Mechanically bonded macromolecules
Lei
Fang
a,
Mark A.
Olson
a,
Diego
Benítez
b,
Ekaterina
Tkatchouk
b,
William A.
Goddard III
b and
J. Fraser
Stoddart
*a
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208-3113, USA. E-mail: stoddart@northwestern.edu; Fax: +1 (847) 491-1009; Tel: +1 (847) 491-3793
bMaterials and Process Simulation Center, California Institute of Technology, 1200 E. California Blvd., Pasadena, California 91125, USA
First published on 18th November 2009
Abstract
Mechanically bonded macromolecules constitute a class of challenging synthetic targets in polymer science. The controllable intramolecular motions of mechanical bonds, in combination with the processability and useful physical and mechanical properties of macromolecules, ultimately ensure their potential for applications in materials science, nanotechnology and medicine. This tutorial review describes the syntheses and properties of a library of diverse mechanically bonded macromolecules, which covers (i) main-chain, side-chain, bridged, and pendant oligo/polycatenanes, (ii) main-chain oligo/polyrotaxanes, (iii) poly[c2]daisy chains, and finally (iv) mechanically interlocked dendrimers. A variety of highly efficient synthetic protocols—including template-directed assembly, step-growth polymerisation, quantitative conjugation, etc.—were employed in the construction of these mechanically interlocked architectures. Some of these structures, i.e., side-chain polycatenanes and poly[c2]daisy chains, undergo controllable molecular switching in a manner similar to their small molecular counterparts. The challenges posed by the syntheses of polycatenanes and polyrotaxanes with high molecular weights are contemplated.
 Lei Fang | Lei Fang was born in Poyang, China, in 1983. He received both his BS (2003) and MS (2006) degrees in chemistry from Wuhan University, China while carrying out research under the supervision of Professor Yong-Bing He. During his graduate studies, Lei spent one and a half years at the Hong Kong Baptist University in the laboratories of Professor Wing-Hong Chan studying cholesterol-derived molecular sensors. Presently, he is pursuing his PhD in chemistry with Professor Stoddart at Northwestern University. During his PhD program, Lei has conducted research on interdisciplinary topics in the broad areas of nanoscience as well as organic and polymer chemistry. |
 Mark A. Olson | Mark A. Olson received his BS degree in chemistry from Texas A&M University-Corpus Christi in 2005. During his undergraduate studies, Mark experienced a short stay at the California Institute of Technology in the laboratories of Dr Jack Beauchamp. Presently a fourth year graduate student, after spending his first three years in UCLA, Mark is now finishing up his PhD in organic chemistry under the tutelage of Professor Stoddart at Northwestern University. His focus is on the synthesis of exotic molecular switchable materials including bistable side-chain poly[2]catenanes, reconfigurable (Au, Pt, Pd, Ag) nanoparticle assemblies, and reprogrammable self-assembling polymer blends. |
 Diego Benítez | Diego Benítez obtained his PhD in chemistry from the California Institute of Technology in 2005 conducting research in the laboratories of Robert H. Grubbs and William A. Goddard III. He then joined the laboratory of J. Fraser Stoddart at UCLA as a Research Associate. In 2009, he returned to the California Institute of Technology as the Director of Nanomaterials Technology in the Materials and Process Simulation Center. |
 Ekaterina Tkatchouk | Ekaterina Tkatchouk obtained her BS degree in chemistry from the Universidad Nacional Autonoma de Mexico and her PhD in Materials Science and Engineering from the same institution. After working for some time in industry, she spent a year in the laboratory of Kendall N. Houk at UCLA as a UC MEXUS-CONACYT Postdoctoral Research Fellow. Since late 2007, she has been a Postdoctoral Scholar in the laboratory of William A. Goddard III where she conducts computational research on topics related to homogeneous catalysis and mechanically interlocked molecules. |
 William A. Goddard III | William A. Goddard III obtained his BS Engr. from UCLA in 1960 and his PhD in Engineering Science and Physics from California Institute of Technology (Caltech) in Oct. 1964. Since Nov. 1964, he has been a member of the Chemistry faculty at Caltech where he is now the Charles and Mary Ferkel Professor of Chemistry, Materials Science, and Applied Physics. His current research interests include new methodology for quantum chemistry, reactive force molecular dynamics, mesoscale dynamics, statistical mechanics and electron dynamics. |
 J. Fraser Stoddart | Fraser Stoddart received all (BSc, PhD, DSc) of his degrees from the University of Edinburgh, UK. Presently, he holds a Board of Trustees Professorship in the Department of Chemistry at Northwestern University. His research has opened up a new materials world of mechanically interlocked molecular compounds and, in doing so, has produced a blueprint for the subsequent growth of functional molecular nanotechnology. |
Introduction
Molecules containing mechanical bonds, such as catenanes,1 rotaxanes,2 knots3 and Borromean rings,4 have captured the attention of the scientific community, not only because of their intriguing architectures and topologies, but also as a result of the ability of their components to undergo controllable intramolecular movement. Interlocked molecules which contain one or more mechanical bonds have been identified in biomacromolecules, e.g., catenated circular DNA5 and protein chainmail6 (Fig. 1a and b), to cite but two examples from mother nature. Although the idea7 of incorporating mechanical bonds into synthetic polymers is more than five decades old, it was not until relatively recently that the exquisitely planned formation of mechanical bonds at the macromolecular level was realised in the laboratory. Mechanical bonds are created in a haphazard manner, however, in an already cross-linked polymer: further cross-linking of a linear polymer in the presence of the former can produce (Fig. 1c) a random interpenetrating network.8 Copolymerisation of lipoic acid and 1,2-dithiane gives9 a randomly interlocked cyclic polymer, which possesses dramatically different mechanical properties compared to its non-interlocked counterpart. In a somewhat more precise manner, infinite interweaving of three-dimensional frameworks has been identified10 and characterised (Fig. 1d) in metal–organic frameworks (MOFs). The physical properties—e.g., dynamic and rheological characteristics, mechanical strengths, and surface areas—of interpenetrating polymers and MOFs, are dictated by the mechanical entanglement of the polymer networks or the catenation present in MOFs.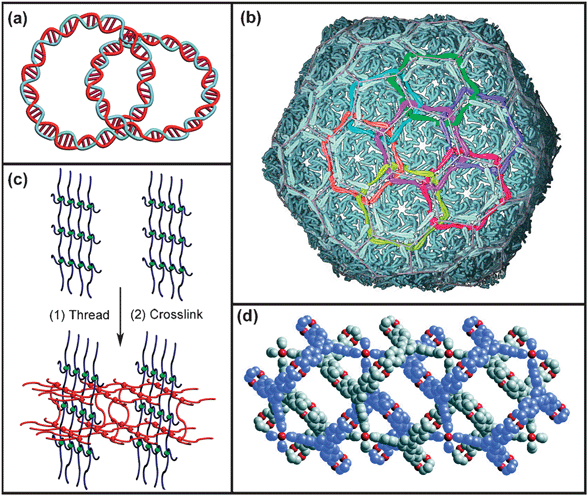 | ||
| Fig. 1 Examples of natural and artificial macromolecular mechanically interlocked systems: (a) graphical representation of catenated circular DNA; (b) crystal structure of the bacteriophage HK97 capsid chainmail,6 with the subunits that are cross-linked into rings colored identically (reprinted with permission of the American Association for the Advancement of Science); (c) the construction of an interpenetrating polymer network at a conceptual level; (d) crystal structure of MOF-14 showing10 a pair of interwoven 3-D porous frameworks (reprinted with permission of the American Association for the Advancement of Science). | ||
Rapid advances in the synthesis of mechanically-interlocked molecules (MIMs) in recent times have enabled11 precise control of the architectures and topologies of the molecules; hence, it has enabled chemists to develop specifically desired functions based on these unique structures. For example, the application of switchable, mechanically-interlocked, small molecules has been widely demonstrated12–14 in solid-state electronic devices,12 mechanised nanoparticles,13 and nanoelectromechanical systems.14 From the perspective of an organic chemist, engineering mechanical bonds into macromolecular scaffolds, by employing organic/polymer synthetic protocols, is becoming an area of considerable contemporary interest. The importance and attraction of this field originated from the fact that (i) device fabrication of MIMs could benefit enormously from the macromolecular materials’ processability if the mechanically interlocked structures could be incorporated into polymer/dendrimer scaffolds, and (ii) intramolecular motion of mechanically interlocked units in a polymer/dendrimer network could induce accumulative, macroscopic property changes in the material itself. In fact, back in the early 1990s, soon after the field of MIMs began to blossom, the development of polyrotaxanes and polycatenanes—the two most commonly sought after mechanically interlocked macromolecules—had been identified by synthetic chemists as important and challenging targets in synthesis. The challenge is a combination of the difficulty in preparing MIMs themselves and the huge entropy cost in making high-molecular weight macromolecules. It has to be admitted that mechanically bonded macromolecules with precisely controlled structures are not yet producible on a gram scale, using routine synthetic procedures. Several comprehensive review articles have been published15–17 on mechanically bonded macromolecules, in addition to the extensive review literature11,18 now available on MIMs themselves.
This review describes our own efforts—covering the past two decades—to introduce mechanical bonds into macromolecules. Fig. 2 shows the graphical representations of some of these mechanically interlocked macromolecules—namely, (a) main-chain [n]catenanes, (b) bridged poly[2]catenanes, (c) pendant poly[2]catenanes, (d) side-chain poly[2]catenanes, (e) main-chain [n]rotaxanes, (f) linear poly[c2]daisy chains, as well as (g) mechanically interlocked dendrimers. Various approaches and synthetic strategies have been employed in synthesising these constitutionally and topologically diverse targets. In general, these synthetic strategies can be described under three categories: (I) The formation of the mechanical bonds spontaneously while constructing the macromolecular scaffold; (II) Coupling already-made MIMs on to polymer/oligomer/dendrimer scaffolds by covalent linking; (III) Incorporating mechanical bonds onto an already existing polymeric/oligomeric/dendritic scaffold.
![Architectures of the mechanically bonded macromolecules that are covered in this review: (a) main-chain [n]catenanes; (b) bridged poly[2]catenanes; (c) pendant poly[2]catenanes; (d) side-chain poly[2]catenanes; (e) main-chain [n]rotaxanes; (f) linear poly[c2]daisy chains; (g) mechanically interlocked dendrimers.](/image/article/2010/CS/b917901a/b917901a-f2.gif) | ||
| Fig. 2 Architectures of the mechanically bonded macromolecules that are covered in this review: (a) main-chain [n]catenanes; (b) bridged poly[2]catenanes; (c) pendant poly[2]catenanes; (d) side-chain poly[2]catenanes; (e) main-chain [n]rotaxanes; (f) linear poly[c2]daisy chains; (g) mechanically interlocked dendrimers. | ||
Linear and branched oligo[n]catenanes
As a challenging target in unnatural product synthesis,19 the construction of linear main-chain [n]catenanes (Fig. 2a) has proved elusive. The only practical synthetic strategy one can use is the Strategy I in making such chain-like structures. In our early forays into oligocatenanes, the structures of the [n]catenanes 1·4PF6, 2·12PF6, 3·20PF6, (n = 3, 5, 7, respectively) were designed20,21 and targeted based on a template-directed protocol, in which the cooperativity of weak noncovalent bonding interactions—e.g., aromatic π–π stacking, [C–H⋯π] and [C–H⋯O] interactions—between complementary components were exploited. The strategy employed in the synthesis of a linear [5]catenane (Olympiadane) and a branched [7]catenane involved a two-step self-assembly route (Scheme 1) making use of templation at each step. Firstly, the construction of a [3]catenane 1·4PF6, which is composed of one cyclobis(paraquat-4,4′-biphenylene) ring and two tris-1,5-naphtho[57]crown-15 macrocycles, was achieved in 10% yield by templating the formation of the large tetracationic cyclophane in MeCN–DMF (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in the presence of an excess of the oligoether macrocycle. These three rings are mechanically interlocked together such that two of the dioxynaphthalene (DNP) units of the [57]crown-15 derivatives thread through the cyclophane, forming an acceptor–donor–donor–acceptor π–π complex, which can be clearly identified (Fig. 3a) in the X-ray crystal structure of 1·4PF6. Secondly, by employing ultrahigh pressure (12 kbar) to the reaction mixture, another two cyclobis(paraquat-p-phenylene) (CBPQT4+) rings could be self-assembled around each of the [57]crown-15 macrocycles in DMF after 6 days to give Olympiadane in 30% yield. The branched [7]catenane 3·20PF6, a byproduct in which all the DNP units in the macrocyclic oligoether are encircled by CBPQT4+ rings, could be isolated in 26% yield from the same reaction mixture. The molecular structures of both 2·12PF6 and 3·20PF6 were confirmed (Fig. 3b, c) unambiguously by X-ray crystallography.
:1) in the presence of an excess of the oligoether macrocycle. These three rings are mechanically interlocked together such that two of the dioxynaphthalene (DNP) units of the [57]crown-15 derivatives thread through the cyclophane, forming an acceptor–donor–donor–acceptor π–π complex, which can be clearly identified (Fig. 3a) in the X-ray crystal structure of 1·4PF6. Secondly, by employing ultrahigh pressure (12 kbar) to the reaction mixture, another two cyclobis(paraquat-p-phenylene) (CBPQT4+) rings could be self-assembled around each of the [57]crown-15 macrocycles in DMF after 6 days to give Olympiadane in 30% yield. The branched [7]catenane 3·20PF6, a byproduct in which all the DNP units in the macrocyclic oligoether are encircled by CBPQT4+ rings, could be isolated in 26% yield from the same reaction mixture. The molecular structures of both 2·12PF6 and 3·20PF6 were confirmed (Fig. 3b, c) unambiguously by X-ray crystallography.
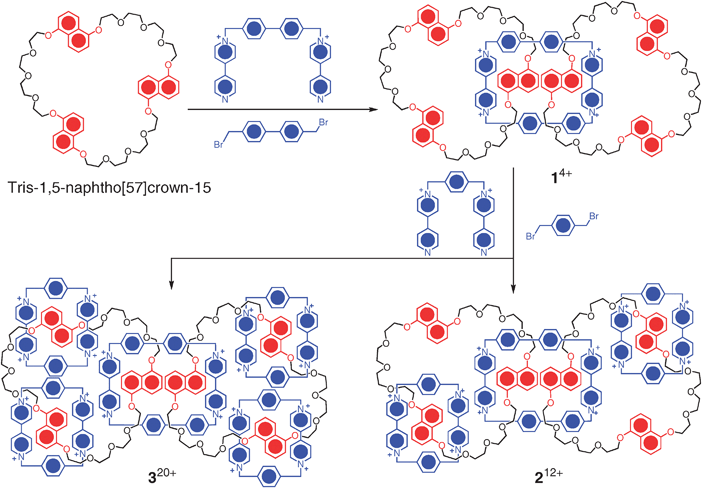 | ||
| Scheme 1 Assembly of the main-chain oligocatenanes 14+, 212+ and 320+. | ||
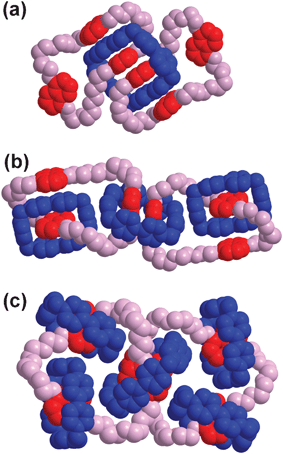 | ||
| Fig. 3 Space-filling representations of the solid-state structures of (a) 14+, (b) 212+ and (c) 320+. | ||
In an attempt to uncover higher synthetic efficiencies in the synthesis of the main-chain [n]catenane, a one-pot self-assembly process of oligocatenanes, using a similar template-directed strategy, gave22 linear [2]-, [3]-, [5]-, and [7]catenanes in yields of 5, 14, 4, and 2%, respectively. In this reaction, a tetra-1,5-naphtho[76]crown-20 was employed as the macrocyclic template, while the cyclobis(paraquat-O,O′-diethylenehydroquinone) rings were formed along the DNP units in the templates, to generate the oligocatenated topologies in one pot. Since the [5]- and [7]catenanes isolated in this reaction could only be characterised by electrospray mass spectroscopy, their topologies and molecular structures remain uncertain. Higher oligocatenanes could not be detected and perhaps did not form on account of their entropically unfavourable nature. Although the use of ultrahigh pressure and the careful choice of templates greatly facilitates the self-assembly of the oligocatenanes, both the stepwise and the one-pot methods suffer from low yields, hampering the further development of higher molecular weight main-chain poly[n]catenanes.
It is apparent that a much more efficient synthetic strategy is necessary for the generation of higher oligo- or polycatenanes using a “bottom-up” approach. Recently, a success23 in highly efficient thermodynamic assembly of a [3]catenane has raised our hopes of making main-chain polycatenanes in high yields. In this reaction, the cyclobis(paraquat-4,4′-biphenylene) ring underwent dynamic nucleophilic substitution in the presence of iodide (I−) at high temperature (>80 °C) which allowed24 the ring to open and close reversibly. This dynamic process enabled formation of the thermodynamically stable product when a π-electron-rich dinaphtho[38]crown-10 macrocycle served as a template in solution, to afford the expected [3]catenane, in 91% yield! It is not difficult to believe that a high molecular weight polycatenane can be obtained by assembling cyclobis(paraquat-4,4′-biphenylene) with a carefully designed π-electron-rich macrocycle in a similar thermodynamically controlled reaction. In principle, under exchange conditions (I− and T > 80 °C) the system should behave as a step-growth dynamic covalent polymerisation.25 The driving force for the polymerisation will be the enthalpic release from the donor–acceptor catenation step. As with every dynamic polymerisation,26 a ring-chain equilibrium will be established and will be highly dependent of the concentration of the reactants. In the wake of these considerations, a reasonable approach would be to use exact molar amounts of reactants with saturated solutions of the highly soluble electron-poor and electron-rich macrocycles. Lower monomer concentrations might shift the ring-chain equilibrium towards cyclic oligomers, producing cyclic oligocatenanes.
Bridged and pendant poly[2]catenanes
The step-growth polymerisation of bifunctional molecules produces linear main-chain polymers. Using this approach (Strategy II), we have achieved27 (Scheme 2) the synthesis of two main-chain poly(bis[2]catenane)s 4·8nPF6 and 5·8nPF6, a main-chain poly([2]catenane) 6·4nPF6, and a pendant poly[2]catenane 7·4nPF6 with degrees of polymerisation up to as far as 25. The monomer of 6·4nPF6 is composed of two catenated macrocyclic components—namely, a chloromethyl-functionalised cyclophane and a carboxylic acid-functionalised polyether ring. On heating in the presence of LiBr and 2,6-lutidine, these two complementary functional groups undergo esterification, inducing an AB + AB type step-growth polymerisation to produce 6·4nPF6. A similar strategy was applied to the synthesis of poly(bis[2]catenane) 4·8nPF6, where the monomers are composed of a “handcuff”-like bis(macrocyclic polyether) and two “terminal” catenated cyclophanes, functionalised with hydroxymethyl groups. In the presence of a bis(isocyanate), the AA + BB type step-growth polymerisation of this bis[2]catenane gave 4·8nPF6 as a result of the highly efficient carbamation. As an analogue of polymer 4·8nPF6, compound 5·8nPF6, with bis(diethyleneglycol) hydroquinone acting as the bridge between the polyether rings, was prepared using the same method. In order to construct a pendant polycatenane, a [2]catenane incorporating two reactive functional groups on one of its two macrocyclic components had to be used. In one example, two hydroxymethyl groups were installed on the macrocyclic polyether component of the monomeric [2]catenane, while the CBPQT4+ component remained unfunctionalised. Reaction of this [2]catenane with bis(4-isocyanatophenyl)methane yielded the pendant poly[2]catenane 7·4nPF6. Chloride salts of all these polycatenanes (4·8nCl–7·4nCl) were analysed in aqueous solution by gel permeation chromatography (GPC). The number-average molecular weights (Mn) of 4·8nCl, 5·8nCl, 6·4nCl and 7·4nCl were revealed to be 35, 45, 45, and 27 kDa, respectively. These Mn values correspond to degrees of polymerisation from 15 up to 25. Compared to Olympiadane 2·12PF6, 4·8nPF6–7·4nPF6 have much higher molecular weights, but are polydisperse, i.e., they are mixtures of species with different molecular weights, on account of the nature of the step-growth polymerisation process.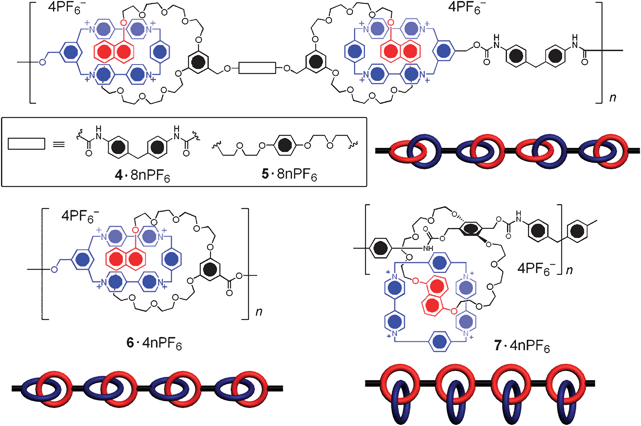 | ||
| Scheme 2 Structural formulae and cartoon representations of the bridged polycatenanes 4·8nPF6, 5·8nPF6, 6·4nPF6 and the pendant polycatenane 7·4nPF6. | ||
Side-chain switchable poly[2]catenanes
The side-chain poly[2]catenane topology (Fig. 2d) has remained28 an elusive synthetic target for some time despite the fact that simple retrosynthetic analysis suggests29 only one obvious approach—that is, using Strategy II. Incorporating a reactive functional group onto one of the macrocyclic components of a catenated species allows for the covalent attachment—by way of a post-polymerisation modification—to polymer bearing side-chain functional groups which act as the points of attachment. This post-polymerisation reaction30 which is arguably stepwise in nature with respect to the attachment of the catenated components needs to be a relatively high yielding one in order to achieve any appreciable increases in polymer molecular weights, while preserving low PDIs. Towards this end, we recently reported31 the synthesis and redox controlled actuation of a bistable side-chain poly[2]catenane (Scheme 3) by way of a post-polymerisation modification of a methacrylate based polymer with azide-terminated side-chains.32 Covalent modification of the π-electron deficient CBPQT4+ ring at the single ortho-position on one of the para-xylyl rings allows for the incorporation33 of a pendant alkyne group. Thus, a donor–acceptor type bistable [2]catenane containing the alkyne-derivatised CBPQT4+ ring can then be subjected to the Cu(I)-catalysed Huisgen 1,3-dipolar cycloaddition34 with the azide-terminated side-chains of polymer (Mw = 55000 g mol−1 and Mn = 39000 g mol−1, PDI = 1.4), obtained by atom transfer radical polymerisation (ATRP).32 Combining these two reactants in DMF-d7 at 60 °C for 3 h with a stoichiometric amount of CuI gives the bistable side-chain poly[2]catenane 8·4nPF6 with >95% conversion—as determined from 1H NMR spectroscopy—following precipitation of the polymer from a saturated aqueous solution of NH4PF6. End-group analysis of 8·4nPF6 reveals an Mn value of 128000 g mol−1, while an Mw value of 1300000 g mol−1 and an Mn value of 870000 g mol−1 (PDI = 1.5) were determined by size-exclusion chromatography with multi-angle light scattering analysis (SEC-MALS). A battery of analytical techniques, including SEC-MALS, chronocoulometry, static-light scattering, photon correlation spectroscopy, and ultimately transmission electron microscopy revealed that the bistable side-chain poly[2]catenane 8·4nPF6 forms spherical aggregates in solution, most likely with the [2]catenanes residing at the peripheries. Encouragingly, this material still behaves as a molecular switch that can be addressed in an “on” and “off” fashion both chemically and electrochemically in solution by virtue of the redox properties of the tetrathiafulvalene (TTF) recognition units incorporated into the π-electron rich macrocycle. Oxidation of the TTF unit to its radical dication initiates a Coulombic repulsion-induced circumrotation within the [2]catenanes along the polymer side-chains (Scheme 3). This process is fully reversible upon reduction of the TTF unit back to the neutral state. This satisfying outcome highlights the feasibility of incorporating molecular switches into polymeric scaffolds where, even in a situation where the polymer adopts a spherical aggregate superstructure, the bistable [2]catenanes still retain their switching properties in the bulk material.
![(a) Synthesis and redox-driven switching of sidechain poly[2]catenane 8·4nPF6; (b) Graphical representation showing the switching process of the polymer 8·4nPF6.](/image/article/2010/CS/b917901a/b917901a-s3.gif) | ||
| Scheme 3 (a) Synthesis and redox-driven switching of sidechain poly[2]catenane 8·4nPF6; (b) Graphical representation showing the switching process of the polymer 8·4nPF6. | ||
Oligomeric and polymeric main-chain [n]rotaxanes
Main-chain oligo- and poly[n]rotaxanes (Fig. 2e) are one of the most extensively studied categories of mechanically bonded macromolecules and have been well investigated as insulated molecular wires.35 Many examples of main-chain oligo- and polyrotaxanes, relying on the threading of cyclodextrins36,37 (Fig. 4), macrocyclic oligoethers,38 and cucurbiturils,39 have been reported in the literature. There are two general synthetic methods for the construction of main-chain [n]rotaxanes—namely, the “clipping” and the “threading-followed-by-stoppering” approaches, both of which fall under Strategy III. These two approaches have been applied successfully to the synthesis of main-chain oligo- and polyrotaxanes recently.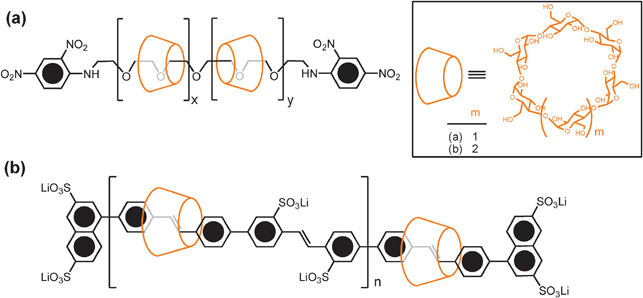 | ||
| Fig. 4 (a) An early example of an α-cyclodextrin-based polyrotaxane, in which a polyethylene glycol chain acts36 as the thread and the terminal 2,4-dinitrophenyl groups serve as the stoppers; (b) the chemical structure of a β-cyclodextrin-threaded conjugated polyrotaxane which represents37 an insulated molecular wire. | ||
Formation of dynamic covalent bonds were employed40 in the “clipping” approach to afford main-chain oligorotaxanes. The reversible nature of the reactions introduces41 the prospects of “error checking” and “proof-reading” into synthetic processes where dynamic covalent chemistry operates. Since the formation of products occurs under thermodynamic control, product distributions depend only on the relative stabilities of the final products. The dynamic covalent chemistry involved in this system, is the highly efficient clipping42 of a diamine and a dialdehyde to form a [24]crown-8 diimine-containing macrocycle, templated by a dialkylammonium ion recognition site. In this event (Scheme 4a), oligomeric dialkylammonium stalks terminated by two bulky dimethoxybenzyl stoppers were used as the dumbbell components for the oligorotaxane and as the template for the thermodynamic controlled clipping reaction. A series of such dumbbell-shaped molecules (9a–9f·nPF6), containing n (n = 2, 3, 4, 6, 10, 14, respectively) dialkylammonium centres, were synthesised firstly by reductive amination with derivatives of benzylamine and benzaldehydes, then by protonation of the secondary amines followed by counterion exchange. The clipping reaction of 2,6-dipyridinedicarboxaldehyde and tetraethyleneglycol bis(2-aminophenyl)ether, templated by 9a–9d·nPF6, gave the thermodynamically stable [3]-, [4]-, [5]-, and [7]rotaxanes (10a–10d·nPF6) in nearly quantitative yields in MeNO2. When n = 10, 4-octyloxypyridinedicarboxaldehyde was used, in a 21-component self-assembly reaction in order to improve the solubility of the product 10e·nPF6. Although the assembly of the [11]rotaxane 10e·nPF6 was successful using the octyloxyl solublising group, unfortunately, when n = 14, the poor solubility of the dumbbell 9f·nPF6 (n = 14) once again put a limit on the template-directed synthesis of the [15]rotaxane, using this approach. On account of their readily hydrolysable imine bonds, the dynamic rotaxanes 10a–10c·nPF6 can be fixed by reducing with BH3·THF. The fixed [3]-, [4]-, and [5]rotaxanes 11a–11c·nPF6 were isolated pure in 77, 74, and 40% yields, respectively. For the higher rotaxanes 10d–10e·nPF6 (n = 6, 10), the reductions were unsuccessful on account of partial cleavage and dissociation of the macrocycles from the dumbbell compounds during the reductions.
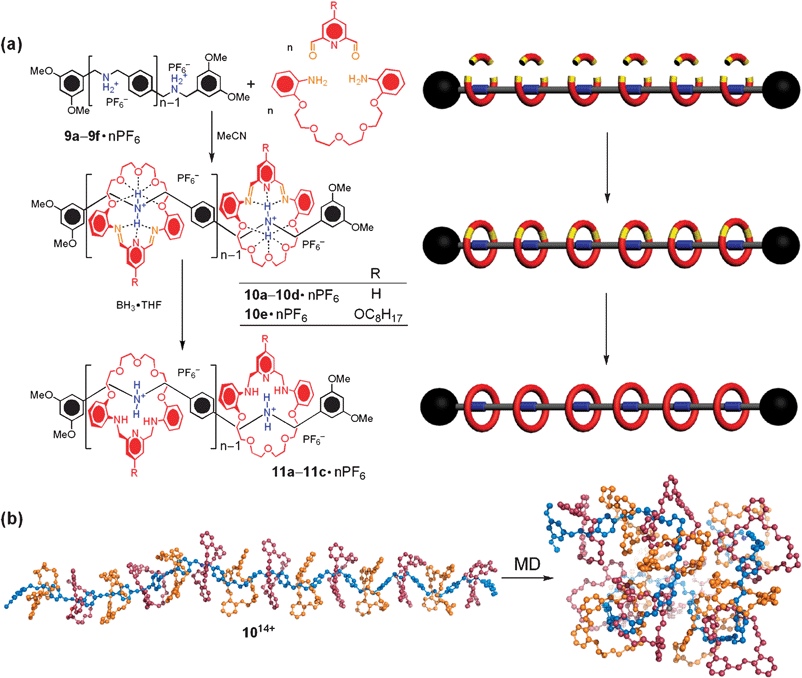 | ||
| Scheme 4 (a) The “clipping” approach to the dynamic oligorotaxanes 10a–e·nPF6, and the “fixed” oligorotaxanes 11a–c·nPF6. The architectures of these structures are shown as cartoon graphs on the right. The dynamic covalent bonds are highlighted in orange. (b) Molecular dynamics simulation for the folding of the oligorotaxane 1014+ into a random coil conformation. The backbone is shaded blue, while alternating rings (for visual ease) are colored red and orange. | ||
Meanwhile, a “threading-followed-by-stoppering” approach had also been developed (Scheme 5) for the synthesis of the π-donor–acceptor polyrotaxane 13·4nPF6. Such [n]rotaxanes are composed of a linear polymeric dumbbell-shaped component, containing 2n DNP units as the π-electron donors, and n CBPQT4+ cyclophanes as the π-electron acceptors. The template-directed “clipping” methodology used traditionally for the installation of CBPQT4+ rings on dumbbell-shaped components was not efficient enough to obtain a high coverage of rings encircling the polymer backbone. Instead, the “threading-followed-by-stoppering” method, employing Cu(I)-catalysed Huisgen 1,3-dipolar cycloadditions, proceeded43 with high efficiencies when it was applied in the kinetically controlled synthesis of [2]rotaxanes. In this context, the dumbbell component 12 was fed with 0.6 equivalents of CBPQT4+ rings relative to the total number of DNP units of 12 in DMF. After 24 h at room temperature, UV/vis absorption spectroscopy demonstrated that the slow threading process had gone to completion. The resulting pseudopolyrotaxane [12 ⊂ nCBPQT4+] could be converted to the polyrotaxane 13·4nPF6 by covalently attaching two bulky 2,6-diisopropylphenyl stoppers onto the chain ends using Cu(I)-catalysed Huisgen 1,3-dipolar cycloadditions. The Mn value of 13·4nPF6 reached up as far as 181 kDa with a PDI of 1.71, indicating that there were about 94 repeating units in each polymer chain on average. The polyrotaxane 13·4nPF6 adopts an alternating stacking pattern that is induced by secondary noncovalent bonding interactions between the CBPQT4+ rings and the alongside DNP units, an interaction which was confirmed by 1H NMR spectroscopy by comparing the polyrotaxanes with a simple folded [2]rotaxane model compound. Another piece of evidence for the folding of 13·4nPF6 came from GPC investigation which showed that the polymer dimensions of poly[n]rotaxane 13·4nPF6 were significantly smaller than that of the polymer thread 12, despite a near two-fold molecular mass increase.
 | ||
| Scheme 5 Structural formulae and graphical representations of the “threading-followed-by-stoppering” approach to the self-folding polyrotaxane 13·4nPF6. | ||
In the absence of experimental data, the prediction of the secondary structure of higher order donor–acceptor [n]rotaxanes (n > 10) is a challenging exercise that awaits a thorough analysis. Although in the solid-state, infinite stacks are clearly possible (Scheme 5), in solution the dynamic nature of the oligomers/polymers is likely to produce a distribution of π-stack lengths in equilibrium with each other and dictated by the enthalpy gain for the stacking being weighed against the entropy loss associated with a rigid rod-like molecule. Quantum mechanical calculations carried out on short donor–acceptor stacks with the constitution of 13·4nPF6 support the folded secondary structure anticipated44 to exist in the solid state. The entropic contributions, however, from unfolding are difficult to predict in such large molecules—especially in solution. Indirect data—1H NMR spectroscopy and hydrodynamic radii—on 134n+ revealed evidence for a folded structure. It is not easy, however, to establish the most abundant length of π-stacks—analogous to the conjugated critical length in polyacetylenes45—using NMR spectroscopy and light scattering alone. Molecular mechanical simulations—although attractive for their performance/cost for large systems—fail to describe the sandwiched, parallel, displaced and T-shaped geometries of stacked and interacting aromatics with the required accuracy to make their use applicable to large systems.
On the other hand, in systems such as 10n+ and 11n+ where [π⋯π] stacking is not a primary consideration, the secondary structures can be modeled using popular force-fields,46 such as the recent OPLS-2005. We have performed a combination of molecular dynamics (MD) and molecular mechanics (MM) minimisations using the OPLS force-field on a [15]pseudorotaxane to obtain (Scheme 4b) a low energy conformation of an analogue of 10n+. The starting geometry for the [15]pseudorotaxane, which was generated from the X-ray crystal structure44 of the monomeric [2]pseudorotaxane, was subjected to a 2000 ps MD simulation at 300 K until an equilibrium geometry was obtained. This analogue is predicted to adopt a random coil conformation similar to that of the dumbbell 9f14+, indicating that the installment of the polyether macrocycles on the dumbbell molecules exerts little significant effect on the secondary structure in such hydrogen-bond interaction-based systems, compared to the [π⋯π] stacking-based structure 134n+.
Poly[c2]daisy chains
The term “daisy chain” is defined as “a string of daisies with stems linked to form a chain, … such a chain carried by chosen students on a class day or other celebrations in some women’s colleges.” A molecular daisy chain is an array of self-complementary plerotopic molecules, linked together by mechanical bonds or noncovalent bonding interactions between the complementary sites in the monomer. These two complementary sites in the monomer, however, must be obliged to recognise each other intermolecularly, rather than intramolecularly—that is, the monomer must be prevented from forming a “pretzelane”47,48 and encouraged to form a “daisy chain” instead.As an entropically unfavourable process, the assembly of a high molecular weight daisy chain polymer requires a high concentration of the monomer and a strong binding affinity between the complementary recognition sites. Although supramolecular [an]daisy chains, composed with two carefully designed heteroditopic monomers, were reported49 recently, the mechanically interlocked macromolecular [an]daisy chain still remains a challenging synthetic target. Instead, cyclic dimers ([c2]daisy chains) were easy to isolate in high yields and show remarkable stabilities both in solution and in the solid-state. On account of its C2h symmetry and linear intramolecular motion, the [c2]daisy chain topology has been employed50,51 in the construction of molecular actuators that have been designed to undergo contraction/extension molecular movements with the appropriate stimuli, e.g., redox and pH. Consequently, the polymerisation of [c2]daisy chains became an obvious target in attempts to create muscle-like materials (Fig. 2f). In this context, a bifunctional [c2]daisy chain was synthesised52 by employing ring-closing metathesis of a bisolefin-containing polyether templated by a dialkylammonium unit, on account of the strong hydrogen bonds formed between oxygen atoms in the macrocyclic component and the NH2+ cationic centre. This bisalkene-functionalised compound was subjected to acyclic diene metathesis polymerisation in the presence of Grubbs’ ruthenium catalyst, to afford a poly[c2]daisy chain with a molecular weight of 13 kDa.
Encouraged by the success in polymerising a [c2]daisy chain, we have synthesised53 a bistable [c2]daisy chain 14·6PF6 functionalised with dialkyne terminal groups (Scheme 6). The macrocycle is dibenzo[24]crown-8 (DB24C8) and it has two recognition sites—a dialkylammonium centre and a bipyridinium unit—appended to it. The mutual association of two of these components during templation prior to stoppering leads to the formation of the [c2]daisy chain. In MeCN, the DB24C8 ring encircles the dialkylammonium unit predominantly as a result of [N+–H⋯O] hydrogen bonding primarily. Since the binding preference of the DB24C8 ring can be reversed by deprotonation of the dialkylammonium ion with a non-nucleophilic base and restored by reprotonation of the resulting dialkylamine function with acid, the daisy chain molecule can be made to undergo acid/base controlled contraction/extension movements. Functionalised with bisalkynyl terminal groups, 14·6PF6 was subjected to an AA + BB type step-growth polymerisation with an appropriate diazide employing the highly efficient Huisgen type 1,3-dipolar cycloaddition.34 The resulting bistable poly[c2]daisy chain 15·6nPF6 has a molecular weight of 33 kDa and a polydispersity of 1.85. This polymer undergoes a similar quantitative, efficient and fully reversible switching process in solution, triggered by base/acid, i.e., DABCO and trifluoroacetic acid. Stop-flow kinetics measurements demonstrated that the extension/contraction movements of 15·6nPF6 are actually faster than those occurring in its monomeric counterpart 14·6PF6. Although the reason for this phenomenon is still not clear to us, the robust switchability of the polymer provides an avenue through which correlated molecular motions can lead to changes in macroscopic properties. In a similar system reported54 recently, the radius of gyration of a poly[c2]daisy chain polymer was observed to increase by 48% after expansion of the [c2]daisy chain monomer had been induced by external stimuli.
![Polymerisation of the bisfunctionalised [c2]daisy chain 14·6PF6 and the acid–base induced switching of the resulting poly[c2]daisy chain 15·6nPF6. Graphical representations of the switchable poly[c2]daisy chain architectures are shown.](/image/article/2010/CS/b917901a/b917901a-s6.gif) | ||
| Scheme 6 Polymerisation of the bisfunctionalised [c2]daisy chain 14·6PF6 and the acid–base induced switching of the resulting poly[c2]daisy chain 15·6nPF6. Graphical representations of the switchable poly[c2]daisy chain architectures are shown. | ||
Mechanically interlocked dendrimers
During the past decade, we have been pursuing the construction of mechanically interlocked dendrimers (Fig. 2g) by employing a convergent templation approach. An approach which involves “threading-followed-by-stoppering”, and then, thereafter, “stopper-exchange”, has been used55 in a template-directed synthesis of a precursor bis[2]rotaxane. It requires that a bisdibenzo[24]crown-8 core is threaded with a bis(bromomethyl)-substituted dibenzylammonium ion derivative before stoppering is achieved with an excess of Ph3P. Subsequent treatment of the bis[2]rotaxane with Fréchet-type wedge-shaped aldehydes (G1 and G2) effected the Wittig reaction, affording isomeric mixtures of tetraolefins, which were hydrogenated catalytically. In this four-step synthesis, in which the first step is the lowest yielding one, the best yield that has been obtained for dendrimers with rotaxane-like mechanical branching is below 30%. In an attempt to find a more efficient route to mechanically interlocked dendrimers, we turned our attention56 to the “slippage” approach. It is known that in CH2Cl2 at 40 °C bis(cyclohexylmethyl)ammonium hexafluorophosphate (50 mM) is converted 98% of the way to a [2]rotaxane in the presence of 150 mM of DB24C8. In the event, this thermodynamically controlled self-assembly process lost all its remarkable efficiency on going from the model system to one that involves a Fréchet-type benzyl ether wedge and a DB24C8 unit which links another two such wedges. The best yield of a mechanically interlocked dendrimer that could be obtained in this single slippage experiment was 19%, and this result involved a 90-day reaction period, followed by chromatography!Not until the dynamic covalent chemistry mentioned42 above was employed, was the assembly of Fréchet-type dendrons into a mechanically interlocked dendrimer achieved57,58 in a nearly quantitative yield (Scheme 7). In order to construct the mechanically interlocked dendritic structure, a tripod molecule 16·3PF6, with three dialkylammonium unit containing arms, was used as the dendrimer core. 1H NMR spectroscopy reveals >90% conversion 5 min after the dendritic dialdehydes ([G1]–[G3]), the diamine, and 16·3PF6 were mixed in a 3:3:1 molar ratio in CD3CN or CD3NO2. This seven-component dynamic assembly process shows remarkable efficiency and the mechanically interlocked dynamic products 17a–17c·3PF6 are thermodynamically stabilised by numerous [N+–H⋯O] hydrogen bonds and [C–H⋯O] interactions. The formation of 17a–17c·3PF6 could be monitored not only by 1H NMR spectroscopy quantitatively, but also by electrospray ionisation mass spectrometry, since the molecular ions [17a]3+, [17b]3+, and [17c]3+ can be clearly identified. In common with the oligomeric rotaxanes 10a–10e·nPF6, the kinetically labile dendrimers 17a–17b·3PF6 ([G1]–[G2]) could be fixed by reduction (BH3·THF), followed by deprotonation to give the kinetically stable, neutral dendrimers 18a–18b·3PF6, in ∼80% yields. These reductions went smoothly with [G1]–[G2], but yielded a mixture of degraded dendrimers with the [G3] dendron, as a result of the massive steric hindrance of the [G3] dendron. In summary, by taking advantage of dynamic covalent chemistry,41 we have developed a practical way of making mechanically bonded dendrimers, in which the thermodynamically controlled seven-component self-assembly process proceeds quantitatively by overcoming the massive steric hindrance of three dendrons as large as the [G3] Fréchet-type wedge-shaped dendrons.
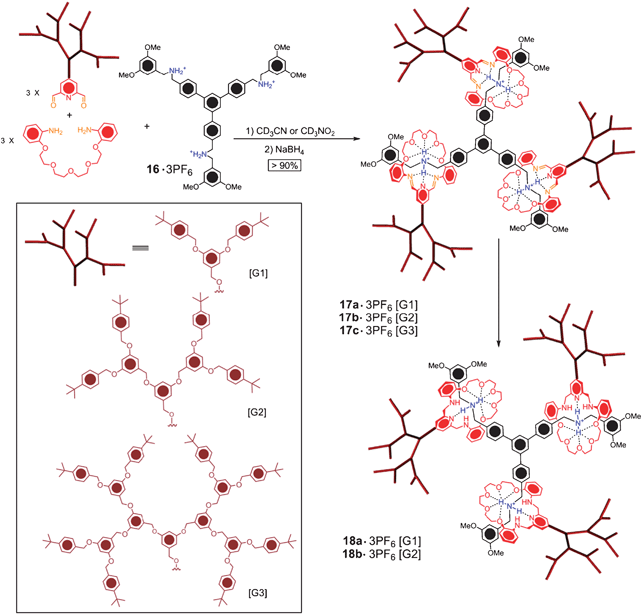 | ||
| Scheme 7 Dynamic covalent assembly of the mechanically interlocked dendrimers 17a–17c·3PF6 and the fixed dendrimers 18a–18b·3PF6. | ||
Conclusions
The elaboration of the structural features of mechanically interlocked molecules into macromolecular materials could be a means by which molecular motion impacts their bulk properties. In this tutorial review, we have described the recent development of mechanically bonded macromolecules with a significant structural and topological diversity. The use of template-directed protocols and highly efficient condensation/conjugation reactions has facilitated the synthetic evolution of mechanically interlocked macromolecules, e.g., from non-switchable oligomers to switchable high molecular weight polymers. In these mechanically interlocked macromolecules, the mechanical bonds not only link the components together in the same way that covalent bonds do, but they also provide the possibility of controlling molecular motion. These unique properties will enable the future development of actuating materials, conductivity/absorbance switchable polymers, and eventually the fabrication of functional devices on the basis of mechanically bonded macromolecules.Acknowledgements
This work was supported by the US Air Force Office of Scientific Research (AFOSR: FA9550-08-1-0349 and FA9550-07-1-0534) and by the National Science Foundation (CHE-0924620), and the Microelectronics Advanced Research Corporation (MARCO) and its Focus Center Research Program (FCRP) on Functional Engineered NanoArchitectonics (FENA). L. F. acknowledges the support of a Ryan Fellowship from Northwestern University and E. T., a UC MEXUS-CONACyT Fellowship.References
- C. O. Dietrich-Buchecker, J.-P. Sauvage and J.-P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS.
- G. Schill and H. Zollenko, Justus Liebigs Ann. Chem., 1969, 721, 53 CAS.
- C. O. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1989, 28, 189–192 CrossRef.
- K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS.
- B. Hudson and J. Vinograd, Nature, 1967, 216, 647–652 CAS.
- W. R. Wikoff, L. Liljas, R. L. Duda, H. Tsuruta, R. W. Hendrix and J. E. Johnson, Science, 2000, 289, 2129–2133 CrossRef CAS.
- H. Mark, Monatsh. Chem., 1952, 83, 545 CrossRef CAS.
- Interpenetrating Polymer Networks, ed. D. Klempner, L. H. Sperling and L. A. Utracki, American Chemical Society, Washington DC, 1994 Search PubMed.
- K. Endo and T. Yamanaka, Macromolecules, 2006, 39, 4038–4043 CrossRef CAS.
- B. L. Chen, M. Eddaoudi, S. T. Hyde, M. O’Keeffe and O. M. Yaghi, Science, 2001, 291, 1021–1023 CrossRef CAS.
- J. F. Stoddart and H. M. Colquhoun, Tetrahedron, 2008, 64, 8231–8263 CrossRef CAS.
- J. E. Green, J. W. Choi, A. Boukai, Y. Bunimovich, E. Johnston-Halperin, E. DeIonno, Y. Luo, B. A. Sheriff, K. Xu, Y. S. Shin, H.-R. Tseng, J. F. Stoddart and J. R. Heath, Nature, 2007, 445, 414–417 CrossRef CAS.
- K. Patel, S. Angelos, W. R. Dichtel, A. Coskun, Y. W. Yang, J. I. Zink and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 2382–2383 CrossRef CAS.
- B. K. Juluri, A. S. Kumar, Y. Liu, T. Ye, Y.-W. Yang, A. H. Flood, L. Fang, J. F. Stoddart, P. S. Weiss and T. J. Huang, ACS Nano, 2009, 3, 291–300 CrossRef CAS.
- F. M. Raymo and J. F. Stoddart, Chem. Rev., 1999, 99, 1643–1663 CrossRef CAS.
- T. Takata, N. Kihara and Y. Furusho, Polymer Synthesis, 2004, 171, 1–75 Search PubMed.
- F. H. Huang and H. W. Gibson, Prog. Polym. Sci., 2005, 30, 982–1018 CrossRef CAS.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS.
- J. F. Stoddart, Nature, 1988, 334, 10–11 CrossRef.
- D. B. Amabilino, P. R. Ashton, V. Balzani, S. E. Boyd, A. Credi, J. Y. Lee, S. Menzer, J. F. Stoddart, M. Venturi and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 4295–4307 CrossRef CAS.
- D. B. Amabilino, P. R. Ashton, A. S. Reder, N. Spencer and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1994, 33, 1286–1290 CrossRef.
- P. R. Ashton, V. Baldoni, V. Balzani, C. G. Claessens, A. Credi, H. D. A. Hoffmann, F. M. Raymo, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, Eur. J. Org. Chem., 2000, 1121–1130 CrossRef CAS.
- K. Patel, O. Š. Miljanić and J. F. Stoddart, Chem. Commun., 2008, 1853–1855 RSC.
- O. Š. Miljanić and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12966–12970 CrossRef CAS.
- T. Maeda, H. Otsuka and A. Takahara, Prog. Polym. Sci., 2009, 34, 581–604 CrossRef CAS.
- C. W. Bielawski, D. Benítez and R. H. Grubbs, Science, 2002, 297, 2041–2044 CrossRef CAS.
- C. Hamers, F. M. Raymo and J. F. Stoddart, Eur. J. Org. Chem., 1998, 2109–2117 CrossRef CAS.
- H. W. Gibson, M. C. Bheda and P. T. Engen, Prog. Polym. Sci., 1994, 19, 843–945 CrossRef CAS.
- S. Menzer, A. J. P. White, D. J. Williams, M. Belohradsky, C. Hamers, F. M. Raymo, A. N. Shipway and J. F. Stoddart, Macromolecules, 1998, 31, 295–307 CrossRef CAS.
- M. Bria, J. Bigot, G. Cooke, J. Lyskawa, G. Rabani, V. M. Rotello and P. Woisel, Tetrahedron, 2009, 65, 400–407 CrossRef CAS.
- M. A. Olson, A. B. Braunschweig, L. Fang, T. Ikeda, R. Klajn, A. Trabolsi, P. J. Wesson, D. Benítez, C. A. Mirkin, B. A. Grzybowski and J. F. Stoddart, Angew. Chem., Int. Ed., 2009, 48, 1792–1797 CrossRef CAS.
- B. S. Sumerlin, N. V. Tsarevsky, G. Louche, R. Y. Lee and K. Matyjaszewski, Macromolecules, 2005, 38, 7540–7545 CrossRef CAS.
- A. B. Braunschweig, W. R. Dichtel, O. Š. Miljanić, M. A. Olson, J. M. Spruell, S. I. Khan, J. R. Heath and J. F. Stoddart, Chem.–Asian J., 2007, 2, 634–647 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- M. J. Frampton and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 1028–1064 CrossRef CAS.
- A. Harada, J. Li, T. Nakamitsu and M. Kamachi, J. Org. Chem., 1993, 58, 7524–7528 CrossRef CAS.
- F. Cacialli, J. S. Wilson, J. J. Michels, C. Daniel, C. Silva, R. H. Friend, N. Severin, P. Samori, J. P. Rabe, M. J. O'Connell, P. N. Taylor and H. L. Anderson, Nat. Mater., 2002, 1, 160–164 CrossRef CAS.
- C. G. Gong and H. W. Gibson, Macromolecules, 1996, 29, 7029–7033 CrossRef CAS.
- D. Whang, Y. M. Jeon, J. Heo and K. Kim, J. Am. Chem. Soc., 1996, 118, 11333–11334 CrossRef CAS.
- J. Wu, K. C. F. Leung and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17266–17271 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- P. T. Glink, A. I. Oliva, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2001, 40, 1870–1875 CrossRef CAS.
- W. R. Dichtel, O. Š. Miljanić, J. M. Spruell, J. R. Heath and J. F. Stoddart, J. Am. Chem. Soc., 2006, 128, 10388–10390 CrossRef CAS.
- D. B. Amabilino, P.-L. Anelli, P. R. Ashton, G. R. Brown, E. Cordova, L. A. Godinez, W. Hayes, A. E. Kaifer, D. Philp, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, M. S. Tolley and D. J. Williams, J. Am. Chem. Soc., 1995, 117, 11142–11170 CrossRef.
- C. B. Gorman, E. J. Ginsburg and R. H. Grubbs, J. Am. Chem. Soc., 1993, 115, 1397–1409 CrossRef CAS.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- C. Reuter, A. Mohry, A. Sobanski and F. Vögtle, Chem.–Eur. J., 2000, 6, 1674–1682 CrossRef CAS.
- Y. Liu, P. A. Bonvallet, S. A. Vignon, S. I. Khan and J. F. Stoddart, Angew. Chem., Int. Ed., 2005, 44, 3050–3055 CrossRef CAS.
- F. Wang, C. Y. Han, C. L. He, Q. Z. Zhou, J. Q. Zhang, C. Wang, N. Li and F. H. Huang, J. Am. Chem. Soc., 2008, 130, 11254–11255 CrossRef CAS.
- M. C. Jimenez, C. Dietrich-Buchecker and J. P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284–3287 CrossRef CAS.
- J. S. Wu, K. C. F. Leung, D. Benítez, J. Y. Han, S. J. Cantrill, L. Fang and J. F. Stoddart, Angew. Chem., Int. Ed., 2008, 47, 7470–7474 CrossRef CAS.
- E. N. Guidry, J. Li, J. F. Stoddart and R. H. Grubbs, J. Am. Chem. Soc., 2007, 129, 8944–8945 CrossRef CAS.
- L. Fang, M. Hmadeh, J. Wu, M. A. Olson, J. M. Spruell, A. Trabolsi, Y.-W. Yang, M. Elhabiri, A.-M. Albrecht-Gary and J. F. Stoddart, J. Am. Chem. Soc., 2009, 131, 7126–7134 CrossRef CAS.
- P. G. Clark, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 13631–13633 CrossRef CAS.
- A. M. Elizarov, S.-H. Chiu, P. T. Glink and J. F. Stoddart, Org. Lett., 2002, 4, 679–682.
- A. M. Elizarov, T. Chang, S.-H. Chiu and J. F. Stoddart, Org. Lett., 2002, 4, 3565–3568 CrossRef CAS.
- K. C.-F. Leung, F. Aricó, S. J. Cantrill and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 5808–5810 CrossRef CAS.
- K. C.-F. Leung, F. Aricó, S. J. Cantrill and J. F. Stoddart, Macromolecules, 2007, 40, 3951–3959 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |