. E-mail: andrews@chem.gla.ac.uk; Fax: +44 (0)141 330 4888; Tel: +44 (0)141 330 5936
First published on 1st September 2010
Abstract
The development of positron emission tomography (PET) and single photon emission computed tomography (SPECT) imaging continues to grow due to the ability of these techniques to allow the non-invasive in vivo visualisation of biological processes at the molecular and cellular levels. As well as finding application for the diagnosis of disease, these techniques have also been used in the drug discovery process. Crucial to the growth of these techniques is the continued development of molecular probes that can bind to the target biological receptor with high selectivity. This tutorial review describes the use of PET and SPECT for molecular imaging and highlights key strategies for the development of molecular probes for the imaging of both cancer and neurological diseases.
 Sally L. Pimlott | Sally Pimlott was born in 1979 in Manchester, UK. After completing her B.Sc. in Neuroscience at the University of Sheffield in July 2000, she moved to the University of Glasgow and the field of radiochemistry where she undertook a PhD under the supervision of Dr Jonathan Owens on the development of novel acetylcholine receptor radioligands. In 2003, she was appointed to a training clinical scientist post at the West of Scotland Radionuclide Dispensary at North Glasgow University Hospitals NHS Trust and was made an Honorary Research Fellow at the University of Glasgow. In 2004, Sally was promoted to Head of Radiopharmacy Research and Development and in 2007 she became a state registered clinical scientist with the Health Professional Council. Her research group's interests are on the development of novel radiopharmaceuticals for use in clinical research studies. |
 Andrew Sutherland | Andrew Sutherland was born in 1972 in Wick, in the north of Scotland. After completing his B.Sc. in chemistry at the University of Edinburgh in 1994, he moved to the University of Bristol where he undertook a PhD under the supervision of Professor Christine Willis on the chemoenzymatic synthesis of α-hydroxy and α-amino acids for natural product synthesis. In 1997, he joined the research group of Professor John Vederas at the University of Alberta where he studied diaminopimelate metabolism for the design of novel antibiotics as well as the characterisation of the LNKS protein, the first purified Diels-Alderase. He then returned to the University of Bristol to take up a junior research fellowship position working with Professor Timothy Gallagher on the design and synthesis of neuronal nicotinic receptors as well as developing new Pd(0)-catalysed methodology for the synthesis of small libraries of highly substituted 2-fluoropyridines and 2-pyridones. In January 2003, he was appointed to a lectureship in the department of chemistry at the University of Glasgow and in 2008 promoted to the position of senior lecturer. His research group's interests are on the development of new synthetic methodology for the synthesis of chiral, biologically active and medicinally important compounds. |
1. Introduction
In the 20th century, molecular imaging technologies such as magnetic resonance imaging (MRI) or the radionuclide techniques of positron emission tomography (PET) and single photon emission computed tomography (SPECT) have had a significant impact on many aspects of healthcare.1,2 For example, these techniques are used routinely for the diagnosis and prognosis of many diseases while novel imaging strategies have been utilised to non-invasively track pathophysiological processes. Molecular imaging has also been used in various aspects of drug development such as understanding drug action and establishing dosage regimens and treatment strategies.In basic terms, molecular imaging effectively allows the non-invasive visualisation, characterisation and measurement of biological processes at the molecular, cellular, whole organ or body level using specific imaging probes. These molecular imaging probes (sometimes called tracers due to the sub-pharmacological amounts administered) provide an analytical signal which is detected by a particular method resulting in either a two- or three-dimensional image. Imaging probes, acquisition protocols and image processing techniques for molecular imaging techniques can be used in animal models and in humans making these truly translational modalities.
Of the non-invasive imaging technologies available PET and SPECT are the most sensitive techniques for imaging function in vivo (Table 1). However, the spatial resolution achieved using PET and SPECT imaging is relatively low where anatomical detail is poor. Often computed tomography or MRI imaging is performed to provide high resolution structural images that can be fused with the functional PET or SPECT image. The development of hybrid imaging techniques such as PET/CT and SPECT/CT allows both anatomical and functional information to be obtained in a single scanning session.
| Imaging technique | Spatial resolution | Sensitivity/mole L−1 | Amount of molecular probe used |
|---|---|---|---|
| PET | 1–2 mm | 10−11 to 10−12 | Nanograms |
| SPECT | 1–2 mm | 10−10 to 10−11 | Nanograms |
| MRI | 25–100 μm | 10−3 to 10−5 | Micrograms to milligrams |
A number of excellent comprehensive reviews have been published on PET and SPECT imaging.1,2 In this review which is aimed at the non-specialist, a general outline of how PET and SPECT are used for molecular imaging is provided. The review then describes the general approach used for designing molecular probes as well as the most efficient methods for their radiosynthesis. Examples of how these strategies and methods are used in practice for neuroimaging and the imaging of cancer are also discussed.
2. Positron emission tomography
Imaging agents used for PET are radiolabelled with radionuclides that decay by the emission of a positively charged particle called the positron. On emission from the nucleus, the positron travels a short distance in the surrounding tissue before it annihilates by combining with an electron (Fig. 1). This distance traveled by the positron is known as the positron range. On annihilation, the mass of the positron and electron are converted into energy producing two 511 keV γ-rays which are emitted simultaneously at approximately 180° to each other. The pair of γ-rays is detected by surrounding detectors which allow information about the positron annihilation to be recorded. The acquisition of a large number of coincidence events provides data which allow reconstruction into an image with information on the spatial distribution of radioactivity as a function of time. The technique used is called electronic collimation.3 An important feature of this whole process is the energy of the positron. Too high an energy and the positron will travel further in the tissue before annihilation resulting in overall loss of spatial resolution.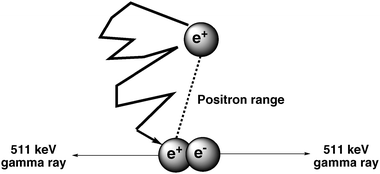 | ||
| Fig. 1 Annihilation of a positron by an electron producing two γ-rays. | ||
2.1 PET radionuclides
A significant number of PET nuclides exist for incorporation into biomolecules. The isotopes generally selected for PET imaging have half-lives comparable to the half-life of the process being imaged. The primary mode of decay should be positron emission. Other modes of emission can be tolerated as long as the energies are not close to the 511 keV produced during annihilation of the positron. The most commonly used nuclides for PET imaging are shown in Table 2. While most of these in theory can be used for PET imaging, it is mainly 11C or 18F labelled molecular probes which are employed. The main advantage of PET imaging over SPECT is that the radiolabelled imaging agent is essentially indistinguishable from its nonradioactive counterpart. Carbon is the main constituent of naturally occurring compounds and thus, replacement of carbon-12 with carbon-11 produces only a negligible isotope effect. Fluorine is not normally found in biomolecules, but the substitution of a hydrogen atom or a hydroxyl group by a fluorine atom is a commonly applied bioisosteric replacement.4Fluorine and hydrogen are similar in size (van der Waal's radii of hydrogen and fluorine are 1.20 and 1.35 Å, respectively) and thus, this replacement induces only a slight steric perturbation. Fluorine is considerably more electronegative than hydrogen but this change in the electronic properties of the molecule can quite often be advantageous producing molecular probes with improved potency.5 Other favourable features of fluorine-18 are that fluorine can accept a hydrogen bond and the carbon–fluorine bond is the strongest in organic chemistry (105.4 kcal mol−1) and therefore, unlikely to be metabolised leading to loss of the isotope.6Imaging agents which are labelled with 18F have the most favourable nuclear properties for PET analysis.7 The 18F half-life of 110 minutes allows for more complex radiosynthesis, longer in vivo studies and from a practical point of view, can be transported to clinical PET centres that lack radiochemistry facilities. Furthermore, the high percentage of β+ emission (97%) and relatively low positron energy (see Table 2) result in the generation of high-resolution images. Carbon-11, due to its relatively short half-life, is more suited for radiopharmaceuticals with short biological half-lives and studies in the same patient can be repeated after short intervals. However, 11C-labelled compounds can be produced and utilised only in facilities with a cyclotron.
| Nuclide | Half-life/min | Type of emission | Max. energy/MeV |
|---|---|---|---|
| 11C | 20.3 | β+ | 0.97 |
| 13N | 10 | β+ | 1.20 |
| 15O | 2 | β+ | 1.74 |
| 18F | 110 | β+ | 0.64 |
| 64Cu | 762 | β+/electron capture | 0.66 |
| 68Ga | 68.1 | β+/electron capture | 1.90 |
| 76Br | 972 | β+/electron capture | 4.00 |
| 124I | 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 192 192 |
β+/electron capture | 2.14 |
2.2 PET radiochemistry
Irrespective of the isotope used, all synthetic routes to isotopically labelled compounds are designed such that incorporation of the expensive and/or unstable isotope is carried out at a late stage of the synthesis thus, maximising the utilisation of the isotope.8 This is very much the case for the main PET isotopes 18F and 11C which have particularly short half-lives. In general, the synthesis and purification timeframe should not exceed two to three times the half-life of the radionuclide in use. To drive the reaction to completion as quickly as possible, a large excess of the unlabelled precursor is typically used. Other techniques such as microwave heating9 and microfluidic devices10,11 produce beneficial effects which allow the fast and efficient generation of PET imaging agents.For the introduction of carbon-11, [11C]methyl iodide is the most widely used reagent and can be easily incorporated either by C-, N-, O- or S-methylation under basic conditions.12,13Carbon-11 is generated in a cyclotron by the bombardment of nitrogen-14 with protons. Addition of oxygen then produces [11C]CO2 which can be transformed into [11C]methyl iodide using either the “wet” method or an alternative approach called the “gas phase” or “dry” method.1 Both approaches involve only two synthetic transformations from [11C]CO2, however the latter method is now more widespread as it produces PET imaging probes with higher specific activities.
[11C]Methyl iodide has also been used to generate PET imaging agents via Sonogashira, Stille or Suzuki cross-coupling reactions.14,15 For example, a Stille reaction was used for the generation of a radioligand for PET imaging of the metabotropic glutamate 1 receptor (Scheme 1).15 Reaction of organotin derived quinoline 1 with [11C]methyl iodide in the presence of a palladium(0) catalyst gave the radioligand 2 in a decay-corrected radiochemical yield of 47 ± 17% and with a specific activity of 607 ± 228 Ci mmol−1.16
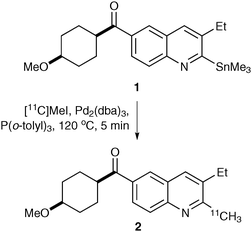 | ||
| Scheme 1 Radiosynthesis of PET imaging agent 2 using a Stille reaction. | ||
The two main strategies for introducing fluorine-18 involve either electrophilic or nucleophilic radiofluorinations.13,17 Electrophilic fluorinations are mediated using [18F]F2 and reagents derived from [18F]F2 such as [18F]acetyl hypofluorite.7 The [18F]F2 gas can be generated by bombardment of neon with deuterons, however [18F]F2 is now generally produced via the 18O(p,n) process using [18O]water. The main reactions of [18F]F2 include direct electrophilic substitution and demetallation reactions as for the synthesis of 6-[18F]fluoro-L-dopa 4, a PET tracer for probing cerebral dopamine metabolism in humans (Scheme 2).18 Fluorodestannylation of 3 with [18F]F2 was complete after only ten minutes. Rapid deprotection using hydrogen bromide gave 6-[18F]fluoro-L-dopa 4 in a decay-corrected radiochemical yield of 25%.
![Synthesis of 6-[18F]fluoro-l-dopa 4 by electrophilic fluorination.](/image/article/2011/CS/b922628c/b922628c-s2.gif) | ||
| Scheme 2 Synthesis of 6-[18F]fluoro-L-dopa 4 by electrophilic fluorination. | ||
The major limitation using [18F]F2 is that only one of the fluorine atoms carries the 18F label and as such the maximum achievable radiochemical yield is 50%. Low specific radioactivities which stem from the presence of unlabelled [19F]F2 carrier gas are also a problem when using [18F]F2 and this can restrict the application of imaging agents generated in this way. For these reasons, nucleophilic substitution using [18F]fluoride is the main strategy for introducing fluorine-18.
The [18F]fluoride ion is generally produced by irradiation of [18O]water and isolated using anion exchange chromatography.19Fluoride ions are poor nucleophiles, however, these reactions can be enhanced by activation of the fluoride ion. This is achieved by adding alkali salts and cryptands such as the aminopolyether Kryptofix® 5 (K222) which complexes the cation producing the “naked” reactive form of fluoride. The most common reaction for aliphatic compounds is displacement of a good leaving group such as a tosylate or a triflatevia an SN2 mechanism. This was the approach used for the preparation of 7, a new PET imaging agent for the translocator protein (Scheme 3).20 The tosylate 6 was reacted with [18F]potassium fluoride in the presence of Kryptofix® 5 at 120 °C. Purification by HPLC gave radiolabelled ligand 7 with over 99% radiochemical purity.
![Radiosynthesis of PET imaging agent 7 using [18F]potassium fluoride.](/image/article/2011/CS/b922628c/b922628c-s3.gif) | ||
| Scheme 3 Radiosynthesis of PET imaging agent 7 using [18F]potassium fluoride. | ||
Nucleophilic aromatic substitution reactions using [18F]fluoride ion can be done but requires highly activated aromatic rings with electron-withdrawing substituents in ortho or para positions.21 Activating groups are not required for electron-poor heterocyclic aromatic systems such as the pyridine rings commonly found in a number of neurological imaging tracers.22 A [6-pyridinyl-18F]-labelled derivative of WAY-100635, a potential PET imaging agent for brain 5-HT1A receptors, was prepared by nucleophilic aromatic substitution of 6-nitropyridinyl analogue 8 (Scheme 4).23 Reaction of 8 with [18F]potassium fluoride and Kryptofix® 5 was found to proceed more rapidly under microwave heating rather than conventional heating and this generated 9 in an excellent chemical yield of 93% and with a decay corrected radiochemical yield of 15–25%.
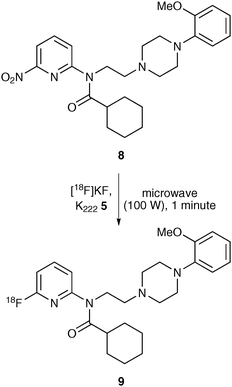 | ||
| Scheme 4 Microwave synthesis of 9 using nucleophilic aromatic substitution. | ||
As highlighted above, nucleophilic aromatic substitution reactions using [18F]fluoride ion are restricted to electron-deficient arenes. More recently, new methodology has been developed involving the use of diaryliodonium salts which allows the fluorination of both electron-deficient and electron-rich arenes.13,17 The diaryliodonium salts are easily prepared by reaction of Koser's reagent, hydroxy(tosyloxy)iodobenzene, with either aryltrialkylsilanes, aryl boronic acids or organostannanes.17,24 These are then reacted with [18F]fluoride ion under standard conditions (Scheme 5).25 An interesting feature of this process is that the regioselectivity of the fluorination can be directed using the “ortho effect” where an ortho substituent directs attack to that ring. In the absence of an ortho substituent, the [18F]fluoride ion is incorporated into the least electron-rich ring. Thus, the use of diaryliodonium salts where one of the aryl groups is an electron-rich p-methoxyphenyl or 2-thienyl ring results in highly selective fluorination of the other ring (Scheme 5).26 As shown by the examples in Scheme 5, one of the benefits of using this approach is that it produces radiolabelled compounds with higher radiochemical yields compared to other more established approaches.25
![[18F]Fluorination using diaryliodonium salts.](/image/article/2011/CS/b922628c/b922628c-s5.gif) | ||
| Scheme 5 [18F]Fluorination using diaryliodonium salts. | ||
2.3 The generation of molecular probes for imaging
The general strategy for designing and synthesising imaging probes is very similar to generating drug-like molecules in that the probes are required to have high affinity and high selectivity for the target receptor or organ. Production of compounds that satisfy these requirements means that only small amounts of the radioactive probe are required, generating precise and well-defined images of the receptor. In practice, compounds from the scientific literature which are known to bind to the receptor with high affinity and have good pharmacokinetics are initially chosen as leads. Synthetic routes for the preparation of a small library of analogues are then designed which incorporate the isotopic label at a late stage of the synthesis. The “cold” (non-radioactive) compounds are initially tested for affinity with the receptor. The most active compound with the highest affinity and the best log p value (according to Lipinski's rules27) that can easily cross cell membranes is then re-synthesised incorporating the radionuclide using the most efficient radiochemical techniques. Depending on the quality of the images generated, further analogues may be required which have greater selectivity for the target receptor. This is the general strategy used for preparing the majority of molecular imaging agents as shown by the key examples highlighted in the following sections.2.4 PET imaging of cancer
The most widely used tracer for imaging cancer is the glucose analogue 2-[18F]fluoro-2-deoxyglucose (FDG) 12.28 [18F]FDG enters cellsviaplasma membrane glucose transporters. During the first step of the glycolytic pathway it undergoes phosphorylation and is irreversibly trapped intracellularly in most tissues. As many tumour cells have upregulated glucose transport and glycolysis, [18F]FDG accumulates in tumour cells rather than in normal tissues. [18F]FDG has been used as a biomarker of many human malignancies including lung cancer, breast cancer, lymphoma, colorectal cancer (Fig. 2) and melanoma.![[18F]FDG image of a patient with colorectal cancer (left, CT image; middle, fused PET–CT image; right, PET image).](/image/article/2011/CS/b922628c/b922628c-f2.gif) | ||
| Fig. 2 [18F]FDG image of a patient with colorectal cancer (left, CT image; middle, fused PET–CT image; right, PET image). | ||
The synthesis of [18F]FDG using an electrophilic fluorination was first published in the 1970's,29 but it was not until much later when a nucleophilic fluorination method involving highly active [18F]fluoride ion was developed did it find more widespread application.30 The key intermediate is 1,3,4,6-tetra-O-acetyl-2-trifluoromethanesulfonyl-β-D-mannopyranose 11 which was prepared in two steps from D-mannose 10 (Scheme 6). Introduction of the 18F-label involved the SN2 reaction of 11 with [18F]fluoride ion in the presence of Kryptofix® 5. This was followed by rapid acid deprotection of the hydroxyl groups which gave [18F]FDG 12 in a high radiochemical yield.
![Synthesis of 2-[18F]fluoro-2-deoxy-d-glucose 12 (FDG).](/image/article/2011/CS/b922628c/b922628c-s6.gif) | ||
| Scheme 6 Synthesis of 2-[18F]fluoro-2-deoxy-D-glucose 12 (FDG). | ||
While [18F]FDG is very widely used as a clinical research tool and as a diagnostic agent in oncology and neurology it does have some limitations. These include high uptake in some normal tissues such as brain and bladder which restricts its use for imaging gliomas and prostate cancers. Furthermore, inflammatory conditions can lead to increased local uptake of [18F]FDG which leads to false positive results. These limitations have prompted research into more specific biomarkers of cancer. Recently, Vasdev and co-workers reported the preparation of [18F]-1-deoxy-1-fluoro-scyllo-inositol 15 as a potential PET imaging agent for both Alzheimer's disease and breast cancer.31 A protected triflate derivative of myo-inositol 13 was reacted with potassium fluoride in the presence of Kryptofix® 5 which gave the [18F]fluoride analogue 14 with high specific activity (Scheme 7). Acid deprotection of the diacetals and base hydrolysis of the benzoyl group gave [18F]-1-deoxy-1-fluoro-scyllo-inositol 15 with an uncorrected radiochemical yield of 16%. Small animal imaging studies showed that compound 15 had very low brain penetration and thus, unlikely to be used for studying the central nervous system. However, preliminary investigation of 15 for the detection of human breast cancer xenografts in athymic mice showed that tumour uptake of 15 was similar to that for [18F]FDG 12. With the relatively low specificity of FDG PET, [18F]-labelled inositols such as 15 may prove more effective for oncology imaging studies.
![Synthesis of [18F]-1-deoxy-1-fluoro-scyllo-inositol 15.](/image/article/2011/CS/b922628c/b922628c-s7.gif) | ||
| Scheme 7 Synthesis of [18F]-1-deoxy-1-fluoro-scyllo-inositol 15. | ||
α-Amino acids are important nutrients for the proliferation of cells through protein synthesis. It is well known that the transport of amino acids and subsequent protein synthesis is upregulated in many tumour types and thus, labelled α-amino acids such as L-[11C]methionine, 2-[18F]fluoro-L-tyrosine, L-[18F]fluoro-α-methyl tyrosine and O-(2-[18F]fluoroethyl)-L-tyrosine have been selectively incorporated into tumours for PET imaging.28,32 For example, [18F]fluoro-L-dopa 4 in animal models has been shown via a transport mechanism to accumulate in melanomas and breast tumours and is therefore, potentially complementary to [18F]FDG.28 Due to the importance of [18F]fluoro-L-dopa 4 for neuroimaging (see Scheme 2) and as an oncological radiotracer, a number of electrophilic and nucleophilic syntheses have been reported. In 2004, a highly efficient and rapid approach for the enantioselective synthesis of 4 was reported using a phase transfer alkylation (Scheme 8).33 The [18F]fluoroveratraldehyde derivative 17 was synthesized from 16 using a nucleophilic fluorination in the presence of Kryptofix® 5. Sodium borohydride reduction of 17 followed by bromination of the resulting alcohol under standard conditions gave [18F]fluorobenzyl bromide 18. To avoid time-consuming operations such as evaporation, extraction, drying and heating, these two reactions were performed using a solid support. The key step was then achieved involving alkylation of glycine derivative 20 with 18 in the presence of the chiral phase transfer catalyst 19. This gave alkylated product 21 in >90% yield and with 96% enantiomeric excess. Finally deprotection of 21 followed by HPLC purification gave [18F]fluoro-L-dopa 4 in a synthesis time of 100 minutes and in a decay corrected radiochemical yield of 25–30%. While this synthetic approach does generate [18F]fluoro-L-dopa 4 in slightly lower ee compared to other methods, it is well adapted for automation and routine production.
![Radiosynthesis of [18F]fluoro-l-dopa 4.](/image/article/2011/CS/b922628c/b922628c-s8.gif) | ||
| Scheme 8 Radiosynthesis of [18F]fluoro-L-dopa 4. | ||
The majority of new anti-cancer agents target molecular pathways to inhibit cell proliferation rather than active cell death. As a consequence, the development of new PET imaging agents for cancer has focused on the production of biomarkers for cell proliferation and apoptosis. A widely used radiopharmaceutical for cell proliferation is the nucleoside [18F]fluorothymidine 23 (FLT).28 FLT enters cells by diffusion and by a carrier-mediated mechanism and is then metabolised by thymidine kinase 1 (TK1). TK1 is overexpressed in tumour cells (up to 15 fold higher than normal cells) and thus is an effective target for imaging cancer cell proliferation. One of the most efficient syntheses of [18F]FLT involved the nucleophilic displacement of a nosyl leaving group using [18F]tetrabutylammonium fluoride in the protic solvent, tert-butanol (Scheme 9).34 Normally, SN2 reactions are carried out in aprotic solvents which enhance the nucleophilicity and thus, the rate of the reaction. However, it has been found that the use of tertiary alcohols as solvents for nucleophilic fluorination dramatically increased the rate of the reaction. This process was particularly useful for the synthesis of [18F]imaging agents including [18F]FLT 23 which was formed after deprotection in a radiochemical yield of 65% and in a total synthesis time of 70 minutes (Scheme 9). As well as producing PET imaging agents rapidly and in high radiochemical yields, this approach for nucleophilic fluorination was found to be amenable to automated synthesis.
![Radiosynthesis of [18F]fluorothymidine 23 using protic solvents.](/image/article/2011/CS/b922628c/b922628c-s9.gif) | ||
| Scheme 9 Radiosynthesis of [18F]fluorothymidine 23 using protic solvents. | ||
[18F]FLT 23 is strictly not a true proliferation marker as it is not incorporated into DNA. A compound which is involved in DNA synthesis and is preferentially accumulated by rapidly proliferating tumour cells is 1-(2′-deoxy-2′-fluoro-β-D-arabinofuranosyl)thymine (FMAU).28 Studies have shown that [18F]FMAU 29 is a direct marker and selective tracer for the PET imaging of tumours in the brain, prostate, thorax and bone.35 As with many other syntheses of [18F]imaging agents, [18F]FMAU is synthesised using a nucleophilic fluorination reaction in the presence of Kryptofix® 5 (Scheme 10).36 Thus, triflate 24 was reacted with [18F]fluoride anion and the resulting substitution product was then treated with hydrobromic acid to give bromide 25. This was then condensed with 2,4-bis-O-(trimethylsilyl)thymine 26 which gave a mixture of the desired β-isomer 27 and α-isomer 28 in a ratio of approximately 6∶1. Deprotection of the isomeric mixture was then done using sodium methoxide and [18F]FMAU 29 was isolated using HPLC. Using this approach, the overall radiochemical yield for 29 was 42% and was prepared in a synthesis time of approximately 3 hours.
![Synthesis of [18F]FMAU 29.](/image/article/2011/CS/b922628c/b922628c-s10.gif) | ||
| Scheme 10 Synthesis of [18F]FMAU 29. | ||
2.5 PET imaging of neurodegenerative diseases
One area where PET finds many applications is in the imaging of neurodegenerative and neuropsychiatric disorders.7,13,17,19 A wide range of molecular imaging agents labelled with carbon-11 or fluorine-18 have been developed for specific receptors including the noradrenaline, serotonin and dopamine transporters, the metabotropic glutamate receptor and the translocator protein 18 kDA. The widespread application of PET for neuroimaging is mainly due to the fact that radiotracer techniques are inherently suited to receptor studies, generating good quality images by the administration of sub-pharmacological doses of the molecular probe. Below are some examples of how radiolabelled molecular probes have been used to image the translocator protein, β-amyloid plaque in Alzheimer's disease and dopamine D2 receptors.The translocator protein 18 kDA (TSPO, formerly known as the peripheral benzodiazepine receptor) is expressed within the brain by reactive glial cells and is implicated in a number of nervous system disorders such as cerebral ischaemia, epilepsy, nerve injury and neurodegenerative diseases.37 Expression of brain TSPO is dramatically increased during chronic neurodegenerative diseases and following acute brain injury and thus, TSPO is an attractive target for imaging human neurodegenerative diseases such as Alzheimer's disease and in stroke-induced brain injury.38 The most widely used ligand for imaging TSPO is the isoquinoline carboxamide PK11195 37 which binds with nanomolar affinity. A highly efficient synthesis of PK11195 37 was recently reported involving only six steps, utilising palladium(0) chemistry to effect the key transformations (Scheme 11).39 A Heck-type reaction was used to couple methyl 2-iodobenzoate 31 and amidoacrylate 32 which gave oxoisoquinoline 33 in 65% yield. Formation of bromide 34 followed by a Suzuki reaction allowed introduction of the pendent chlorophenyl moiety. Hydrolysis of the ester and introduction of the amide side chain under standard conditions completed the six-step synthesis of PK11195 37.
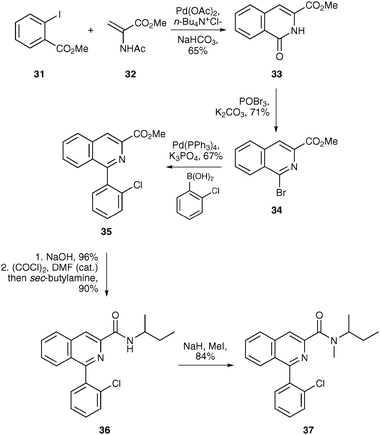 | ||
| Scheme 11 Synthesis of PK11195 37. | ||
Various radiolabelled analogues of PK11195 have been used to image neurodegenerative pathologies such as Alzheimer's disease, Parkinson's disease, Huntington's disease and multiple sclerosis.37 These studies have led to a greater understanding of each of these diseases and allowed better approaches for treatment. [11C]PK11195, which is commonly used for PET imaging of TSPO, was first synthesised in 1984.40 This straightforward approach involved the methylation of desmethyl analogue 36 using [11C]methyl iodide under basic conditions (Scheme 12). This generated [11C]PK11195 in 45 minutes with a specific activity of 3.54 Ci μmol−1.
![Radiosynthesis of [11C]PK11195 37.](/image/article/2011/CS/b922628c/b922628c-s12.gif) | ||
| Scheme 12 Radiosynthesis of [11C]PK11195 37. | ||
As well as using methylation reactions to introduce [11C]-labels for PET imaging, carbonylation and carboxylation are also widely utilised.13 For example, Långström and co-workers used a palladium(0)-mediated carbonylation reaction for the preparation of [11C-carbonyl]PK11195 (Scheme 13).41Triflate 38, prepared in six steps from 2-chlorobenzophenone, was reacted in a micro-autoclave with tetrakis(triphenylphosphine)palladium(0) and N-methyl-sec-butylamine in the presence of [11C]carbon monoxide. This gave [11C-carbonyl]PK11195 37 in a radiochemical yield of 55% after a 10 minute reaction time.
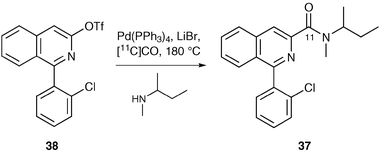 | ||
| Scheme 13 Use of palladium(0)-mediated carbonylation chemistry. | ||
While PK11195 is widely used to study TSPO it does have some limitations including a high level of nonspecific binding, low brain uptake and low sensitivity. For these reasons, a number of new PET radioligands for TSPO have been developed. Phenoxyarylacetamides such as PBR28 45 have been developed as a new class of ligand for TSPO and shown to be promising candidates for human PET studies.42 These compounds have been used to study traumatic brain injury with further human trials ongoing.38 As well as having higher affinity for TSPO than PK11195, phenoxyarylacetamides also have a higher uptake ratio into the brain (up to six times higher). A fully automated synthesis of [11C]PBR28 45 has recently been developed (Scheme 14).42 Nucleophilic aromatic substitution of 4-chloro-3-nitropyridine 39 with phenol gave 3-nitro-4-phenoxypyridine 40. Reduction of the nitro group and condensation of the resulting amine with o-salicylaldehyde 42 under reducing conditions gave 43 in high yield. Acetylation of the amine and phenolic hydroxyl groups followed by base hydrolysis gave desmethyl-PBR28 44. Radiosynthesis was then carried out using methyl triflate under basic conditions which gave [11C]PBR28 45 in a decay corrected radiochemical yield of 70–80%.
![Fully automated synthesis of [11C]PBR28 45.](/image/article/2011/CS/b922628c/b922628c-s14.gif) | ||
| Scheme 14 Fully automated synthesis of [11C]PBR28 45. | ||
2-Phenylimidazo[1,2-a]pyridineacetamides have been shown to be potent and selective ligands for TSPO.43 Among this class of compounds, CLINME 47 has been radiolabelled and used for PET imaging of TSPO. Dollé and co-workers reported the first radiosynthesis of [11C]CLINME by the methylation of precursor 46 using methyl iodide and potassium hydroxide (Scheme 15).44 Careful control of the reaction temperature allowed the preparation of [11C]CLINME in 27 minutes with a decay corrected radiochemical yield of 16–23%.
![Radiosynthesis of [11C]CLINME 47.](/image/article/2011/CS/b922628c/b922628c-s15.gif) | ||
| Scheme 15 Radiosynthesis of [11C]CLINME 47. | ||
Alzheimer's disease (AD) accounts for 60–70% of all dementia cases with a prevalence rate of approximately 30% by 85 years.45 AD is characterised by the presence of two abnormal aggregated proteins in the brain, amyloid plaques which are predominantly composed of insoluble amyloid-β peptides and neurofibrillary tangles. As the insoluble amyloid-β peptides are more prevalent, research has focused on the development of an imaging agent for these peptides which could help accelerate drug development and assist in delivering therapies which delay the onset of AD. Thioflavin-T derivatives such as Pittsburgh Compound B (PiB) 49 have been shown to bind selectively to insoluble amyloid-β deposits but not to soluble amyloid-β forms. As such, a number of PET imaging agents based on PiB have been reported. For example, Solbach and co-workers reported an efficient synthesis of [11C]PiB 49 by alkylation with methyl triflate (Scheme 16).46 This gave [11C]PiB 49 in a radiochemical yield of 58% after a synthesis time of 40 minutes. [11C]PiB has been evaluated in several human PET studies and has shown great potential for imaging of AD.
![Radiosynthesis of [11C]PiB 49.](/image/article/2011/CS/b922628c/b922628c-s16.gif) | ||
| Scheme 16 Radiosynthesis of [11C]PiB 49. | ||
The dopaminergic system is involved in numerous brain disorders such as schizophrenia and Parkinson's disease.1 There are five dopamine receptors classified in two different families. The D1 family consists of the D1 and D5 subtypes while the D2 family includes the D2, D3 and D4 subtypes. Due to the relative abundance of D1 and D2 receptors in the striatum and their involvement in a number of diseases, there have been substantial efforts directed towards the development of a molecular imaging agent for these receptors. One of the most commonly used PET imaging agents for investigation of striatal D2/D3 receptors in humans is the benzamide derivative, [11C]raclopride 54.47 [11C]Raclopride has been used to image patients with Parkinson's disease, Huntington's disease and schizophrenia as well as determining receptor occupancy of antipsychotic drugs. The precursor for the radiosynthesis of [11C]raclopride 54 is prepared in three steps from 3,5-dichloro-2,6-dimethoxybenzoic acid 50 (Scheme 17).48 This is converted to the corresponding acid chloride and coupled with (S)-(−)-2-aminomethyl-1-ethylpyrrolidine 51. The hydrochloride salt is then formed followed by stepwise removal of the methyl ether groups. The resulting hydrobromide salt is neutralized before methylation under basic conditions with [11C]methyl triflate. This gave [11C]raclopride 54 in a decay corrected radiochemical yield of 55–65% and in a total radiosynthesis time of 35 minutes.
![Synthesis of [11C]raclopride 54.](/image/article/2011/CS/b922628c/b922628c-s17.gif) | ||
| Scheme 17 Synthesis of [11C]raclopride 54. | ||
3. Single photon emission computed tomography
SPECT imaging uses radionuclides that directly emit γ-rays, such as iodine-123 (123I) and technetium-99m (99mTc), and are generally of a lower energy than those used for PET (Tables 2 and 3). In SPECT imaging, directional information is obtained by placing a collimator in front of the gamma camera. The collimator defines the angle of incidence of the isotropic γ-rays emitted. The use of a collimator in SPECT results in low detection efficiency compared to PET, which only requires electronic collimation in order to detect the two coincident γ-rays. PET systems are therefore generally more sensitive than SPECT systems (Table 1) which in turn translates into a higher resolution for PET compared with SPECT. However, the use of longer lived radionuclide and the relatively lower costs of gamma cameras make SPECT imaging much more widely available for clinical use than PET scanners.| Nuclide | Half-life/h | Type of emission | Principal photon emission energies/MeV |
|---|---|---|---|
| 123I | 13.2 | Electron capture | 0.16 |
| 99mTc | 6 | Isomeric transition | 0.14 |
| 111In | 67.9 | Electron capture | 0.17/0.25 |
| 67Ga | 78.3 | Electron capture | 0.09/0.19/0.30 |
| 201Tl | 73.1 | Electron capture | 0.17 |
3.1 SPECT radionuclides
Generally, SPECT isotopes have a considerably longer half-life than PET isotopes (Table 3) which makes SPECT tracers more available for imaging and longer radiosynthesis times make them more practical. In contrast, the shorter half-life of the PET isotopes limits the availability of PET imaging, requiring an on-site cyclotron, and makes radiolabelling protocols more challenging.The biological half-life of a tracer should ideally be shorter than the half-life of the radionuclide being utilised. This allows the non-specific signal to be biologically washed out within the imaging timeframe, leaving a cleaner signal of interest. Hence, the longer half-life of SPECT radionuclides also has the advantage of enabling SPECT imaging studies to be conducted over longer time periods, whereas imaging studies using the shorter half-life PET radionuclides may require more complicated modelling.
Imaging with 99mTc is more widely accessible than imaging with other cyclotron-produced radionuclides primarily because 99mTc can be produced from the decay of its longer-lived parent nuclide, 99Mo (half-life = 67 h), using commercially available generator systems. Technetium-99m-labelled imaging agents are therefore the mainstay of nuclear medicine imaging worldwide, where diagnostic imaging agents can be produced using commercially available kits. The incorporation of 99mTc into a molecule requires a chelating agent to produce a metal complex, unlike 123I and 18F that can directly substitute a hydrogen atom of a molecule. 99mTc chemistry therefore requires the development of complex chemistry to allow incorporation of 99mTc into a molecule without affecting binding to the target site. Because of this, the application of 99mTc chemistry for imaging specific target sites can be restricted. For example, the blood–brain barrier may preclude target uptake of complex 99mTc-labelled molecules, thereby limiting the usefulness in neurological applications.
As with 18F, 123I can form a covalent bond with carbon and therefore it is much easier to incorporate 123I into a small molecule, compared to 99mTc. The production of 123I, however, requires a cyclotron, but the relatively longer half-life compared to the positron emitting isotopes enables 123I to be distributed over larger distances. One disadvantage however is that iodine is larger than fluorine (van der Waal's radius of iodine is 2.15 Å) and thus, its placement into a molecule induces a greater steric perturbation. One advantage of iodine is the availability of other isotopes such as 125I, a longer lived low energy δ-emitter that can be used for pre-clinical development, and 131I, a δ- and β-emitter that can be used in radiotherapy. Thus, the same agent can be labelled with different iodine isotopes, increasing its flexibility. For example, 131I-labelled m-iodobenzylguanidine ([131I]-mIBG) is used for the therapy of neuroendocrine tumours, where patients have had a positive 123I-mIBG diagnostic scan.49
3.2 SPECT radiochemistry
As with PET chemistry, the SPECT radionuclide needs to be incorporated at the latest possible stage. Radiolabelling reactions need to be rapid and efficient and the radiopharmaceutical needs to be obtained with a high radiochemical yield and purity.The 99Mo/99mTc generator provides 99mTc in the form of [99mTc]-pertechnetate in its highest oxidation state of +7. There are many technetium imaging agents50 that can be prepared by combining [99mTc]-pertechnetate with commercially available kits, such as [99mTc]-MAG355, [99mTc]-tetrofosmin 56 and [99mTc]-HMPAO 57 (Fig. 3). Radiolabelling of these kits is carried out by reducing [99mTc]-pertechnetate to a lower oxidation state, usually +4, allowing the technetium to complex with a chelating agent. Commercially available kits for technetium labelling commonly use stannous ions (Sn2+) as the reducing agent resulting in high labelling yields (normally greater than 95%), with no purification step necessary.
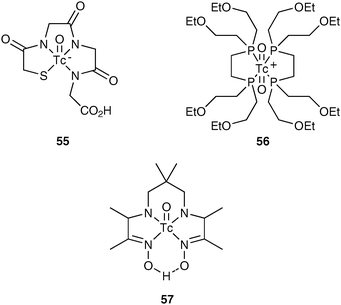 | ||
| Fig. 3 Examples of 99mTc radiopharmaceuticals. | ||
More recently, the use of bifunctional chelating agents has allowed the development of a number of novel target-specific technetium imaging agents. A bifunctional chelating agent not only binds 99mTc securely without dislocation in vivo, but also links to a biological active molecule without affecting its integrity. The hydrazine–nicotinamide HYNIC 58 ligand is an example of a bifunctional chelating agent that reacts with nucleophilic groups in a biomolecule, such as the ε-amine groups of lysine residues, to form a HYNIC–biomolecule complex that can be radiolabelled with 99mTc.51 This versatile linker has been used to label numerous proteins and peptides (for example, HYNIC–annexin A5 59, Fig. 4).52–54
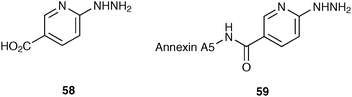 | ||
| Fig. 4 HYNIC 58 and HYNIC–annexin A5 59. | ||
All radioiodine isotopes are produced in the form of radioiodide, commonly sodium iodide in sodium hydroxide (0.01–0.1 M). Hence, there are a variety of methods available for radiolabelling compounds with radioiodide. The nature of the compound (precursor) to be radioiodinated helps determine the most appropriate radiolabelling methodology. There are a number of factors that need to be taken into account when choosing radioiodination methodology:
1. precursor stability and reactivity,
2. precursor toxicity,
3. ease of precursor synthesis,
4. purification of final product from precursor.
Direct radioiodination (replacement of hydrogen atom by radioiodine atom) is generally limited to arenes that are activated for electrophilic substitution and requires an oxidising agent to obtain positively charged iodine (I+). Common oxidising agents include chloramine-T, Iodogen™ and peracetic acid. This direct labelling methodology can give rise to good radiolabelling yields, however, often side-reactions can be problematic. The 123I-labelled amino acid iodo-α-methyltyrosine 61 has been obtained via direct radioiodination (Scheme 18).55
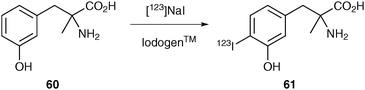 | ||
| Scheme 18 The direct radioiodination of α-methyltyrosine 60. | ||
Other radioiodination reactions require introduction of a leaving group into the precursor that can easily be replaced with radioiodine. These reactions can be either electrophilic or nucleophilic exchange reactions. Electrophilic exchange reactions are mainly based on iodo-demetallation reactions and require in situoxidation as with direct radioiodination. The organometallic precursors required for this radiolabelling methodology can often be difficult to synthesise. However, these precursors have the advantage of allowing regioselective radioiodination under mild conditions, where the carbon–metal bond is much more activated for electrophilic attack than the carbon–hydrogen bond. Common metal precursors include organosilyl and organostannyl compounds. Organosilyl precursors offer the advantage of being more stable, however are less reactive than stannyl precursors. The disadvantage of these organometals is that they are often highly toxic and removal of metal residues after the radiolabelling reaction can be problematic, raising toxicology issues.
No-carrier-added (n.c.a.) [131I]-mIBG 63 has previously been synthesised viaiodo-desilylation of the corresponding trimethylsilyl precursor. This methodology obtains radiolabelled mIBG with a high radiochemical yield.56 However, using this method, large scale production is limited due to the requirement of HPLC purification to obtain the final purified product. More recently the radiosynthesis of mIBG has been developed using a resin-supported tin precursor (Scheme 19).57 This technology allows the purification of n.c.a. [131I]-mIBG using simple solid-phase extraction.
![Radiosynthesis of n.c.a. [131I]-mIBG 63 using a resin supported tin precursor.](/image/article/2011/CS/b922628c/b922628c-s19.gif) | ||
| Scheme 19 Radiosynthesis of n.c.a. [131I]-mIBG 63 using a resin supported tin precursor. | ||
The most common nucleophilic exchange methodology utilised for radioiodination is the halogen exchange reaction. The simplest halogen exchange reaction is via isotropic exchange with a stable iodine atom. The utility of this methodology, however, is limited by the presence of the stable, iodine-127 labelled compound that cannot be separated from the radiolabelled compounds, resulting in products with low specific activity. Bromine and chlorine (to a lesser extent) precursors can also be used in nucleophilic exchange reactions under solid state conditions, however, these reactions often require harsh conditions for radiolabelling to take place.
Both electrophilic and nucleophilic exchange radiolabelling methodologies have been compared for the radioiodination of the TSPO ligand, PK11195 (Scheme 20).58 This study found that electrophilic iododestannylation of the trimethylstannyl precursor 66 produced [123I]-PK11195 67 with a higher radiochemical yield and a higher specific activity, than achievable with halogen exchange.
![Radiosynthesis of [123I]-PK11195 67 from I-PK11195 64, Br-PK11195 65 and Me3Sn-PK11195 66 precursors.](/image/article/2011/CS/b922628c/b922628c-s20.gif) | ||
| Scheme 20 Radiosynthesis of [123I]-PK11195 67 from I-PK11195 64, Br-PK11195 65 and Me3Sn-PK11195 66 precursors. | ||
3.3 SPECT imaging of neurodegenerative diseases and head injury
SPECT imaging, like PET imaging, has many applications across the field of medicine. In this review we have focused on applications in the field of neurology. One of the most successful utilities of SPECT in neurology is cerebral blood flow imaging, using 99mTc-labelled radiopharmaceuticals. Numerous radioiodinated SPECT imaging agents have also been developed targeting various neurotransmitter systems. Efforts have been primarily focused on developing imaging agents for the monoamine system, but there are also imaging agents available for other systems, such as cholinergic and glutamatergic targets. Below are some examples of how SPECT imaging agents have been used to image cerebral blood flow, serotonin and noradrenaline transporters and the α4β2 nicotinic acetylcholine receptor (nAChR).The 99mTc-labeled radiopharmaceuticals, [99mTc]-HMPAO 57 (or Ceretec™, see Fig. 3) and [99mTc]-ethylcysteinate dimer (99mTc-ECD or Neurolite™), have been developed commercially for imaging cerebral blood flow using SPECT. These agents are small lipophilic compounds that cross the intact blood–brain barrier by simple diffusion. Once in the brain, these agents have a fixed distribution that reflects regional brain perfusion for sufficient time to permit image acquisition. Cerebral blood flow imaging with SPECT has been shown to aid the diagnosis and treatment of patients with various different diseases, such as cerebrovascular diseases, dementias, epilepsy and head injury.59
The dopamine transporter imaging agent [123I]-FP-CIT 68 (or DATScan™, GE Healthcare), a cocaine derivative, is the only licensed SPECT or PET radiopharmaceutical for neuroreceptor imaging (Fig. 5).
![[123I]-FP-CIT
68 and [123I]-β-CIT 69.](/image/article/2011/CS/b922628c/b922628c-f5.gif) | ||
| Fig. 5 [123I]-FP-CIT 68 and [123I]-β-CIT 69. | ||
Dopamine transporter imaging is now used routinely in the clinical differential diagnosis of Parkinson's disease60 (Fig. 6) and dementia with Lewy bodies.61
![[123I]-FP-CIT
68
SPECT images in a normal (a) and Parkinson's disease patient (b), showing a reduction of [123I]-FP-CIT 68 binding in the patient with Parkinson's disease.](/image/article/2011/CS/b922628c/b922628c-f6.gif) | ||
| Fig. 6 [123I]-FP-CIT 68 SPECT images in a normal (a) and Parkinson's disease patient (b), showing a reduction of [123I]-FP-CIT 68 binding in the patient with Parkinson's disease. | ||
The structurally similar SPECT imaging agent, [123I]-β-CIT 69 binds to both the dopamine and serotonin transporters (Fig. 5). Due to the different uptake and washout kinetics of [123I]-β-CIT at the dopamine and serotonin transports, both sites can be investigated using this single imaging agent.62 This dual imaging ability of [123I]-β-CIT has enabled imaging studies investigating antidepressant drug effects on both the dopamine and serotonin transporter systems in the same patients with only a single radioactive exposure.63 The tributylstannyl precursor 71 for the radiosynthesis of [123I]-β-CIT 69 has been prepared from cold β-CIT 70 in a palladium catalysed reaction with bis(tributyl)distannane (Scheme 21).64 The radiosynthesis of [123I]-β-CIT 69 was then achieved via electrophilic iododestannylation, using peracetic acid as the oxidising agent. This produced [123I]-β-CIT 69 in a radiochemical yield of 65%.
![Radiosynthesis of [123I]-β-CIT 69.](/image/article/2011/CS/b922628c/b922628c-s21.gif) | ||
| Scheme 21 Radiosynthesis of [123I]-β-CIT 69. | ||
Noradrenergic synaptic dysfunction has been associated with various psychiatric and neurodegenerative disorders, including depression, Alzheimer's disease and Parkinson's disease. Thus, the noradrenaline transporter (NAT) is a major target for drug development. However, in contrast to the serotonin and dopamine transporters, imaging of the NAT has been limited due to the lack of a suitable imaging agent. Recently, several groups have developed radiolabelled derivatives of the noradrenaline selective re-uptake inhibitor, reboxetine, for use in both PET65,66 and SPECT imaging.67,68 A SPECT tracer known as [123I]-INER or [123I]-IPBM 75 has shown good characteristics for in vivo imaging in non-human primates, however further investigation is required to assess its suitability for imaging humans.67,68 Radiolabelled [123I]-INER 75 has been prepared viaiodo-destannylation of the corresponding tin precursor 73 (Scheme 22).67 The tin precursor 73 was synthesized viapalladium catalysed stannylation of Boc-protected INER 72. Once radiolabelled, the Boc-protecting group was removed using trifluoroacetic acid and preparative HPLC was used to obtain purified [123I]-INER 75.
![Radiosynthesis of [123I]-INER 75.](/image/article/2011/CS/b922628c/b922628c-s22.gif) | ||
| Scheme 22 Radiosynthesis of [123I]-INER 75. | ||
[123I]-INER 75 is the (S,S)-enantiomer of iodinated reboxetine. Recent studies have synthesised various (S,S)-, (R,R)-, (R,S)- and (S,R)-stereoisomers of iodinated reboxetine, in order to gain insight into the structure–activity relationship with NAT.69–71 Results found that the (R,S)-enantiomer of INER has a high affinity for the NAT (Ki of 8.4 nM), similar to the reported affinity of the (S,S)-enantiomer 75 (Ki of 0.84 nM67 and 4.22 nM68). Further investigation is required to assess the in vivo imaging characteristics of the radiolabelled (R,S)-enantiomer of INER.
The nicotinic acetylcholine system plays a pivotal role in a number of functional processes including attention, learning and memory. In addition, adaptations of nAChRs in response to repeated and protracted nicotine exposure are likely to contribute to the addiction related to cigarette smoking. The initial search for a α4β2 nAChR imaging agent involved the synthesis of numerous analogues of epibatidine 76 (Fig. 7), an azabicycloheptane alkaloid, which was isolated from the skin of the Ecuadorian poison frog (Epipedobates tricolor).72 These epibatidine analogues all suffered from high toxicity, even at tracer dose levels, preventing their use in humans. More recently, a series of halogenated 3-pyridyl ethers (including 5-I-A85380 77, 2-F-A85380 78 and 6-F-A85380 79, Fig. 7) were radiolabelled and investigated as both PET and SPECT imaging agents for the α4β2 nAChR.73
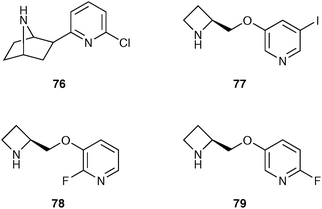 | ||
| Fig. 7 Structure of compounds that bind to the α4β2 nAChR. | ||
These 3-pyridyl ethers have lower toxicity while still maintaining high affinity for the α4β2 nAChR and have progressed to human imaging studies. The radiolabelled iodo analogue, 5-[123I]-A85380 82, has been used in numerous studies investigating the α4β2 nAChR in dementias such as Alzheimer's disease,74 dementia with Lewy bodies75 and also in studies investigating smoking abstinence.76
5-[123I]-A85380 82 has been radiolabelled via electrophilic iododestannylation of the corresponding Boc-protected trimethylstannyl precursor 80, using chloramine-T as an oxidising agent (Scheme 23).77 The Boc-protecting group was removed using acid mediated conditions. 5-[123I]-A85380 82 was obtained with a high radiochemical yield of 61% and a specific activity of 149 Ci μmol−1.
![Radiosynthesis of 5-[123I]-A85380 82.](/image/article/2011/CS/b922628c/b922628c-s23.gif) | ||
| Scheme 23 Radiosynthesis of 5-[123I]-A85380 82. | ||
4. Conclusions
Molecular tracers in combination with the radionuclide techniques of PET and SPECT are now widely established and used routinely for the non-invasive in vivo visualisation of disease. For example, [18F]FDG is used for the PET imaging of various cancers, while [123I]-FP-CIT is a SPECT biomarker for the dopamine transporter. The continued growth and increasing importance of molecular imaging for the diagnosis of disease and for application in the drug discovery process along with the greater availability of PET and SPECT facilities for clinical and research use has driven the need for new radiotracers. To meet this potential, the development of new methods and approaches for the design, synthesis and radiosynthesis of new molecular tracers are required. Studies are underway to develop new methods for the rapid and efficient radiosynthesis of precursors using techniques such as solid phase chemistry, microreactors and flow chemistry. Another key area under current investigation is the development of multi-functional molecular tracers with more than one labelling site that can be applied to several molecular imaging modalities. Such multi-functional molecular tracers could overcome the restrictions that are commonly incurred when using a single technique and allow greater insight and the generation of more detailed information regarding a particular disease.Acknowledgements
We would like to thank Dr Gerry Gillen at the PET imaging department, Gartnavel General Hospital, NHS Greater Glasgow and Clyde for the [18F]FDG image (Fig. 2) and Dr James Patterson at the Institute of Neurological Sciences, Southern General Hospital, NHS Greater Glasgow and Clyde for the [123I]-FP-CIT images (Fig. 6).References
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501 CrossRef CAS , and references therein.
- S. R. Meikle, P. Kench, M. Kassiou and R. B. Banati, Phys. Med. Biol., 2005, 50, R45 CrossRef CAS , and references therein.
- C. S. Levin, Eur. J. Nucl. Med. Mol. Imaging, 2005, 32, 325.
- L. Pauling, Nature of the Chemical Bond, Cornell University Press, New York, 1940 Search PubMed.
- W. Zhang, K. F. Koehler, B. Harris, P. Skolnick and J. M. Cook, J. Med. Chem., 1994, 37, 745 CrossRef CAS.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC.
- M. J. Adam and D. S. Wilbur, Chem. Soc. Rev., 2005, 34, 153 RSC , and references therein.
- N. M. Kelly, A. Sutherland and C. L. Willis, Nat. Prod. Rep., 1997, 14, 205 RSC.
- M. C. Lasne, C. Perrio, J. Rouden, L. Barré, D. Roeda, F. Dollé and C. Crouzel, Top. Curr. Chem., 2002, 222, 203.
- C. C. Lee, G. Sui, A. Elizarov, C. J. Shu, Y. S. Shin, A. N. Dooley, J. Huang, A. Daridon, P. Wyatt, D. Stout, H. C. Kolb, O. N. Witte, N. Satyamurthy, J. R. Heath, M. E. Phelps, S. R. Quake and H. R. Tseng, Science, 2005, 310, 1793 CrossRef CAS.
- J. M. Gillies, C. Prenant, G. N. Chimon, G. J. Smethurst, W. Perrie, I. Hamblett, B. Dekker and J. Zweit, Appl. Radiat. Isot., 2006, 64, 325 CrossRef CAS.
- P. J. H. Scott, Angew. Chem., Int. Ed., 2009, 48, 6001 CrossRef CAS.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998 CrossRef CAS , and references therein.
- L. Samuelsson and B. Långström, J. Labelled Compd. Radiopharm., 2003, 46, 263 CrossRef CAS.
- Y. Huang, R. Narendran, F. Bischoff, N. Guo, Z. Zhu, S.-A. Bae, A. S. Lesage and M. Laruelle, J. Med. Chem., 2005, 48, 5096 CrossRef CAS.
- For the IUPAC definition of specific activity and other terms associated with radioactivity, see: J. J. M. de Goeij and M. L. Bonardi, J. Radioanal. Nucl. Chem., 2005, 263, 13 Search PubMed.
- L. Cai, S. Lu and V. W. Pike, Eur. J. Org. Chem., 2008, 2853 CrossRef CAS , and references therein.
- M. Namavari, A. Bishop, N. Satyamurthy, G. Bida and J. R. Barrio, Appl. Radiat. Isot., 1992, 43, 989 CrossRef CAS.
- R. Bolton, J. Labelled Compd. Radiopharm., 2002, 45, 485 CAS.
- W. Yu, E. Wang, R. J. Voll, A. H. Miller and M. M. Goodman, Bioorg. Med. Chem., 2008, 16, 6145 CrossRef CAS.
- G. Angeli, M. Speranza, A. P. Wolf, C. Y. Shiue, J. S. Fowler and M. Watanabe, J. Labelled Compd. Radiopharm., 1984, 21, 1223.
- F. Dollé, Curr. Pharm. Des., 2005, 11, 3221 CrossRef CAS.
- M. Karramkam, F. Hinnen, M. Berrehouma, C. Hlavacek, F. Vaufrey, C. Halldin, J. A. McCarron, V. W. Pike and F. Dollé, Bioorg. Med. Chem., 2003, 11, 2769 CrossRef CAS.
- V. W. Pike, F. Butt, A. Shah and D. A. Widdowson, J. Chem. Soc., Perkin Trans. 1, 1999, 245 RSC.
- J. Ermert, C. Hocke, T. Ludwig, R. Gail and H. H. Coenen, J. Labelled Compd. Radiopharm., 2004, 47, 429 CrossRef CAS.
- T. Ross, J. Ermert and H. H. Coenen, J. Labelled Compd. Radiopharm., 2005, 48, S153.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3 CrossRef.
- O. Couturier, A. Luxen, J.-F. Chatal, J.-P. Vuillez, P. Rigo and R. Hustinx, Eur. J. Nucl. Med. Mol. Imaging, 2004, 31, 1182 CAS.
- T. Ido, C.-N. Wan, V. Casella, J. S. Fowler, A. P. Wolf, M. Reivich and D. E. Kuhl, J. Labelled Compd. Radiopharm., 1978, 14, 175 CrossRef CAS.
- K. Hamacher, H. H. Coenen and G. Stöcklin, J. Nucl. Med., 1986, 27, 235 CAS.
- N. Vasdev, J. Chio, E. M. van Oosten, M. Nitz, J. McLaurin, D. C. Vines, S. Houle, R. M. Reilly and A. A. Wilson, Chem. Commun., 2009, 5527 RSC.
- J. McConathy and M. M. Goodman, Cancer Metastasis Rev., 2008, 27, 555 CrossRef.
- C. Lemaire, S. Gillet, S. Guillouet, A. Plenevaux, J. Aerts and A. Luxen, Eur. J. Org. Chem., 2004, 2899 CrossRef CAS.
- D. W. Kim, D.-S. Ahn, Y.-H. Oh, S. Lee, H. S. Kil, S. J. Oh, S. J. Lee, J. S. Kim, J. S. Ryu, D. H. Moon and D. Y. Chi, J. Am. Chem. Soc., 2006, 128, 16394 CrossRef CAS.
- H. Sun, A. Sloan, T. J. Manger, U. Vaishampayan, O. Muzik, J. M. Collins, K. Douglas and A. F. Shields, Eur. J. Nucl. Med. Mol. Imaging, 2005, 32, 15 CrossRef CAS.
- T. J. Mangner, R. W. Klecker, L. Anderson and A. F. Shields, Nucl. Med. Biol., 2003, 30, 215 CrossRef CAS.
- A. M. Scarf, L. M. Ittner and M. Kassiou, J. Med. Chem., 2009, 52, 581 CrossRef CAS , and references therein.
- F. Chauveau, H. Boutin, N. Van Camp, F. Dollé and B. Tavitian, Eur. J. Nucl. Med. Mol. Imaging, 2008, 35, 2304 CrossRef , and references therein.
- L. Stevenson, S. L. Pimlott and A. Sutherland, Tetrahedron Lett., 2007, 48, 7137 CrossRef CAS.
- R. Camsonne, C. Crouzel, D. Comar, M. Mazière, C. Prenant, J. Sastre, M. A. Moulin and A. Syrota, J. Labelled Compd. Radiopharm., 1984, 21, 985 CrossRef CAS.
- O. Rahman, T. Kihlberg and B. Långström, J. Chem. Soc., Perkin Trans. 1, 2002, 2699 RSC.
- M. Wang, K. K. Yoder, M. Gao, B. H. Mock, X.-M. Xu, A. J. Saykin, G. D. Hutchins and Q.-H. Zheng, Bioorg. Med. Chem. Lett., 2009, 19, 5636 CrossRef CAS.
- G. Trapani, V. Laquintana, N. Denora, A. Trapani, A. Lopedota, A. Latrofa, M. Franco, M. Serra, M. G. Pisu, I. Floris, E. Sanna, G. Biggio and G. Liso, J. Med. Chem., 2005, 48, 292 CrossRef CAS.
- C. Thominiaux, F. Mattner, I. Greguric, H. Boutin, F. Chauveau, B. Kuhnast, M.-C. Grégoire, C. Loch, H. Valette, M. Bottlaender, P. H. Hantraye, B. Tavitian, A. Katsifis and F. Dollé, J. Labelled Compd. Radiopharm., 2007, 50, 229 CrossRef CAS.
- C. A. Mathis, B. J. Lopresti and W. E. Klunk, Nucl. Med. Biol., 2007, 34, 809 CrossRef CAS.
- C. Solbach, M. Uebele, G. Reisch and H.-J. Machulla, Appl. Radiat. Isot., 2005, 62, 591 CrossRef CAS , and references therein.
- L. Farde, H. Hall, E. Ehrin and G. Sedvall, Science, 1986, 231, 258 CrossRef CAS.
- O. Langer, K. Nagren, F. Dolle, C. Lundkvist, J. Sandell, C.-G. Swahn, F. Vaufrey, C. Crouzel, B. Maziere and C. Halldin, J. Labelled Compd. Radiopharm., 1999, 42, 1183 CrossRef CAS.
- J. B. Bomanji, W. Wong, M. N. Gaze, A. Cassoni, W. Waddington, J. Solano and P. J. Ell, Clin. Oncol., 2003, 15, 193 CrossRef CAS.
- S. Liu, Dalton Trans., 2007, 1183 RSC.
- D. A. Schwartz, M. J. Abrams, M. M. Hauser, F. E. Gaul, S. K. Larsen, D. Rauh and J. A. Zubieta, Bioconjugate Chem., 1991, 2, 333 CrossRef CAS.
- F. G. Blankenberg, P. Katsikis, L. Naumovski, K. Ohtsuki and H. W. Strauss, Radiology, 1997, 205, 1424 Search PubMed.
- Z. F. Su, G. Liu, S. Gupta, Z. Zhu, M. Rusckowski and D. J. Hantowich, Bioconjugate Chem., 2002, 13, 561 CrossRef CAS.
- H. Fonge, M. de Saint Hubert, K. Vunckx, D. Rattat, J. Nuyts, G. Bormans, Y. Ni, C. Reutelingsperger and A. Verbruggen, Bioorg. Med. Chem. Lett., 2008, 18, 3794 CrossRef CAS.
- C. Krummeich, M. Holschbach and G. Stöcklin, Appl. Radiat. Isot., 1994, 45, 929 CrossRef CAS.
- G. Vaidyanathan and M. R. Zalutsky, Appl. Radiat. Isot., 1993, 44, 621 CrossRef.
- D. H. Hunter and X. Zhu, J. Labelled Compd. Radiopharm., 1999, 42, 653 CrossRef CAS.
- S. Pimlott, L. Stevenson, D. J. Wyper and A. Sutherland, Nucl. Med. Biol., 2008, 35, 537 CrossRef CAS.
- E. Camargo, J. Nucl. Med., 2001, 42, 611 CAS , and references therein.
- H. T. S. Benamer, J. Patterson and D. G. Grosset, Movement Disord., 2000, 15, 503 Search PubMed.
- J. T. O'Brien, S. Colloby, J. Fenwick, E. D. Williams, M. Firbank, D. Burn, D. Aarsland and I. G. McKeith, Arch. Neurol., 2004, 61, 919 CrossRef.
- T. Briicke, J. Kornhuber, P. Angelberger, S. Asenbaum, H. Frassine and I. Podreka, J. Neural Transm., 1993, 94, 137 CrossRef.
- J. Cavanagh, J. Patterson, S. Pimlott, D. Dewar, J. Eersels, M. F. Dempsey and D. Wyper, Biol. Psychiatry, 2006, 59, 301 CrossRef CAS.
- R. M. Baldwin, Y. Zea-Ponce, S. S. Zoghbi, M. Laurelle, M. S. Al-Tikriti, E. H. Sybirska, R. T. Malison, J. L. Neumeyer, R. A. Milius, S. Wang, M. Stabin, E. O. Smith, D. S. Charney, P. B. Hoffer and R. B. Innis, Nucl. Med. Biol., 1993, 20, 597 CrossRef CAS.
- A. A. Wilson, D. P. Johnson, D. Mozley, D. Hussey, N. Ginovart, J. Nobrega, A. Garcia, J. Meyer and S. Houle, Nucl. Med. Biol., 2003, 30, 85 CrossRef CAS.
- M. Schou, C. Halldin, J. Sóvágó, V. W. Pike, H. Hall, B. Gulyás, D. Mozley, D. Dobson, E. Shchukin, R. B. Innis and L. Garde, Synapse, 2004, 53, 57 CrossRef CAS.
- G. D. Tamagnan, E. Brenner, D. Alagille, J. K. Staley, C. Haile, A. Koren, M. Early, R. M. Baldwin, F. I. Tarazi, R. J. Baldessarini, N. Jarkas, M. M. Goodmand and J. P. Seibyl, Bioorg. Med. Chem. Lett., 2007, 17, 533 CrossRef.
- N. Kanegawa, Y. Kiyono, H. Kimura, T. Sugita, S. Kajiyama, H. Kawashima, M. Ueda, Y. Kuge and H. Saji, Eur. J. Nucl. Med. Mol. Imaging, 2006, 33, 639 CrossRef.
- N. K. Jobson, R. Spike, A. R. Crawford, D. Dewar, S. L. Pimlott and A. Sutherland, Org. Biomol. Chem., 2008, 6, 2369 RSC.
- N. K. Jobson, A. R. Crawford, D. Dewar, S. L. Pimlott and A. Sutherland, Bioorg. Med. Chem. Lett., 2008, 18, 4940 CrossRef CAS.
- N. K. Jobson, A. R. Crawford, D. Dewar, S. L. Pimlott and A. Sutherland, Bioorg. Med. Chem. Lett., 2009, 19, 4996 CrossRef CAS.
- W. Sihver, A. Nordberg, B. Långström, A. G. Mukhin, A. O. Koren, A. S. Kimes and E. D. London, Behav. Brain Res., 2000, 113, 143 CrossRef CAS , and references therein.
- A. O. Koren, A. G. Horti, A. G. Mukhin, D. Gundisch, A. S. Kimes, R. F. Dannals and E. D. London, J. Med. Chem., 1998, 41, 3690 CrossRef CAS.
- J. T. O'Brien, S. J. Colloby, S. Pakrasi, E. K. Perry, S. L. Pimlott, D. J. Wyper, I. G. McKeith and E. D. Williams, J. Neurol., Neurosurg. Psychiatry, 2007, 78, 356 CAS.
- J. T. O'Brien, S. J. Colloby, S. Pakrasi, E. K. Perry, S. L. Pimlott, D. J. Wyper, I. G. McKeith and E. D. Williams, NeuroImage, 2008, 40, 1056 CrossRef.
- J. K. Staley, S. Krishnan-Sarin, K. P. Cosgrove, E. Krantzler, E. Frohlich, E. Perry, J. A. Dubin, K. Estok, E. Brenner, R. M. Baldwin, G. D. Tamagnan, J. P. Seibyl, P. Jatlow, M. R. Picciotto, E. D. London, S. O'Malley and C. H. van Dyck, J. Neurosci., 2006, 26, 8707 CrossRef CAS.
- S. L. Pimlott, M. Piggott, J. Patterson, J. Cavanagh and D. J. Wyper, J. Labelled Compd. Radiopharm., 2007, 50, 350 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |