Dynamic control over cell adhesive properties using molecular-based surface engineering strategies
Jort
Robertus
,
Wesley R.
Browne
* and
Ben L.
Feringa
*
Stratingh Institute of Chemistry and Centre for Systems Chemistry, Faculty of Mathematics and Natural Sciences, University of Groningen, Groningen, Nijenborgh 4, 9747AG Groningen, The Netherlands. E-mail: w.r.browne@rug.nl; b.l.feringa@rug.nl; Fax: +31 50 363 4296; Tel: +31 50 363 4235
First published on 14th October 2009
Abstract
In complex organisms, cells are often dependent on their extracellular matrix (ECM) for structural integrity, the mechanical properties of tissues, and for signaled regulation of cellular processes including adhesion, migration, growth, secretion, gene expression and apoptosis. Achieving dynamic control, i.e. by using an external stimulus, over the interactions between cells and artificial interfaces holds considerable promise in tissue engineering, medicine, cell biology and immunology. For example, improved spatial control over cell–surface interaction is potentially useful in the design of cell-based screening devices. Dynamic control over SAMs for cell adhesion provides an additional handle to direct and study the attachment of cells to surfaces, e.g., in studying cell spreading from a predetermined pattern in order to screen the cytotoxicity of drug candidates. However, ‘reversible’ control of cell adhesion onto substrates is an area that is still in its infancy. In this critical review recent developments in cell adhesion of mammalian cells to SAM-modified surfaces, the physical properties of which can be controlled by an external stimulus, e.g. by light, electrochemistry, etc., are discussed (118 references).
 Jort Robertus | Jort Robertus is a native of Groningen and completed a BSc in Analytical and Organic Chemistry at the Hanzehoogschool Groningen in 2005, and subsequently a MSc in Organic Chemistry at the University of Groningen (The Netherlands) in 2008. He is currently undertaking a PhD degree in responsive organic self-assembled monolayers under the guidance of Prof. Ben Feringa in the Centre for Systems Chemistry at the Stratingh Institute for Chemistry, University of Groningen. |
 Wesley R. Browne | Wesley R. Browne is an assistant Professor at the Stratingh Institute for Chemistry. He obtained a PhD under the guidance of Prof. Han Vos, in 2002, at Dublin City University (Ireland) followed by postdoctoral research jointly with Prof. J. G. Vos and Prof. John J. McGarvey (Queens University Belfast, UK) 2002–2003. In 2003, he was appointed as a postdoctoral research fellow in the group of Prof. Dr Ben L. Feringa. In 2007, he was awarded a VIDI research grant by the Netherlands Organization for Scientific Research. His current research interests include transition metal based oxidation catalysis, electrochromic materials and responsive surfaces. |
 Ben L. Feringa | Ben L. Feringa obtained his PhD degree in 1978 at the University of Groningen under the guidance of Professor Hans Wynberg. After working as a research scientist at Shell, he was appointed Full Professor at the University of Groningen in 1988 and named the distinguished Jacobus H. van’t Hoff Professor of Molecular Sciences in 2004. He was elected foreign honorary member of the American Academy of Arts and Sciences and member of the Royal Netherlands Academy of Sciences (KNAW). In 2008 he was appointed as Academy Professor of the KNAW. His research interests include stereochemistry, organic synthesis, asymmetric catalysis, molecular switches and motors, self-assembly and nanosystems. |
Introduction
In vivo, most mammalian cells adhere to and spread on a biological matrix called the extracellular matrix (ECM). This matrix is largely comprised of proteins as well as other large biomolecules, such as glycosaminoglycans.1 The ECM functions as a scaffold facilitating transfer of signals2 to adhering cells via specific proteins (such as those presenting the RGD ligand; fibrinogen,3 vitronectin,4 collagen5 and fibronectin6), which are recognized by cellular receptors such as integrins.7 These ligands regulate cellular processes including adhesion, migration,9 growth, secretion, gene expression and apoptosis which are triggered, influenced or controlled by the ECM,8 and permit cells to respond to their immediate environment. The ECM provides structural integrity and determines the mechanical properties of tissues by mediating intercellular adhesion. Without adhesion to a specific surface via integrins, cells typically initiate apoptosis.1The study of cells on surfaces is relevant in several scientific disciplines including cell biology, biotechnology, synthetic biology, systems chemistry and medicine.10 At the point of contact of artificial surfaces, with biotic environments, proteins adsorb to the surface rapidly following immersion.10,11 Subsequently, this can be followed by the attachment of cells to these surfaces.10,11 However, the interactions that lead to attachment are generally secondary and depend on the character of the protein layer adsorbed on the surface.10
The interactions between cells and artificial surfaces are of central importance in the field of medical implantation,10–13 for example, in integrating implants by controlling the adhesion of cells, the non-biofouling of implants and in biosensors.14–16 Hence in this regard it is important to improve our comprehension and control of the interface between cells and artificial surfaces.10,11
A major part of our understanding of mammalian cells is drawn from studying cell cultures, and techniques to anchor cells for culturing are now routine.8a Although culturing cells in vitro is convenient, it is difficult to reproduce the organization and function of the ECM.8a,17 Cell-adhesion studies frequently involve glass or polystyrene substrates coated with ECM proteins.18 Although these substrates are convenient,8a their usefulness in mechanistic studies of the interactions between cells and the ECM is limited,19 mainly due to the complexity of these interactions.20 Conventional substrates have a significant degree of surface heterogeneity and several parameters can contribute to the cellular response, for example, the type and distribution of functional groups, hydrophilic and hydrophobic domains, surface roughness, etc.21 Additionally, cells tend to remodel surfaces to better suit their requirements, by secreting their own ECM proteins and carbohydrates that adsorb non-specifically.11,22 This complicates studies of the mechanisms of cell adhesion on conventional substrates further.
Dynamic surfaces for controlled cell adhesion
Despite these challenges surface chemistry provides a useful approach to prepare customized model substrates.20,23 By using self-assembled monolayers (SAMs), it is possible to design model substrates,24 with sufficient control over surface properties, to potentially mimic cell–ECM interactions.10,20,22,25 Introducing a dynamic aspect, i.e. the ability to modify the surface with an external stimulus, opens up many further opportunities in designer surfaces for cell culture and reversible control over surface properties.Dynamic control over SAMs for cell adhesion might provide an additional handle to direct and study the attachment of cells to surfaces.20 For example, it would enable the study of cell spreading from a predetermined pattern to screen the cytotoxicity of drug candidates.26 However, it is not well understood how to ‘reversibly’ control the adhesion of cells to a dynamic substrate.
In this review molecular approaches taken to study and control mammalian cell behavior on synthetic dynamic substrates that mimic the ECM are explored. Recent developments in cell adhesion to SAM-modified surfaces in which the physical properties of the SAM can be controlled externally, e.g. by light, electrochemistry, etc., will be reviewed.20,27 Additionally several systems based upon thin polymer films will be discussed. Thermoresponsive polymers for cell-adhesion applications are, however, beyond the scope of this review.20
Self-assembled monolayers for cell adhesion
Self-assembled monolayers (SAMs) are spontaneously organized assemblies of molecules formed by adsorption from solution or the gas phase onto the surface of solids or liquids (e.g., mercury).25,28–30 The molecules bear a terminus that has a specific affinity for a substrate which is sufficient to displace preadsorbed materials,25 and, indeed, an extensive toolbox of functional groups that bind to specific metals, metal oxides, SiO2 and semiconductors are available.28–30In vivo most cells adhere to the ECM;1 conversely, in vitro SAMs present a method to generate and study model substrates presenting specific ligands via which cells can adhere.10,11,22,28a The most important benefit of using SAMs for these studies, compared to methods that involve polymer films or adsorbed proteins, is the level of control over the exact composition of the substrate achievable via a predetermined approach.10,11,25 These are key aspects that are necessary for conducting mechanistic studies of cell immobilization and investigating intracellular signaling upon binding.25 Before discussing examples of modified surfaces for cell adhesion it is perhaps pertinent to first discuss briefly several issues that arise in preparing the surfaces themselves.
Bioinert self-assembled monolayers
Surfaces that are able to resist the non-specific (physical) adsorption of biomolecules and cells are generally referred to as “bio-inert” surfaces.11,25 Self-assembling compounds terminated with oligo- or poly(ethylene glycol) (OEG or PEG) units are used extensively to render surfaces inert to cell adhesion.31,32 However, the specific structural requirements essential to resist the adsorption of proteins and cells remain unclear.33 Nevertheless, there are several classes of SAMs that inhibit the adhesion of proteins and cells effectively including SAMs terminated with oligosarcosines, oligosulfoxides, perfluoroalkyls, or oligo(phosphorylcholine) groups.34–36Mixed self-assembled monolayers
One of the potential advantages of using self-assembled monolayers is the possibility of constructing SAMs that consist of distinct mixtures of molecular adsorbents, i.e. “mixed” SAMs.25 Mixed SAMs are useful in presenting ligands to immobilize cells via specific interactions.37,38 Mixed SAMs for the immobilization of cells generally consist of a molecule for adhesion (usually at a 0.01–1% substrate density)22 and bioinert molecules that can resist adsorption of proteins and cells. Mixed SAMs can also be used to form gradients that are especially useful for cell-migration studies.8a,39–41Strategies for cell adhesion to artificial surfaces
Cells carry transmembrane proteins called integrins on their surface.7 Integrins are receptors that link cells to the ECM and to other cells physically, and mediate signaling between the cells and the matrix.1,7 They are heterodimeric proteins consisting of α‴ and β‴ subunits, and short cytoplasmic tails.42,43 Adhesion through these integrins is essential to immobilization and for the healthy functioning of cells.1,7Cells can adhere to SAM covered surfaces through several mechanisms.10,38 Cells can be immobilized through preadsorption of ECM proteins, glycans and other biomolecules onto the SAM.21,44 Cells can also be immobilized by covalently binding the adhesion proteins,45 such as fibronectin, to the SAM or by presenting proteins via chelation, for example, via SAMs presenting Ni-NTA and a His-tagged ligand.46 The absence of such proteins attached to a SAM does not necessarily mean that cells will not adhere.11,47 Cells can, and generally do, excrete their own ECM proteins to adhere and ensure their survival.
Cells can also be immobilized via non-specific interactions48 such as hydrophobic interactions between the cell membrane and a SAM.11,47 However, cells tend to remodel their immediate environment on these types of surfaces over time.11,38 An alternate approach is to present SAMs of integrin binding peptide sequences,49 commonly the arginine-glycine-aspartic acid (RGD) peptide domain is used.10,18,22,38,50 In this approach cells are immobilized onto SAM covered surfaces by targeting integrins with a binding ligand presented on the SAM surface, since this ensures the viability of the cells growing on the surface.51
Dynamic control of cell adhesion on SAMs
Dynamic SAM substrates for immobilizing cells on-demand present new possibilities in studying the mechanistic pathways involved in responses to alterations in cells’ immediate environments.22 Dynamic changes in the ECM affect cell behavior at several critical stages.8 Among these are the migration and differentiation of cells during the growth and spread of cancer.22 Our understanding of the dynamic processes which occur in the ECM is limited, due to the their complexity,7 and hence studying dynamic substrates that allow for explicit control over changes of the cell-adhesion properties may lead to improved understanding of the ECM as well as cell adhesion to surfaces.Developing dynamic SAM substrates starts with the design of a strategy to “switch”52 the composition of cell-binding ligands on a substrate surface,20 between an active and an inactive state. The switching could involve isomerization of an immobilized ligand, or a substrate can be ‘turned on’ by binding a ligand, while it is ‘turned off’ by releasing a ligand from the substrate.22 Dynamic control over cell adhesiveness has been achieved using these strategies employing electro-, photo- or chemical stimuli,20 at the cell–surface interface.53 Non-invasive methods, such as photochemical or electrochemical stimuli, are potentially interesting since the nature of the substrate surface properties can be switched rapidly.22
Dynamic control over cell-surface adhesiveness based on SAMs is of increasing importance in the design of biomolecular materials and biosensors.20,54 Materials that mimic the dynamic properties of the ECM that surround cells in vivo could be capable of changing surface properties in response to an applied stimulus, with the ultimate aim being the control and direction of cell behavior.20 To date, responsive surfaces have been developed to respond to changes in electric field strength, light, temperature, solvent polarity, ionic strength, or the presence of small molecules or biomolecules.20,22 However, modification of a substrate, after the cells have adhered to the surface, is highly challenging.55
Electrochemical desorption
So how can these types of surfaces be applied? The behavior of cells under the influence of bioactive molecules can be studied by monitoring their migration behavior upon release from pre-patterned zones,56 from which they are then free to migrate, to, e.g., determine the cytotoxicity of a drug.26 This can be achieved by removing bioinert barriers that confine cells to a predetermined shape on a surface patterned by microcontact printing (μCP).56,26Whitesides and co-workers have reported a method for screening drug candidates by evaluating the migration rates of pre-patterned micron dimensioned ‘islands’ of bovine capillary endothelial (BCE) cells after electrochemical desorption of the bio-resistant PEG alkanethiol pattern.56 This procedure relies on μCP and readily available alkanethiols such as HS(CH2)17CH3 (C18) and HS(CH2)11(OCH2OCH2)3OH (C11EG3) to confine cells within zones of the monolayer.57,58
Cells were confined by micropatterns in a growth medium for 24 h. Application of a negative potential (<−1.2 V, for 30 s) to the gold substrate resulted in partial or complete desorption of the C11EG3 SAM.56 In the absence of the inert SAMs, adsorption of ECM proteins such as fibronectin onto the gold surface is possible,59 through which cells can attach to, and spread across the, previously, inert areas (Fig. 1). The cells migrated as if migrating over the gold surface with a layer of preadsorbed proteins.56 The BCE cells appeared to be unaffected by the application of a negative potential to the surface.
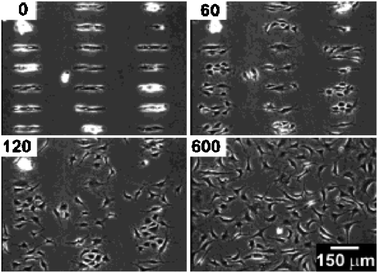 | ||
| Fig. 1 Migration of BCE cells on a mixed SAM consisting of C11EG3 and C18, upon release of the pattern by application of a voltage pulse (−1.2 V for 30 s). Reproduced from ref. 56. Copyright ACS 2003. | ||
Studying the mechanism of cell migration
Most mammalian cell types can polarize and translocate in the absence of an external stimulus.60,61 The teardrop shape is adopted by many mobile mammalian cell types. However, it is unclear whether the cell’s shape determines the direction of the displacement.26The migration of mammalian cells proceeds typically through five individual events (Fig. 2):26
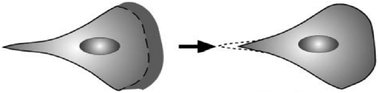 | ||
| Fig. 2 Illustration of a teardrop shaped migrating mammalian cell. Reproduced from ref. 26. Copyright NAS 2005. | ||
(a) Polarization of the cell, e.g. by changing to a teardrop form.
(b) Extension of the membrane in the direction of movement.
(c) Anchoring of the leading edge membrane to the substrate surface.
(d) Displacement of the cell body.
(e) Release of the anchorage from the substrate surface at the now pointed trailing edge.26,60
Electrochemical desorption of inert OEG-terminated thiol SAMs56 allows the influence of cell shape in determining the primary direction of movement in the absence of a stimulant gradient to be addressed.26
From the studies by Whitesides and co-workers it was apparent that, by defining the polarity of cells adhering to SAMs in a teardrop shape by means of micropatterning, and the subsequent release of the asymmetric constraint, the cell’s shape has an effect on the initial direction of cell translation. It was established that cells migrate in the direction of their blunt end, preferably.
Recently, Revzin and co-workers reported a related method to pattern model T-lymphocytes (T-cells) on antibody-presenting SAMs of alkanethiols on gold electrodes.62 Leucocytes are white blood cells which react to the presence of malignancies, infections, and in autoimmune disorders in the body.63 Among the lymphocytes, T-cells are a vital branch as they coordinate the immune response. Diseases such as HIV/AIDS obliterate CD4+ T-cells compromising the immune system of the host and increasing the susceptibility to opportunistic infections.64,65 As CD4+ T-cells are of pivotal importance to the immune system, novel devices capable of rapid capture and characterization of cells are a key goal.62 Generally such cytometric devices utilize antibody functionalized surfaces to capture cells, which can be assessed after immobilization (Fig. 3).66
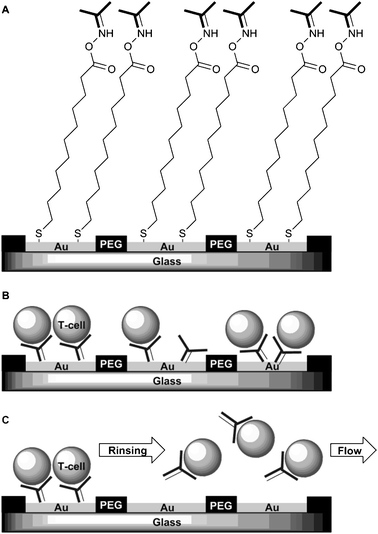 | ||
| Fig. 3 T-cell immobilization and subsequent release. Step A: covalent binding of anti-CD4 antibodies on gold microelectrodes via COOH-terminated alkene thiols. Step B: immobilization of T-cells via the antibody-presenting SAM. Step C: application of a reductive potential (−1.2 V vs. Ag/AgCl reference) desorbs the alkanethiols and results in release of T-cells.62 | ||
The objective of the study by Revzin et al. was to develop a method for releasing captured cells from the surface of the device, as this is of interest for analysis and re-culturing in microfluidic systems.62 Towards this goal, they immobilized T-cells on SAMs presenting anti-CD4 antibodies on the surface. This antibody ensures specific adhesion of T-cells to the gold electrode. Non-specific adhesion to the glass surface surrounding the gold electrodes was prevented by coating with bioinert PEG-terminated alkane silanes.
Application of a reductive potential (−1.2 V vs. Ag/AgCl reference electrode) to the gold electrode resulted in desorption of the antibody functionalized alkanethiols and subsequent release of the T-cells from the surface. By using gold microelectrodes on an insulating glass surface, it is possible to direct cell adhesion on the gold electrodes independently from each other (Fig. 4). This procedure provides a simple method, employing readily available alkanethiols. Microfabricated devices that allow the immobilization of antigen-specific cells and subsequent release from the surface after examination of the adherent cells present a potentially valuable tool in immunology and oncology. In these fields diagnostics often involve the screening for malignant cells or monitoring the numbers and/or the function of customary cells.
 | ||
| Fig. 4 Discharge of T-cells from antibody-modified microelectrodes. (A) Model T-cells seeded onto gold electrodes (300 μm ∅) surrounded by a PEG-terminated SAM on glass. Cells cover both the gold electrode and the bioinert regions. (B) Upon washing, only the antibody-presenting regions are covered by T-cells. (C) Application of a reductive potential (–1.2 V vs. Ag/AgCl reference) for 60 s followed by gentle agitation of the substrate liberated the T-cells. (D) Image of three separately addressable microelectrodes. T-lymphocytes were immobilized on all electrodes, however, the T-cells of the upper and lower electrodes were released by applying a reductive potential, while the central electrode remains in its cell adhesive state. A viability study by staining the released T-cells with trypan blue revealed ±90% of the cells remained viable. Reproduced from ref. 62. Copyright Elsevier 2008. | ||
Although gold surfaces are convenient in cell-adhesive strategies a major drawback is the stability of SAMs. Therefore the use of other electrode materials is desirable. Revzin and co-workers have reported the control of cell adhesion on indium tin oxide (ITO) electrodes by the controlled reductive release of a protein resistant PEG silane SAM from the ITO electrodes (Fig. 5).67 Several hepatic cell lines (HepG2 cells) were immobilized side by side in a sequential fashion on the surface of ITO coated glass. The adjoining PEG covered glass surface retained its bio-resistant character.
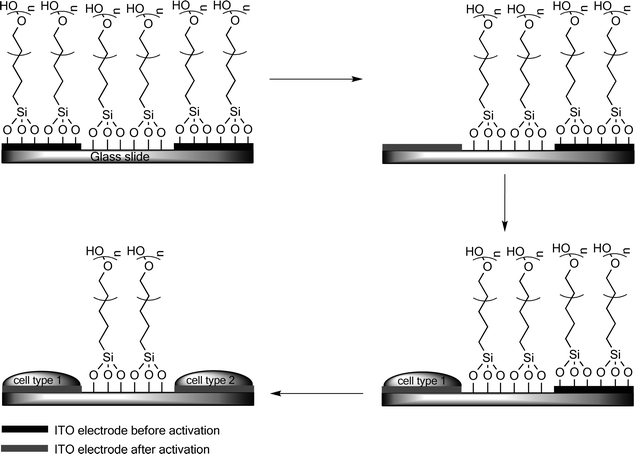 | ||
| Fig. 5 Electrochemical switching of the cell adhesiveness of ITO electrodes. Step 1: glass is modified with micro-ITO electrodes and covered with PEG-terminated silanes to make the surfaces non-adhesive. Step 2: a reductive potential is applied for 60 s to a specific electrode resulting in desorption of the PEG silane SAM from the ITO electrode. The PEG SAM on the untreated ITO electrodes and the glass surface remain intact. Step 3: cells adhere selectively to the exposed ITO surface upon their introduction, while not adhering to the PEG covered glass regions and untreated ITO electrodes. Step 4: subsequent to the immobilization of the first cell line a second electrode can be activated by the application of a negative potential and a second cell line may be introduced on the same surface.62 | ||
Desorption of the PEG-terminated silanes from the ITO electrodes could be detected by cyclic voltammetry with potassium ferricyanide as a redox probe. Disappearance of the redox response subsequent to assembly of the PEG-terminated silane SAM (Fig. 5, step 1) indicated the formation of an insulating layer on the ITO electrode surface. Reduction resulted in a recovery of the redox response of ferricyanide, indicating desorption of the PEG-terminated monolayer. The selective desorption was verified by water contact angle measurements. Furthermore it was possible to verify the “switching” of the surface by adhering collagen labeled with fluorescent units to the electrochemically activated surface of the electrodes. This approach holds considerable potential as a facile approach to immobilizing multiple cell types, with precise control over the geometry on the surface, possibly permitting the construction of complex arrays of multiple cell types in the future.
Most of the examples summarized in this section depend on complete or partial electrochemical desorption of the bioinert monolayer covering the sample surface. A disadvantage of the electrochemical desorption of a bioinert barrier is that it allows the adherent cells to reorganize the surface. This results in reduced control over cell–surface interactions. In the case of patterns formed by electrochemical desorption adherent cells can remodel the bioinert regions, which can lead to unwanted spreading of the adherent cells.
Additionally the methods described above are limited to the size and the shape of the electrode surface. Although examples have been shown where several electrodes decorate a surface, which can be addressed individually, it remains a challenge to alter the shape or size of existing electrodes or add new electrodes (using the same fabrication method), once the electrode substrate is fabricated.
Oxidative release
Wittstock and co-workers have reported an in situ method for manipulating the bioadhesive properties of oligo ethylene glycol (OEG)-terminated thiol SAMs using ultra microelectrodes (UME).68,69 This strategy is based on the switching of bioinert OEG-terminated SAMs to cell-adhesive SAMs by exposure to oxidizing agents such as Br2. The bromine can be generated electrochemically in aqueous media (Fig. 6).70 This overcomes the need to prepattern surfaces with electrode materials, e.g. ITO, gold, etc., and instead a large area electrode can be custom patterned. The limitation in terms of reversibility remains however.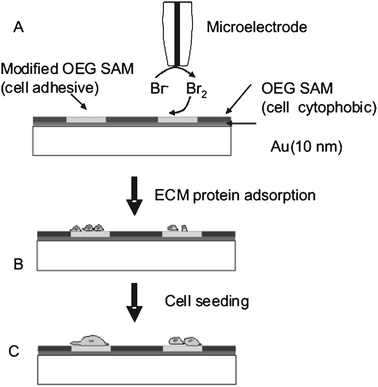 | ||
| Fig. 6 Induced cell adhesion using a microelectrode on an OEG SAM substrate. (A) Modification of the OEG monolayer by electrogenerated Br2. (B) Selective adsorption of fibrinogen-Alexa 488 onto a modified region of the monolayer. (C) Attachment of, e.g., human fibroblasts onto pattered regions. Reproduced from ref. 68. Copyright Wiley 2006. | ||
The dimensions of the micropatterns is strongly dependent on the size of the UME. The electrochemically generated Br2 diffuses to the substrate where it reacts locally with the OEG-terminated SAM. Furthermore the oligo ethylene units are degraded, while the alkyl chains remain intact for a longer period. It has been reported that preadhered cell cultures are not damaged significantly by Br2 or the side products of the reaction.
Recently Wittstock et al. investigated the molecular and electrochemical mechanisms of the degradation of OEG-terminated SAMs by electrochemically generated Br2 by scanning electrochemical microscopy (SECM).71 Polarization-modulation Fourier transform infrared reflection-absorption spectroscopy (PM FTIRRAS)72 indicated that treatment of the monolayer by locally generated Br2 removes the OEG-terminated units from the SAM within the first seconds of the reaction, whereas the alkyl tails on the surface degrade at a slower rate. X-Ray photoelectron spectroscopy indicated that bromine was not quantitatively incorporated in the modified SAMs.
The degradation of the OEG-terminated monolayers was proposed to proceed via a radical mechanism.72 PM FTIRRAS confirmed that the helical crystalline structure of the OEG degrades within seconds. However after rapid initial damage of the monolayer the mechanism changes as the SAM becomes more permeable. At this point the system operates as a galvanic cell, wherein a substantial part of the electrochemically generated Br2 is consumed by heterogeneous electron transfer to the gold surface once the monolayer becomes permeable. This results in a limit to the size of the modified region.
A similar technique was employed by Nishizawa and co-workers, to control cell adhesiveness on a surface pretreated with the cell-adhesion resistant BSA protein on glass and quartz.73–75 The adhesion resistant BSA-coated surface is switched to a cell-adhesive surface by electrochemical generation of oxidizing hypobromous acid, which releases the BSA protein from the surface. Subsequent adsorption of ECM proteins, such as fibronectin, allows for the adhesion of HeLa cells to the surface. In addition physisorbed cell-repellent heparin could be removed with a high degree of spatial control from a glass surface by electrochemically generated HBrO− or Br2 at an UME,76,77 enabling the study of cell motility as described above.71 Although this method enables fast patterning of cell cultures on gold substrates in ways that are not possible by fabricating patterned electrodes, it experiences similar disadvantages as electrochemical desorption methods: specifically the direct interaction between the substrate surface and the cell, which might lead to surface remodeling by the cells.
Light controlled desorption
Controlling cells by removing the bioinert barrier facilitates studies concerning the migration behavior of cells. However, desorption of the bioinert SAMs from a surface allows cells to roam, impeding efforts to determine the interactions that are essential to a cell’s viability.Using irradiation with light to alter the cell adhesiveness of a substrate surface allows for patterning of cell cultures in various shapes and sizes without compromising the bioinert character of the non-irradiated regions. Additionally this permits the alteration and addition of the substrates cell-adhesive regions at will. However, these types of strategies often involve the use of UV light which can damage both monolayers and adherent cells.
Nakanishi et al. have reported a method for dynamic control of cell adhesion to predetermined regions on a substrate by photoactivation under a standard fluorescence microscope, without compromising the overall inertness of the surface.78 Their strategy was based on a silane grafted SAM containing a photocleavable 2-nitrobenzyl group (Fig. 7).79 Additionally they used proteins that either inhibit or promote cell adhesion (e.g., bovine serum albumin and fibronectin, respectively). A glass coverslip was modified with 1-(2-nitrophenyl)ethyl-5-trichloro silylpentanoate (NPE-TCSP).80 Subsequently the SAM substrate was coated with non-specifically adsorbed bovine serum albumin (BSA), to render the surface inert to cell adhesion. Irradiation of the coverslip at λ = 365 nm led to photocleavage of the 2-nitrobenzyl group (Fig. 7), which changes the hydrophilicity of the surface by changing the nitrobenzyl ester-terminated SAM to a carboxylic acid-terminated SAM.
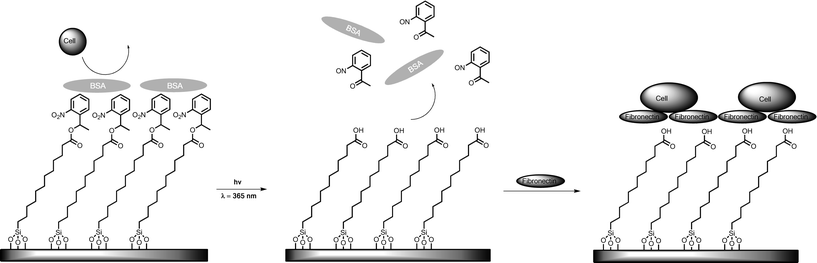 | ||
| Fig. 7 UV-directed elimination of BSA and subsequent adhesion of fibronectin changes surface adhesiveness from a state that prevents cell adhesion to a state that promotes cell adhesion.78 | ||
BSA is expected to be adsorbed via hydrophobic interactions onto the nitrobenzene-presenting SAMs. Photocleavage results in an increase in the hydrophilicity, which diminishes the affinity for BSA to adhere, and results in the protein dissociating from the surface. The addition of fibronectin led to its non-specific adsorption to the carboxylic acid group presenting regions. Subsequently HEK293, COS7 and NIH3T3 cell lines could be adhered. By repeating the technique on another section of the substrate it was possible to pattern an additional cell culture of HEK293 cells onto a sample containing an existing pattern of COS7 cells (Fig. 8).
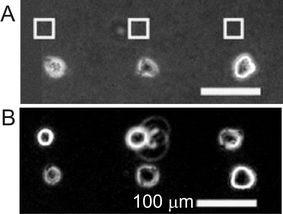 | ||
| Fig. 8 Phase-contrast images of UV-directed placement of individual cells in proximity to pre-attached cells. (A) Substrates before and (B) after seeding of a second cell culture of fluorescent HEK293. Boxes indicate regions that were irradiated. Reproduced from ref. 78. Copyright ACS 2004. | ||
It is useful to form cell-adhesive islands that are smaller than single cells to study the formation of focal adhesions (FA) and organization of cellular cytoskeletal structures in cells on SAMs.8b,81 Cells form FA at the interface with the ECM to convey signals between the ECM and the cytoskeleton.82 Irradiation at λ = 365 nm through a photomask of a SAM-presenting NPE-TCSP groups83 was employed in the formation of subcellular-sized 6 μm2 adhesive islands (Fig. 9). HEK293 cells were found to form FAs shaped as nodes on the adhesive islands, whilst their bulk was extended across the non-adhesive areas, confirming the formation of cell-adhesive islands of subcellular size.
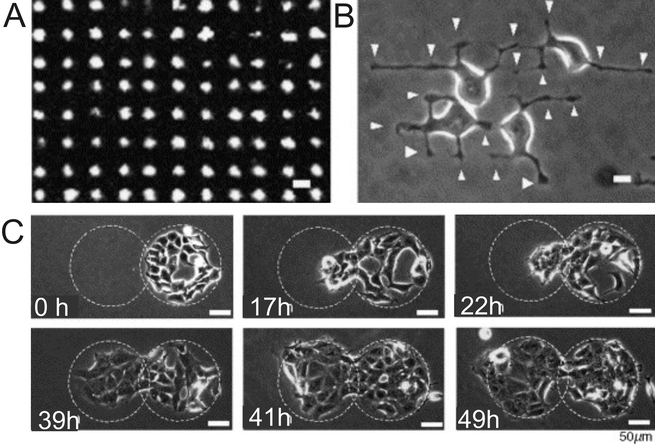 | ||
| Fig. 9 (A) Subcellular size pattern for photoactivation. (B) Phase-contrast image of HEK293 cells 2 h after seeding. Arrows correspond to nodal structures of cells on the irradiated pattern. (C) Phase-contrast images of induced cell migration on the SAM after photoactivation of an adjoining region next to a preadhered culture of HEK293 cells over time (a–f). Reproduced from ref. 83. Copyright Elsevier 2006. | ||
Upon irradiation with UV light, HEK293 cells were seeded and cultured for 28 h on circular islands formed by photo-deprotecting the alkane silanes.83 Irradiating a second circular region adjoining the original circular cellular pattern resulted in release of the spatially constrained cells. This allowed the cells to migrate toward the newly formed cell-adhesive region with the number of cells increasing through proliferation without additional fibronectin (Fig. 9).83
Notably, Nakanishi et al. were able to study the initial step in the migration of single NIH3T3 cells by using a similar method to those described previously for photocleavable SAMs,78,83 permitting quantitative analysis of the rates of extension of cell protrusions.84 For this cell type, extension of wide protrusions is led by lamellipodia85 and thin stretched out protrusions led by filopodia.86
A glass surface was coated with a SAM of 1-(2-nitrophenyl)-ethyl-11-trichlorosilylundecanoate as described above. This SAM was coated with Pluronic F108, a tri-block copolymer surfactant terminated with primary hydroxyl groups. 25 × 25 μm square spots were formed by irradiation of the bioinert alkanethiols through a photomask. This produced small protein and cell-adhesive islands onto which single cells were allowed to immobilize themselves. Migration of individual cells could be induced by irradiating regions adjoining the pre-patterned cells at λ = 365 nm (Fig. 10). Irradiation led to localized desorption of the bioinert SAM. Cell adhesion to the substrate is driven by the increase in surface hydrophilicity, which allows for adsorption of fibronectin. Irradiation of a 25 μm wide rectangular section resulted in the cells spreading through extension of lamellipodia, while 5 μm wide paths resulted in the cells extending filopodia at the leading edges, i.e. developing thin protrusions along the irradiated pathways (Fig. 11).
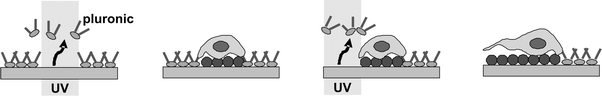 | ||
| Fig. 10 Illustration of the control of cell migration on the photoactivatable cell-culturing substrate. Reproduced from ref. 84. Copyright ACS 2007. | ||
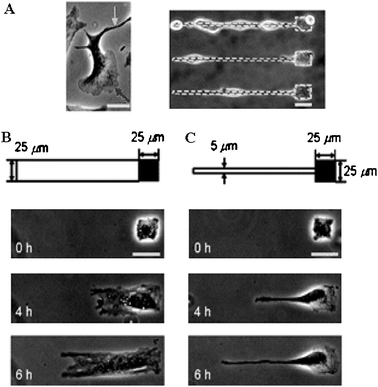 | ||
| Fig. 11 Selective induction of protrusions in NIH3T3 cells, (A) led by a lamellipodium and by a filopodium on a fibronectin-coated non-patterned surface. Selective induction of protrusions by irradiating a path (B) 25 μm or (C) 5 μm wide on the SAM adjoining the pre-attached cells. Reproduced from ref. 84. Copyright ACS 2007. | ||
Compared to the electrochemical and oxidative methods described (vide supra), where the whole surface pattern is disrupted by an external stimulus, this photoactivated method78,83 provides increased spatial control over the patterning of cells on SAM substrates and even allows the size, shape and number of adherent regions to be altered in a fast and relatively simple fashion without disturbing established cultures of cells.
However, these substrates still depend on non-specifically adsorbed proteins for cell immobilization and patterned regions are therefore still susceptible to surface remodeling by adherent cells through secretion of ECM proteins. Del Campo and co-workers recently reported photocontrolled cell adhesion to self-assembled monolayers presenting a cyclic-RGD integrin binding peptide.87 In this particular case the cell-adhesion mediating peptide is caged using a photo labile 3-(4,5-dimethoxy-2-nitrophenyl)-2-butyl ester (DMNPB) protecting group on the carboxylic acid side chain of the aspartic acid residue (Fig. 12). This residue acts as a ligand for the divalent cation binding sites of this integrin.50,88 It was suggested that the caging group might introduce steric hindrance, conformational restriction or changes in the charge distribution of the peptide, resulting in the inhibition of recognition of the peptide by the integrin.
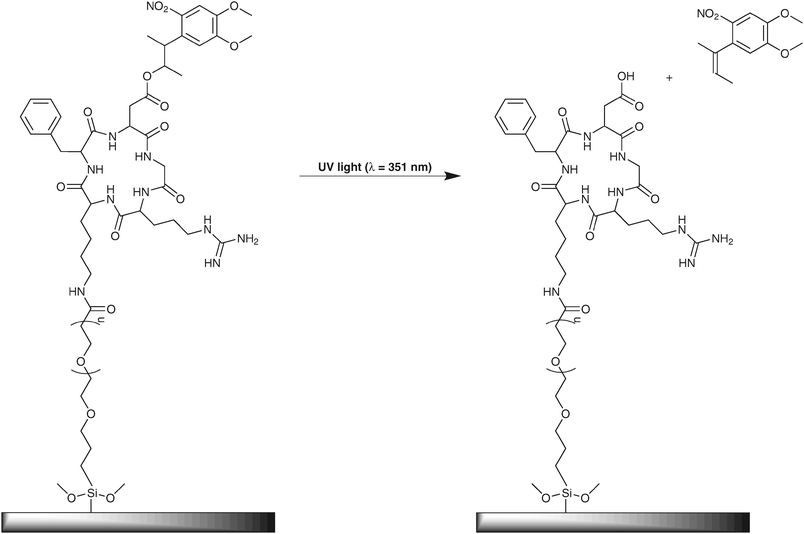 | ||
| Fig. 12 c-(RGD(DMNPB)fK) on a silica gel surface through the tetra oligoethylene glycol spacer. The photo labile caging group is removed upon irradiation at λ = 351 nm.87 | ||
The cyclic pentapeptide c-(Arg-Gly-Asp-D-Phe-Val-), c-(RGDfK), is especially selective at binding the αvβ3 integrin,89 the Lys residue of which can be used to couple the peptide to the OEG silane. This does not affect the activity of the peptide towards the integrin.89 Haubner et al. have employed the ligand c-(RGD(DMNPB)fK) by coupling through the free amine group of the Lys residue to a tetra(ethylene glycol) (TEG) linker that contains a triethoxysilane head group. This was then immobilized on a silica surface (Fig. 12). Coupling of the pentapeptide and the self-assembled monolayer of the TEG-terminated silane on silica was monitored with UV/Vis spectroscopy, contact angle measurements and ellipsometry.
Irradiation of a c-RGD(DMNPB)fK-modified surface at 351 nm resulted in cleavage of the (DMNPB) protecting group. Subsequent rinsing of the surface ensures complete removal of the photocleaved protecting group, however the extent of cleavage was limited to 64% even under prolonged irradiation.
Fibroblast 3T3 cells were incubated for 3, 6 and 21 h on both non-irradiated and irradiated (351 nm, for 10 min) c-RGD(DMNPB)fK monolayers (Fig. 13). Limited adherence to the protected c-RGD(DMNPB)fK-presenting surface was observed, comparable to the number of cells adhering to TEG-modified surfaces, with spreading of the cells beginning only after 21 h of incubation. By contrast the deprotected RGD containing cyclic peptide is active towards the binding of integrins (Fig. 14). Further incubation results in increased cell coverage over the SAM outside of irradiated areas, however this may be due to the absorption of proteins from the medium onto the SAM covered surface.
![Density of fibroblast 3T3 cells on non-patterned and patterned substrates presenting c-[RGD(DMNPB)fK] prior to and after irradiation with UV light at λ = 351 nm for 10 min. The tetra oligo ethylene glycol linker is included for comparison. Reproduced from ref. 87. Copyright Wiley 2008.](/image/article/2010/CS/b906608j/b906608j-f13.gif) | ||
| Fig. 13 Density of fibroblast 3T3 cells on non-patterned and patterned substrates presenting c-[RGD(DMNPB)fK] prior to and after irradiation with UV light at λ = 351 nm for 10 min. The tetra oligo ethylene glycol linker is included for comparison. Reproduced from ref. 87. Copyright Wiley 2008. | ||
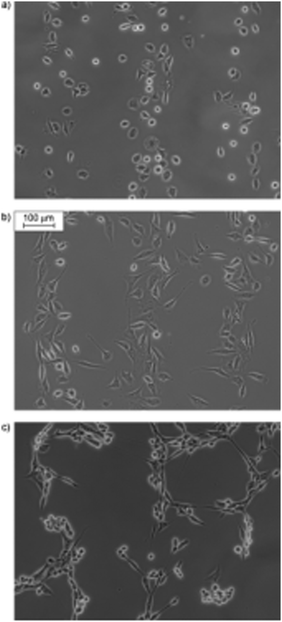 | ||
| Fig. 14 Optical microscopy images of in situ one-way control over cell adhesion to substrates presenting c-(RGD(DMNPB)fK). Cells were seeded onto the substrates presenting the caged peptide. Subsequent irradiation through a photomask (100 μm wide lanes) resulted in deprotection of the aspartic acid residue. Cells adhere preferentially to the irradiated regions, presenting the unprotected cyclic RGD sequence. The fibroblast cells were incubated for 3 (a), 6 (b), and 21 h (c). Reproduced from ref. 87. Copyright Wiley 2008. | ||
This study indicates that the caging of the carboxylic group of the aspartic acid residue is an effective method to inhibit binding of integrins to the RGD sequence. The phototriggered method of cell adhesion described by the del Campo et al. is an effective method to gain spatiotemporal control over the positioning of cell adhesion on the SAM substrate using specific adhesion through RGD-integrin binding. Specific immobilization of cells on a SAM is in some cases preferable to cell adhesion through non-specifically adhered protein, as these latter systems are more vulnerable to surface remodeling by adhered cells. However, as the authors note it is necessary to further increase the selectivity of the binding to the surface by preventing adhesion of cells outside the irradiated patterns over extended incubation times, as can be observed (Fig. 14).
Reversible SAM photoswitching of RGD availability
Recently, Liu et al. reported a SAM system based on azobenzene photochromic switches which allowed for control of the availability of an RGD cell-binding domain to gain light responsive control over the cell adhesiveness of the substrate surface.90 This is the first example where an azobenzene switching unit is used in a SAM to gain dynamic control of the cell adhesiveness of a surface. The design of the RGD containing switching moiety is related to the approach taken earlier by Kessler and co-workers.91 The azobenzene unit can be switched from the thermally stable E-isomer to the thermally unstable Z-isomer, by irradiation of UV light (340–380 nm). This photochemical isomerization usually proceeds to a photostationary state (PSS) of 70–90%.92 Subsequently the Z-isomer can revert back to the E-isomer either thermally or by irradiation with visible light (450–490 nm). In the E-isomer a RGD containing ligand is presented above the SAM-modified surface, whereas isomerization to the Z-isomer masks the cell-binding ligand. Utilizing reversible photochromic switches as a light responsive handle can in principal allow for reversible control of the level of cell adhesiveness of the SAM.Liu et al. fabricated mixed monolayers consisting of N-hydroxysuccinimide(NHS)-presenting azobenzene containing alkenethiol and oligo ethylene glycol-terminated alkenethiol. The NHS-presenting mixed SAMs were subsequently reacted with an GRGDS cell-binding peptide via the NHS-activated ester.90 The monolayers could be prepared presenting different densities of RGD sequence containing ligands (0.01, 0.1 and 1%) by varying the density of the NHS-presenting molecules on the surface. All the chemical processes on the surface were performed under the exclusion of light to ensure the azobenzene unit remained in the E-isomeric form.
In the E-isomer form, these SAMs supported cell adhesion of murine NIH 3T3 cells, while in the Z-isomer the cell binding was diminished significantly. This effect was most notable for SAM surfaces presenting the RGD containing ligand at a 0.1% density. The adherent cells could be detached by adding the GRGDS peptide (1 mg mL−1) at pH 8. The peptide in solution competes with the surface-bound GRDGS peptide for binding to the cell’s integrin receptors. After approximately 30 min the cells detached, and the azobenzene unit could be switched to the unstable Z-isomer by irradiation with UV light for 10 h, effectively masking the RGD containing ligand within the OEG-terminated SAM. Irradiating the now cell-adhesion resistant SAM with visible light results in a photochemical Z to E-isomerization, leading to renewed presentation of the GRGDS peptide above the gross of the SAM surface, once again allowing cell adhesion. This cycle can be repeated several times, reverting the surface to its original state after two irradiation steps.
This approach allows for renewal of the cell-culture surface, by utilizing RGD-modified azobenzene switches in a SAM. Though the control over cell adhesion to the surface is reversible, it still relies on an additional step to detach the adhered cells from the surface. In this way the method does not utilize the full potential of the azobenzene switch, that is, to utilize the mechanical motion that is associated with the E to Z-isomerization of the photochromic azobenzene switch to detach the adhered cells from the surface as was demonstrated by Kessler et al. (vide infra).91 Nevertheless, this approach demonstrates the potential of using SAMs to achieve control over the distance and orientation at which a ligand is presented.
Using light to alter the cell adhesiveness of the surface, although offering considerable freedom in the choice of substrate, shape and size, is limited only by the availability of a photomask or multipulse patterning technique to generate patterns. This circumvents the need for microfabrication, however, long exposure of monolayers to UV light may be problematic. Employing UV light to alter cell adhesiveness of photoresponsive SAMs near or adjacent to preadhered cells or cell cultures could lead to damage and/or mortality of irradiated cells. Moving from UV photochromes to photoswitches which utilize visible light, however, will certainly circumvent this issue.52,93
Electrochemically controlled tethering of peptide ligands
By designing and synthesizing dynamic SAM covered substrates that present peptide ligands, it is possible to obtain more control over the interactions between cells and SAM-coated surfaces. However, generating SAMs from large and complex adsorbents allows for only limited control over surface density and defects in the ordering of the surface will be present by default. Such defects would facilitate adhesion of proteins and cells, limiting the value of information and level of control obtained on cell–surface interactions. An alternative approach to achieving control over surface density is to first generate a SAM terminated with a small reactive group, followed by immobilization of the desired ligand.Electrochemically controlled cell adhesion
In their pioneering work in the field of electrochemical adhesion control Mrksich and co-workers reported a dynamic substrate for cell adhesion that can “switch on” adhesion ligands selectively under an electrical stimulus (Fig. 15 and 16), inducing the migration and proliferation of Swiss 3T3 fibroblast cells.94 This system is based on electrochemical generation of a quinone dieneophile for RGD peptide binding via a Diels–Alder cycloaddition.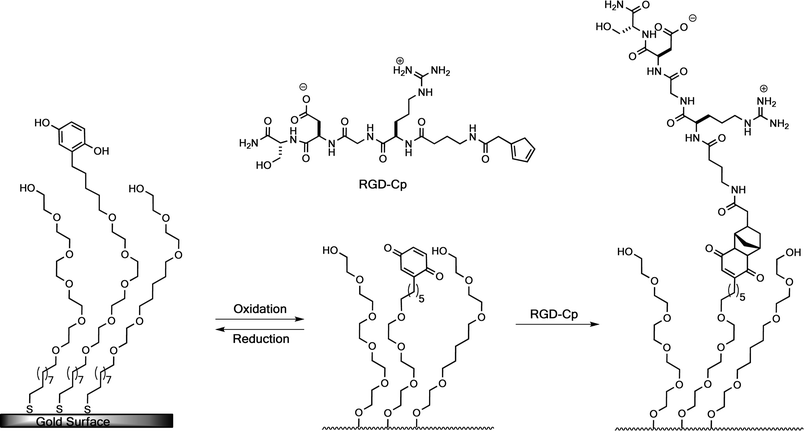 | ||
| Fig. 15 Electrochemical switching of cell adhesion. A SAM-presenting hydroquinone and OEG groups (left) is converted to a SAM-presenting quinone groups (center) by application of a potential. Both monolayers are inert to the adhesion of cells. Addition of a cyclopentadiene-functionalized peptide allows immobilization of the RGD peptide through a Diels–Alder reaction (right) to yield a cell-adhesive surface.94 | ||
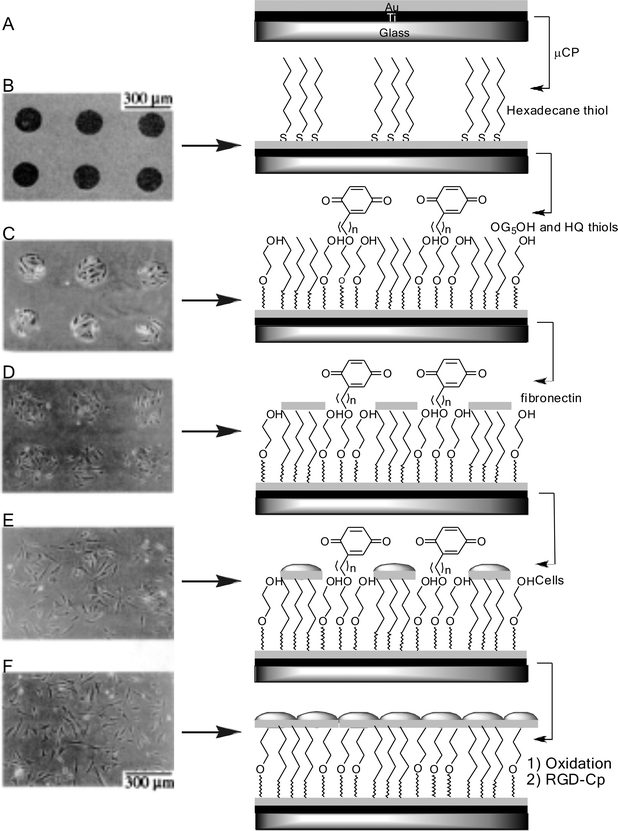 | ||
| Fig. 16 Switching cell migration and growth on an electroactive substrate. (A) The substrate was prepared by evaporating titanium and gold onto a glass coverslip. (B) Microcontact printing was used to pattern hexadecane thiolate onto the gold surface. (C) A second mixed monolayer was assembled onto the exposed regions by immersing the substrate in a solution of hydroquinone-terminated (HQ) and oligo ethylene glycol (OEG)-terminated alkanethiol. (D) Fibronectin was physisorbed to the CH3-terminated SAM. (E) 3T3 fibroblast cells adhered exclusively to the circular regions presenting fibronectin. (F) Electrochemical oxidation of the HQ SAM in the presence of serum-free media containing RGD–Cp (2 mM) led to the immobilization of the peptide and subsequent migration of cells from the circular regions. Adapted from ref. 94. Copyright Wiley 2001. | ||
Specific interaction of cells with the surface could be demonstrated through release of the adherent cells from the substrate by adding soluble Gly-Arg-Gly-Glu-Ser (GRGDS) to the growth medium. This substrate holds potential as a novel screening technique for the detection of pro-migratory and anti-migratory compounds.
Furthermore, this method offers an excellent opportunity to investigate the mechanisms involved in cell adhesion to surfaces in a controlled environment. Cell adhesion to surfaces consisting of either low-affinity or high-affinity ligands for binding integrin receptors has been compared. However, it is essential to present the ligands at a uniform density, given that small variations in ligand density can affect cell immobilization and proliferation dramatically.95 The customary methods for preparing mixed monolayers are restricted due to the ratios of alkane thiolates in solution typically not comparing well to the ratio of alkanethiols in the monolayer. This is especially the case where the ratio of the alkane thiolates in the monolayer depends heavily on the structure of the terminal group.
Using the method described above, Kato and Mrksich prepared RGD and cyclic RGD-presenting substrates. This strategy allows peptides to be presented at a uniform density across the substrate. The Diels–Alder immobilization of peptide–diene conjugates onto SAMs presenting electrochemically derived quinone groups allow the dependence of the affinity for integrin–ligand complexes on the formation of focal adhesions to be examined.96
By combining analogous electrochemical and Diels–Alder cycloadditions Feng and Mrksich immobilized peptide ligands such as RGD and Pro-His-Ser-Arg-Asn (PHSRN) and demonstrated that 3T3 Swiss and IMR 90 fibroblasts adhere to and spread on monolayers presenting RGD and PHSRN.97 A blocking experiment using anti-integrin antibodies confirmed that IMR 90 fibroblast attachment to PHSRN can be inhibited by either anti-integrin α5 or anti-integrin β1 antibodies. This demonstrated that both PHSRN and RGD peptide sequences can maintain the integrin-mediated attachment of cells. Furthermore it was established that the two ligands bind competitively.
Chan and Yousaf have increased the scope of electrochemically controlled immobilization,98 in a “switch on” system for cell adhesiveness to a surface. This was achieved by coverage with a mixed SAM of hydroquinone and tetra(ethylene glycol) alkanethiols (1 : 99) to generate surface patterns via an electrochemical-microfluidic strategy.99 The application of a low positive potential to the gold surface did not damage the bioinert OEG components of the SAM, and oxidized only the hydroquinone to the reactive quinone. Instead of adding a cyclopentadiene-terminated peptide ligand as described above by Mrksich and co-workers, Chan and Yousaf reacted a soluble oxyamine-terminated RGD ligand,98,100 chemoselectively, with the surface tethered quinone, resulting in a pattern of immobilized ligands. Specific cell adhesion was obtained, by culturing Swiss 3T3 fibroblast cells on the surface in serum-free media. Cell immobilization was only observed on the regions patterned with the electrochemically active hydroquinone-terminated SAM.
Mrksich et al. have reported the patterning of Swiss 3T3 fibroblast cells by combining their dynamic electrochemical SAM covered surfaces with photopatterning.101 A bioinert mixed SAM of nitroveratryloxy-carbonyl (NVOC)-protected hydroquinone-terminated and tri(ethyleneglycol)-terminated alkanethiol (1 : 99) was prepared on a gold substrate. The NVOC-protected SAM was then deprotected, by irradiation at λ = 365 nm, for 2 min, exposing the electroactive hydroquinone (Fig. 17). A pattern of deprotected islands could be formed by irradiating through a mask or an objective while non-irradiated regions remained bioinert.
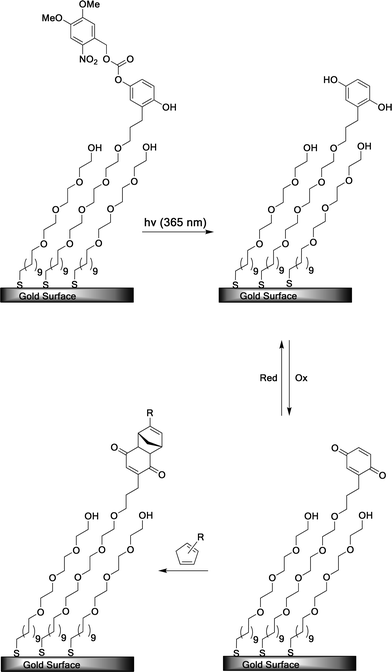 | ||
| Fig. 17 A SAM-presenting NVOC-protected hydroquinone is irradiated at λ = 365 nm to photodeprotect the hydroquinone, which can then be oxidized reversibly to the quinone. The quinone undergoes a Diels–Alder reaction with a Cp-ligand derivative to immobilize the ligand.101 | ||
Electrochemical oxidation leads to formation of a benzoquinone-terminated SAM (Fig. 17), which was used to immobilize a Cp-presenting RGD ligand via the Diels–Alder reaction. The combination of photo- and electrochemistry allows for control over the patterning of bioinert and adhesive sections and the ligand density to create multiple cell-adhesive islands side by side on a substrate (Fig. 18).
 | ||
| Fig. 18 Phase contrast microscopy image of Swiss 3T3 fibroblast cells patterned on a SAM by photo-deprotection (UV light) and subsequent oxidation (+400 mV, 15 s) followed by Diels–Alder immobilization of a RGD–Cp ligand (5 mM, 4 h). The interaction between cell and substrate is specific so that regions that were not illuminated do not present RGD ligands. Reproduced from ref. 101. Copyright ACS 2004. | ||
By combining bioinert SAMs with dynamic electrochemical immobilization of peptide ligands, increased control can be obtained over surface density and patterning of cell-adhesion ligands. Additionally, the surface density can be analyzed quantitatively with electrochemistry. These methods provide more control over the SAM environment for studying cell–surface interactions, and could lead to surface designs that are more representative of the ECM.
In addition to electrochemical activation of cell adhesiveness of SAM-presenting substrates, Mrksich and co-workers reported another dynamic approach based on a mixed SAM of alkanethiolates presenting RGD peptide ligands via an electroactive quinone ester (<0.1%) and tri(ethylene glycol) groups on gold (Fig. 19).102 The quinone ester was reduced to the corresponding hydroquinone electrochemically. Subsequent rapid cyclization to the lactone takes place and the RGD peptide is released.103 Patterning two regions that differ only in the linkage of the RGD peptide to the monolayer enabled the selective release of Swiss 3T3 fibroblast cells (Fig. 20). The application of a potential did not influence cells that were adherent to the adjacent regions.
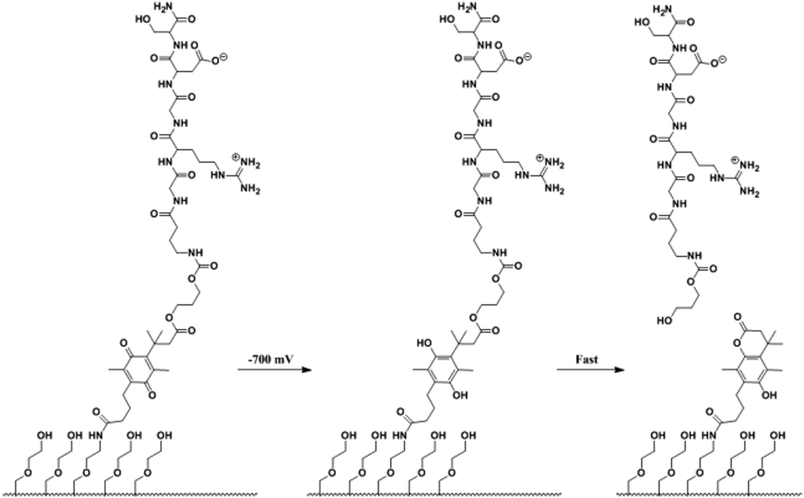 | ||
| Fig. 19 A SAM of alkane thiolates on gold was used in the selective release of adherent cells. RGD is present at a density of less than 0.1% in the SAMs. The SAM releases the RGD ligand upon electrochemical reduction of the quinone to the corresponding hydroquinone via cyclization to the lactone.102 | ||
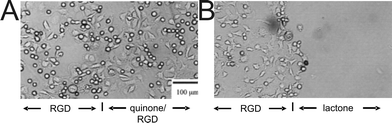 | ||
| Fig. 20 Optical micrographs of 3T3 Swiss fibroblast cells interacting with a patterned SAM. The left side of the substrate presents covalently bound RGD peptide, while the right side presents electroactive RGD ligands. (A) Cells adhere and spread uniformly on both sides of the substrate. (B) After application of a potential (−700 mV for 4 min), the quinone groups were reduced to the hydroquinone with subsequent lactonization and release of RGD. Only the cells attached to the region presenting the electroactive RGD ligand were released from the substrate. Reproduced from ref. 102. Copyright Wiley 2001. | ||
A region was patterned with the quinone ester tethered peptide, together with a second region of RGD peptide tethered with a non-electrochemically active linker.104 A suspension of 3T3 Swiss fibroblast cells was added and, upon incubation of the substrate in a cell-culture medium, the cells attached to both regions evenly. After culturing (37 °C, 30 min), a potential (−700 mV vs. Ag pseudo-reference, 4 min) was applied to the gold substrate. Over 70% of the cells on the electroactivated region adopted a rounded morphology immediately after the treatment, indicating that they were lying unattached on the substrate. In contrast cells attached to the non-electroactive region of the monolayer remained unaffected (Fig. 20).
Although approaches to switching cell adhesiveness on or off selectively might be useful for patterning cell cultures and in studies investigating the mechanisms involved in cell adhesion and migration, it is equally interesting to combine the two methods to create true on–off–on switching of the cell adhesiveness of surfaces.
Electrochemical switching of cell adhesiveness
Mrksich et al. have combined the electroactive immobilization of peptide ligands with the electroactive release of peptide ligands creating cell adhesive substrates that can be switched on–off–on.105 They demonstrated that, by using two different electrochemically active SAMs each presenting peptide ligands, the adhesion of Swiss 3T3 fibroblast cells could be modulated. The dynamic interface was based on SAMs that incorporated an O-silyl hydroquinone functionality presenting an RGD ligand (Fig. 21).105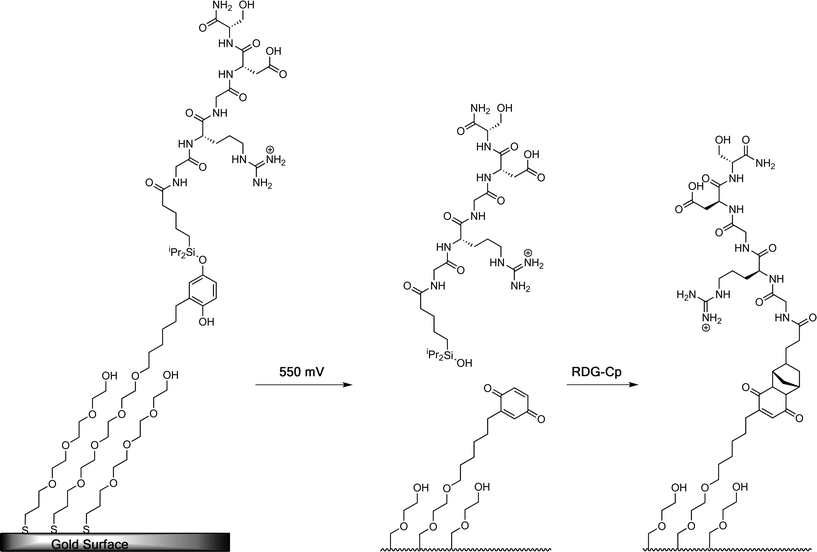 | ||
| Fig. 21 A SAM-presenting the O-silyl hydroquinone is oxidized to benzoquinone with resulting hydrolysis of the silyl ether and selective release of the RGD ligand by application of a potential (550 mV). The resulting benzoquinone reacts with RGD–Cp via a Diels–Alder reaction, which selectively immobilizes the second ligand.105 | ||
Hexadecane thiol was patterned in circular regions (∅ 200 μm) on a gold substrate. The remaining substrate surface was functionalized with a mixed SAM of RGD alkanethiols (at a density of 0.02%) and tri(ethylene glycol)-terminated alkanethiolate. Fibronectin (FN) was non-specifically adhered onto the circular hexadecane thiol regions. Cells were dispersed equally across the regions exhibiting fibronectin and RGD peptide after incubation. The O-silyl hydroquinone ether allows for selective release of the RGD peptide from the substrate via electrochemical oxidation (550 mV potential, 5 min), which yields the corresponding benzoquinone (Fig. 21). The cells detached from the RGD functionalized regions of the monolayer, while cells on the fibronectin-coated regions were unaffected, indicating that the RGD ligand was liberated from the SAM. Subsequently the benzoquinone group could be used to immobilize, selectively, a cyclopentadiene-tagged peptide (RGD–Cp) through a Diels–Alder reaction, by replacing the medium with a serum-free medium containing the RGD–Cp ligand. The cells migrated from the fibronectin-coated circular regions, with cells dispersed evenly over the entire surface after 14 h (Fig. 22).
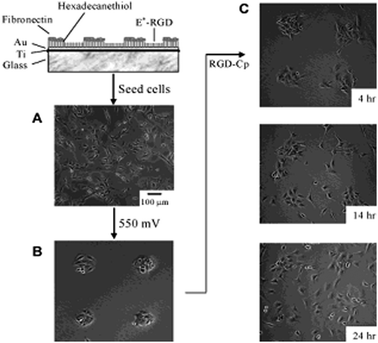 | ||
| Fig. 22 A substrate combining two dynamic properties: (I) releasing cells from the surface and (II) the immobilization, migration and growth of cells. A hexadecane thiol SAM was patterned into circular regions and covered with fibronectin. The circular regions were surrounded by RGD ligands tethered via an electroactive linker (E*-RGD). (A) Swiss 3T3 fibroblast cells adhered and spread evenly over the entire substrate. (B) A potential was applied to the substrate (550 mV for 5 min and incubated for 4 h). Cells were released selectively from the E*-RGD regions. (C) Treatment of the monolayer with RGD–Cp resulted in immobilization of the RGD ligand and initiated cell migration from fibronectin covered regions. After 24 h, cells were distributed evenly over the substrate. Reproduced from ref. 105. Copyright ACS 2003. | ||
Recently, this line of research was extended by patterning a monolayer with distinct regions that present cell-adhesive ligands, by way of two different redox-active tethers (quinone ester and O-silyl hydroquinone), that respond to negative (−650 mV) or positive (650 mV) applied potentials, respectively. In this way cell adhesion of various cell populations can be manipulated separately (Fig. 23).106 Although these dynamic SAM systems allow for on–off–on interchange of cell-adhesion properties of surfaces by external stimuli, they cannot be labeled rigorously as switchable, since it is not possible to reverse the changes, and revert to the original system. Overall changing the cell adhesiveness of a surface typically involves the addition of new ligands for tethering.
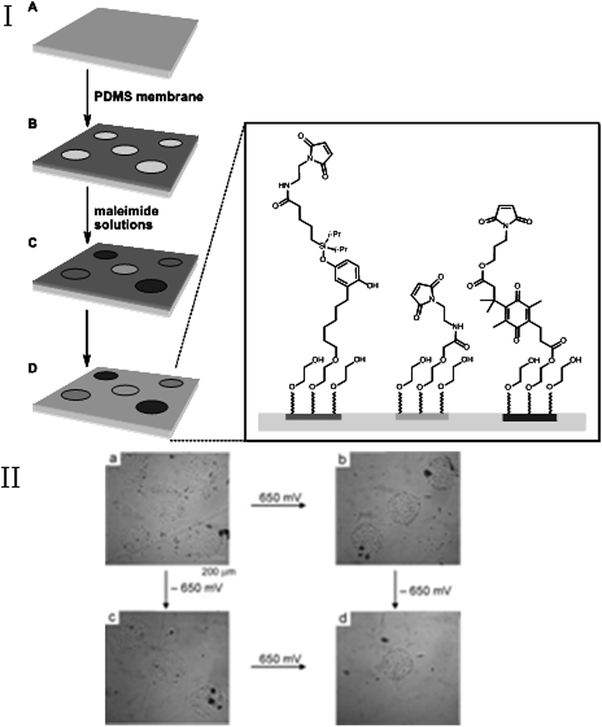 | ||
| Fig. 23 (I) Preparation of an electroactive substrate combining two dynamic functions. (II) Demonstration of the selective release of adherent cells under electrochemical control. (a) Swiss 3T3 fibroblast cells adhere to circular regions presenting RGD ligands. (b) Electrochemical (650 mV) release of the cells from the patterned regions presenting the electroactive O-silyl hydroquinone. (c) Electrochemical (−650 mV) release of cells from regions presenting the electroactive quinone ester. The subsequent application of a potential of −650 mV (panel b) or 650 mV (panel c) results in an additional release of cells (panel d). Reproduced from ref. 106. Copyright ACS 2006. | ||
Recently Yousaf and co-workers were able to extend this methodology using an additional pH controlled release step, thus creating a fully catalytic surface which can be renewed a number of times (Fig. 24).107 The release step is governed by a functional group transformation of the oxyamine group to a primary alcohol upon application of an electrochemical potential.
![Reaction of soluble oxyamine with a quinone-terminated SAM. Electrochemical oxidation [O] of hydroquinone presented on the mixed SAM. Subsequently the quinone reacts selectively with a soluble oxyamine-tagged RGD ligand (R–ONH2) providing the redox-active oxime conjugate. The oxime is chemically stable, but undergoes a reversible redox reaction. Electrochemical reduction [R] of the SAM (pH > 0) reverts the oxime to the hydroquinone by liberating the RGD containing ligand as an alcohol. Adapted from ref. 107. Copyright Wiley 2008.](/image/article/2010/CS/b906608j/b906608j-f24.gif) | ||
| Fig. 24 Reaction of soluble oxyamine with a quinone-terminated SAM. Electrochemical oxidation [O] of hydroquinone presented on the mixed SAM. Subsequently the quinone reacts selectively with a soluble oxyamine-tagged RGD ligand (R–ONH2) providing the redox-active oxime conjugate. The oxime is chemically stable, but undergoes a reversible redox reaction. Electrochemical reduction [R] of the SAM (pH > 0) reverts the oxime to the hydroquinone by liberating the RGD containing ligand as an alcohol. Adapted from ref. 107. Copyright Wiley 2008. | ||
The approach is based upon the same hydroquinone-terminated SAMs as described earlier. Coupling of an oxyamine ligand to the quinone tethered SAM results in an oxime conjugate. The oximine conjugate is stable at pH 1–14, however, upon application of an applied potential at low pH (1 M HClO4, pH 0) it can undergo a reversible redox reaction. Electrochemical reduction at pH 7 in phosphate buffered saline (PBS) results in the regeneration of the hydroquinone and release of the ligand from the surface.
This approach was used in cell biology to prepare surfaces for cell adhesion and the subsequent release of patterned cells. Using the NVOC-protected hydroquinone reported by Mrksich and co-workers101 it was possible to photo-deprotect a predetermined pattern of hydroquinone molecules on the surface. Subsequent oxidation provided a quinone-presenting SAM, and a RGD peptide containing oxyamine was coupled to this quinone-presenting surface (Fig. 25). The addition of the RGD containing peptide allows the adhesion of fibroblast cells to the SAM. If desirable the cell-adhering ligand can be removed selectively by mild electrochemical reduction (−50 mV) of the gold substrate, even in the presence of cells adhering to hydrophobic SAMs without affecting them. The release of the RGD containing sequence renders the remaining surface bio-resistant once again.
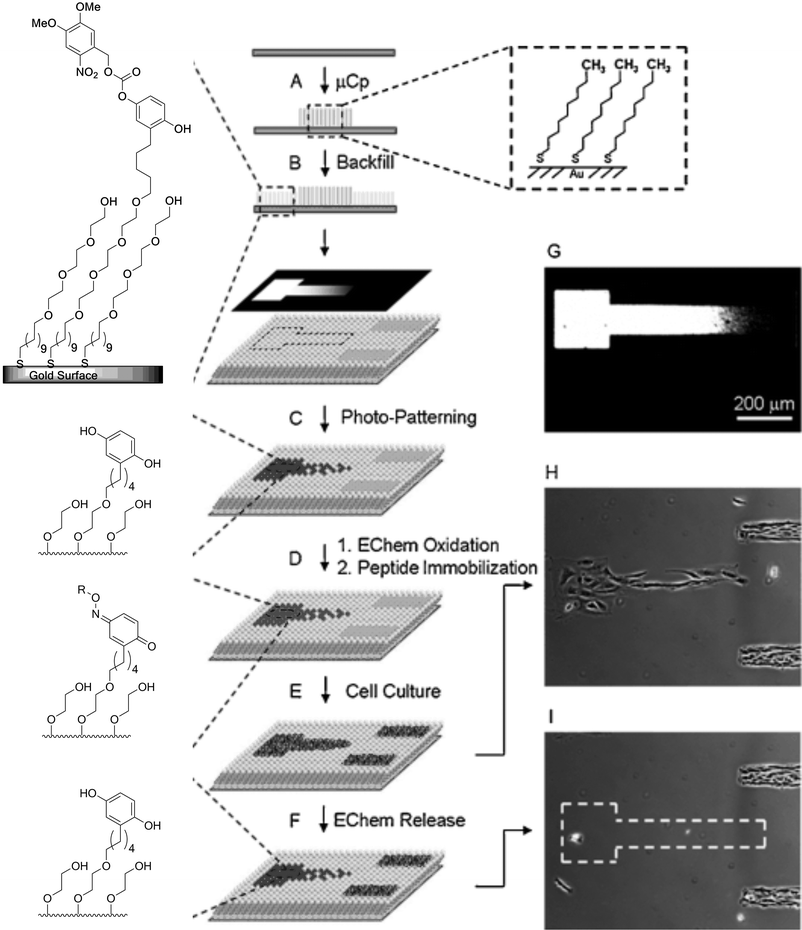 | ||
| Fig. 25 Electrochemical release of cells, patterned on an RGD-presenting SAMs. (A) Hexadecane thiols were μCP on a gold substrate to generate hydrophobic lined patterns. (B) The residual exposed gold surface was filled with a mixed monolayer of NVOC hydroquinone and tetra(ethylene glycol)-presenting alkane thiols. (C) A second pattern presenting hydroquinone was formed by UV irradiation through a photomask. (D) The hydroquinone-presenting substrate was oxidized to the corresponding quinone. Subsequent addition of soluble RGD-oxyamine provides a peptide-presenting monolayer by oxime formation, switching the irradiated area from bioinert to cell adhesive. (E) Seeding of fibroblast cells on the monolayer resulted in cell adhesion to both the μCP and photopatterned regions. (F) Electrochemical reduction of the gold substrate results in selective liberation of the cells from the RGD-presenting regions, whereas cells adhering to the hexadecane thiol SAMs remain attached. (G) Microscopy image of a gradient containing photomask for the preparation of photopatterned RGD-peptide-presenting SAMs. (H) Fibroblast cells patterned on a RGD gradient and on μCP lines. (I) Application of a reductive electrochemical potential to the monolayer results in detachment of the cells on the RGD-presenting gradient, cells patterned on hydrophobic regions remain adherent. Reproduced from ref. 107. Copyright Wiley 2008. | ||
Dynamic cell-responsive SAMs
Control over the interface between cells and a synthetic material is essential to many fields, including the development of cell-based screening devices. These types of cell-based sensors are potentially applicable in drug discovery and in developing analytic screening devices that can recognize biohazard agents in environmental samples.54 Although the following system is not able to change the adhesiveness of cells to the surface via external stimuli, such as the dynamic SAM substrate described above, the system described by Collier and Mrksich discussed here is an illustrative example of how responsive SAMs can be used in microelectronic screening devices. In this system the surface can be switched from an electropassive state to an electroactive state by the modification of the substrate surface through adhesion of engineered Chinese hamster ovary cells that present the enzyme cutinase (Fig. 26).54,108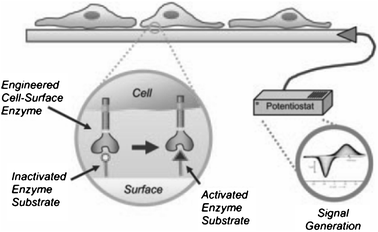 | ||
| Fig. 26 Schematic representation of the transfer of cellular activity to an electrical output. Adherent cells presenting cutinase interact with synthetic ligands presented on SAMs. Enzymatic switching of the electrode surface can be followed using cyclic voltammetry. Reproduced from ref. 54. Copyright NAS 2006. | ||
Cutinase is a fungal esterase that is not expressed in mammalian cells. The enzyme is able to catalyze the hydrolysis of acyl groups from surface-bound hydroquinone derived ester substrates. SAMs were prepared containing 4-hydroxy-(3-mercaptopropyl)phenyl valerate and tri(ethylene glycol) moieties in densities ranging from 70% to 95% (Fig. 27). SAMs presenting 4-hydroxyphenyl valerate could be hydrolyzed by cutinase to yield a hydroquinone. The switch to an electroactive state results in an electrical signal when the enzyme covered cell surface is placed in proximity to a Ag/AgCl electrode. This deprotection allows for reversible oxidation to take place, yielding benzoquinone. This redox cycle can be detected directly using cyclic voltammetry.
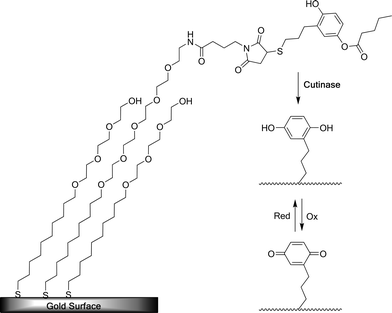 | ||
| Fig. 27 4-Hydroxy-(3-mercaptopropyl)phenyl valerate-protected maleimide-presenting SAM is deprotected by addition of cutinase. This enables electrochemical switching between the hydroquinone and quinone states.54 | ||
This system54 demonstrates how responsive SAMs might be incorporated into microscale devices, which are capable of performing a sensing function using living cells. Developing new strategies that utilize cell activity by directly interfacing the cells with synthetic materials will facilitate many categories of hybrid microsystems in which living cells are combined with synthetic machinery.
Dynamic control of cell adhesion on thin polymeric films
Photoresponsive control over cell adhesion
Kessler and co-workers reported the use of a polymer system which incorporates an azobenzene switch (Fig. 28) for switching between a cell adhesive and non-adhesive state of the polymer.91 Their system was based on SAMs presenting RGD peptides in which photoswitchable azobenzene derivatives were incorporated to control the availability of the RGD peptide ligand presented on the PMMA surface. Cell adhesion on different surfaces is affected by the spacer length between the peptide ligand and the surface.109 4-[(4-Aminophenyl)azo] benzoic acid was employed as a light controlled switch, as it retains its photochemical activity and stability when immobilized in the polymer.109b A key feature of azobenzene derivatives is that, upon switching, the length of the azobenzene moiety is varied. The E-isomer is ca. 3 Å longer than the Z-isomer.91 The azobenzene can be switched to the Z-isomer by irradiation at 360 nm, however, at the photostationary state (PSS) only between 70 and 90% of the unstable Z-isomer is obtained.110![Cyclic RGD peptides presented on a photoswitchable 4-[(4-aminophenyl)azo]benzocarbonyl unit], where c-(RDfK) is a cyclic pentapeptide.91](/image/article/2010/CS/b906608j/b906608j-f28.gif) | ||
| Fig. 28 Cyclic RGD peptides presented on a photoswitchable 4-[(4-aminophenyl)azo]benzocarbonyl unit], where c-(RDfK) is a cyclic pentapeptide.91 | ||
Several of the photoswitchable RGD peptide-presenting units provided enhanced cell adhesion on PMMA disks in the E-isomer state. Irradiation reduced the proximity of the RGD ligands to the surface via E/Z isomerization, resulting in a minor reduction in cell adhesion in the Z-configuration. On acryloyl-Gly-[4-(4-aminophenyl)azo] benzocarbonyl-c(-RGDfK-) units, cell adsorption could be reduced to the level of uncoated PMMA disks. Adsorption was enhanced by 17% in the E-isomer state.
Switching of the substrate does not yield a high contrast in cell adhesiveness and has limited influence on the cells’ adhesiveness to the surface. Systems that incorporate azobenzene derivatives should in principal allow the cell adhesiveness to be switch on and off by controlling the binding peptides through mechanical movement. However this characteristic has yet to be demonstrated. Additionally, quantum yield and photobleaching might present difficulties under conditions of high numbers of switching cycles.
Higuchi et al. coated (poly)NSP-co-MMA, a copolymer of photoresponsive nitrospiropyran and methyl methacrylate, on glass111 to direct the cell adhesiveness of a copolymer. Irradiation at 365 nm switches the cell adhesive apolar state of the nitrospirobenzopyran switch to its non-cell adhesive polar zwitterionic state (Fig. 29). Switching of the nitrospirobenzopyran results in an increase in the hydrophilicity of the surface as determined through changes in surface contact angle. Platelets and mesenchymal stem (KUSA-A1) cells detached from the (poly)NSP-co-MMA surface after UV irradiation of the polymer-modified surface (Fig. 30). The viability of the detached cells was found to be 98%.
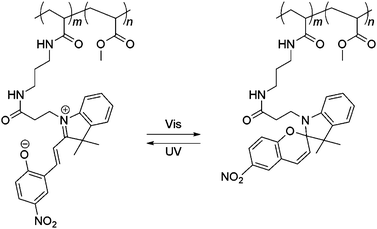 | ||
| Fig. 29 Transition of poly(NSP-co-MMA) to its non-cell adhesive zwitterionic state upon exposure to UV irradiation.112 | ||
 | ||
| Fig. 30 Light-triggered desorption of KUSA-A1 cells from poly(nitrospiropyran-co-MMA)-coated glass substrate. Reproduced from ref. 111. Copyright ACS 2004. | ||
Control experiments using PMMA-coated glass surfaces did not exhibit photoresponsive cell detachment, indicating a change in the surface energy and/or the switching characteristics of the nitrospirobenzopyran unit from one state to the other was responsible for the effects observed. Additional experiments using enzyme-linked immunosorbent assay (ELISA) confirmed that UV exposure (of the surface) resulted in a decrease in the amount of preadsorbed fibrinogen. However, experiments using glass substrates coated with (poly)NSP-co-MMA in its apolar and polar zwitterionic states revealed the photoactivated regions contained elevated concentrations of fibrinogen (up to 1.2 fold), compared to the non-irradiated regions.
Yoshimi and co-workers have recently reported a photocontrolled substrate based upon a thermally responsive poly(N-isopropylacrylamide) polymer tailored with a photoresponsive spirobenzopyran switch.112 Cell adhesion of the polymer could be increased upon irradiation at 365 nm. Non-adherent cells on the non-irradiated regions could be removed by cooling and subsequent washing. Irradiation of the surface between 400 and 440 nm and subsequent annealing (2 h, 37 °C) returns the material to its original non-adhesive state. Utilizing this sequence the cell adhesiveness of the surface could be repeatedly switched between a cell adhesive and non-adhesive state (Fig. 31).
 | ||
| Fig. 31 Microscopy images of CHO-K1 cell culture on a nitrospiropyran containing photoresponsive surface. Before (A) and after (B) spatially controlled UV irradiation, (C) subsequently followed by low-temperature washing switch the cell adhesiveness of the surface. Reproduced from ref. 112. Copyright ACS 2005. | ||
Yoshimi and co-workers also described a glass surface that was coated by means of a mixture of bioinert PEG (wt 4%) and a photoresponsive copolymer of nitrospirobenzopyran functionalized methacrylamide and methyl methaacrylate.113 Irradiation, at 365 nm, of the cell adhesion resisting coated surface resulted in switching of the nitrospirobenzopyran unit to its zwitterionic form, allowing BALB/3T3 fibroblast cells to adhere to the surface. In contrast to the previous example this system cannot be switched reversibly between a cell adhesive and a non-adhesive state due to loss of PEG from the surface upon UV irradiation. It was suggested that the ability of PMN to retain PEG chains in its polymer matrix was decreased upon switching to the zwitterionic state.
Tatsu and Ohmuro-Matsuyama reported a method to direct the cell adhesiveness of commercially available culture dishes by coating the surface with poly-L-lysine using crosslinked PEG and a photoresponsive 2-nitrobenzene protecting group to cage the integrin binding RGD sequence (Fig. 32).114 The photoresponsive 2-nitrobenzene protecting group was positioned on the amide functionality connecting the Gly and Arg residues of the RGD containing ligand.
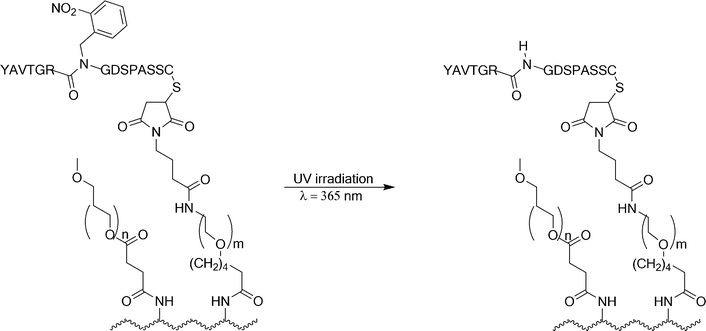 | ||
| Fig. 32 Photochemical reaction of a caged RGD peptide attached to a culture dish. The caged RGD peptide was linked to poly-L-lysine (depicted as a wavy line).114 | ||
HeLa cells did not adhere when they were incubated on the surface containing the caged RGD sequence. The HeLa cells adopted a rounded morphology and did not exhibit formation of focal adhesions. Upon irradiation of half of the substrate surface coated with the photoresponsive layer at 365 nm for 1 min, followed by 20 min incubation at 20 °C and rinsing with PBS, adherent cells were observed only on the region exposed to UV irradiation (Fig. 33). The HeLa cells extend pseudopods and the cell periphery expands while their morphology flattens over the irradiated region. The photocontrolled system presented here allows for analysis of adhesion events on a sub-minute timescale.
 | ||
| Fig. 33 Spatial control of cell adhesion. The left half of the dish was irradiated during cell cultivation. The micrograph was recorded after washing and fixing. Reproduced from ref. 114. Copyright Wiley 2008. | ||
Enzyme controlled cell adhesion
In biological tissues dynamic processes are controlled by molecular feedback systems involving on-demand activation of biomolecules by enzymes. For example, integrin binding peptide regions can be hidden due to the complex folding of the protein scaffold in the ECM, which results in inactivity. The binding domains can be exposed by the alteration of the ECM via enzyme cleavage or mechanical distortion.115Mimicking responsive biomaterials is of contemporary interest, in particular, through the use of enzymes. This approach has several advantages: (I) high selectivity, (II) mild conditions (aqueous, pH 5–8, 37 °C) and (III) key association with biological pathways.115 Ulijn and co-workers have reported the design of a dynamic cell adhesive surface based on the enzyme-triggered activation of chemically inactivated cell-adhesive substrates, i.e. a poly(ethylene glycol) acrylamide (PEGA) surface, presenting RGD peptides to control cell adhesion on command.116 The peptide ligand was rendered inactive by using a bulky (9-fluorenyl-methyoxycarbonyl phenylalanine (Fmoc–F) group (Fig. 34). The group contained a phenylalanine enzyme recognition sequence. This allows for an enzyme (chymotrypsin) to be used to hydrolyze the Fmoc–FQRGD peptide link, effectively forming an ‘OFF’ to ‘ON’ switch.
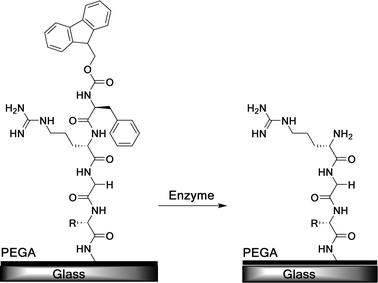 | ||
| Fig. 34 PEGA spin coated on a glass slide presenting a Fmoc-protected RGD sequence, which is released by addition of chymotrypsin.116 | ||
Chymotrypsin (TF-Ch) was used to hydrolyze the Fmoc protecting group from the RGD peptide with high selectivity.117 Human osteoblast cells were seeded and cultured on Fmoc–FRGD–PEGA. Minimal spreading was observed, demonstrating that the Fmoc–F functionality deactivates the RGD peptide effectively. After exposure to TFCh, cell spreading increased to approximately 50% (±5%) (Fig. 35). Although Fmoc-amino acids are known for their anti-inflammatory activity,118 no adverse side effects were observed in the proliferation of the osteoblast cells. Applying similar dynamic biomolecular strategies to SAM covered surfaces may lead to improved understanding of the dynamic cell-adhesion properties of the ECM.
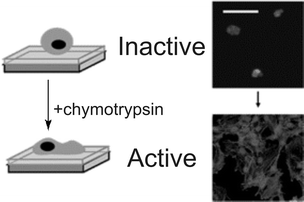 | ||
| Fig. 35 Schematic representation of inactive surface (Fmoc–FRGD–PEGA) and fluorescence images and schematic representation of an activated surface (Fmoc–FRGD–PEGA after chymotrypsin treatment) stained with DAPI (nucleus) and phalloidin (cytoskeleton). Reproduced from ref. 116. Copyright RSC 2007. | ||
Concluding remarks
In this review recent advances in the application of SAMs to achieve spatial control over the growth of cells on surfaces have been discussed. The central theme is the ability to control or switch cell-adhesive properties dynamically by application of an external stimulus. The application of dynamic SAMs in hybrid microdevices has already seen success and holds considerable promise for application of this methodology in controlling cell growth.Over the last decade several approaches to activate or deactivate the cell adhesiveness of surfaces on-demand have stimulated the development of dynamic self-assembled monolayers. These SAMs have been used primarily to pattern cells on a surface or release cells from their confinement on-demand. This is mainly due to the inherent limitations in the majority of current systems in which one way switching is employed. Nevertheless, these systems have proven invaluable in studying the mechanisms of cell adhesion, as well as the migratory behavior of cells.
Some of the approaches discussed can be combined to obtain “reversible” switching of the cell adhesive properties of the SAMs. These methods are technically challenging, in particular regarding the level of reversibility that is achievable, as they involve a complete elimination or addition of the cell adhesive ligands presented. Proceeding to the next level requires responsiveness to different external stimuli. A further key goal is to make the systems employed robust enough to overcome the effects of surface remodeling by secreted ECM proteins whilst maintaining a highly dynamic response to external stimuli. It is apparent that the challenge remains in achieving more comprehensive control over the cell adhesiveness of SAMs and the search for a truly reversibly switchable SAM has only begun.
Acknowledgements
We thank Dr A. Kocer for discussion and the Dutch Organisation for Scientific Research (NWO-VIDI, WRB) and NanoNed for financial support.References
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, London, 2002 Search PubMed.
- (a) F. G. Giancotti and E. Ruoslahti, Science, 1999, 285, 1028 CrossRef CAS; (b) M. A. Schwartz, M. D. Schaller and M. H. Ginsberg, Annu. Rev. Cell Dev. Biol., 1995, 11, 549 CrossRef CAS; (c) D. D. Schlaepfer and T. Hunter, Trends Cell Biol., 1998, 8, 151 CrossRef CAS; (d) A. Howe, A. E. Alpin, S. K. Alahari and R. L. Juliano, Curr. Opin. Cell Biol., 1998, 10, 220 CrossRef CAS; (e) M. G. Coppolino and S. Dedhar, Int. J. Biochem. Cell Biol., 2000, 32, 171 CrossRef CAS.
- R. Pytela, M. D. Pierschbacher, M. H. Ginsberg, E. F. Plow and E. Ruoslahti, Science, 1986, 231, 1559.
- R. Pytela, M. D. Pierschbacher and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 5766 CAS.
- S. Dedhar, E. Ruoslahti and M. D. Pierschbacher, J. Cell Biol., 1987, 104, 585 CrossRef CAS.
- Fibronectins, Springer series in Microbiology, ed. R. O. Hynes, Springer Verlag, New York, 1990 Search PubMed.
- Cell Adhesion, Frontiers in Molecular Biology 39, ed. M. C. Beckerle, Oxford University Press, New York, 2001 Search PubMed.
- (a) D. B. Weibel, P. Garstecki and G. M. Whitesides, Curr. Opin. Neurobiol., 2005, 15, 560 CrossRef CAS; (b) C. S. Chen, M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber, Science, 1997, 276, 1425 CrossRef CAS; (c) S. Huang, C. S. Chen and D. E. Ingber, Mol. Biol. Cell, 1998, 9, 3179 CAS; (d) R. McBeath, D. M. Pirone, C. M. Nelson, K. Bhadriraju and C. S. Chen, Dev. Cell, 2004, 6, 483 CrossRef CAS.
- A. Huttenlocher, R. R. Sandborg and A. F. Horwitz, Curr. Opin. Cell Biol., 1995, 7, 697 CrossRef CAS.
- M. Mrksich and G. M. Whitesides, Annu. Rev. Biophys. Biomol. Struct., 1996, 25, 55 CrossRef CAS.
- E. Ostuni, L. Yan and G. M. Whitesides, Colloids Surf., B, 1999, 15, 3 CrossRef CAS.
- J. M. Anderson, A. Rodriguez and D. T. Chang, Semin. Immunol., 2008, 20, 86 CrossRef CAS.
- (a) A. de Mel, G. Jell, M. M. Stevens and A. M. Seifalian, Biomacromolecules, 2008, 9, 2969 CrossRef CAS; (b) E. S. Place, N. D. Evans and M. M. Stevens, Nat. Mater., 2009, 8, 457 CrossRef CAS.
- D. G. Castner and B. D. Ratner, Surf. Sci., 2002, 500, 28 CrossRef CAS.
- J. L. Brash, J. Biomater. Sci., Polym. Ed., 2000, 11, 1135 CrossRef CAS.
- K. R. Patel, H. Y. Tang, W. E. Grever, K. Y. S. Ng, J. Xiang, R. F. Keep, T. Cao and J. P. McAllister, Biomaterials, 2006, 27, 1519 CrossRef CAS.
- A. Folch and M. Toner, Annu. Rev. Biomed. Eng., 2000, 2, 227 CrossRef CAS.
- M. Mrksich, Curr. Opin. Chem. Biol., 2002, 6, 794 CrossRef CAS.
- X. Y. Jiang and G. M. Whitesides, Eng. Life Sci., 2003, 3, 475 CrossRef CAS.
- P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512 RSC.
- N. Faucheux, R. Schweiss, K. Lutzow, C. Werner and T. Groth, Biomaterials, 2004, 25, 2721 CrossRef CAS.
- M. Mrksich, Chem. Soc. Rev., 2000, 29, 267 RSC.
- Y. Guo, M. Y. Li, A. Mylonakis, J. J. Han, A. G. MacDiarmid, X. S. Chen, P. I. Lelkes and Y. Wei, Biomacromolecules, 2007, 8, 3025 CrossRef CAS.
- M. Mrksich, Acta Biomater., 2009, 5, 832 CrossRef CAS.
- A recent review on SAMs in nanotechnology: J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitsides, Chem. Rev., 2005, 105, 1103 Search PubMed.
- X. Jiang, D. A. Bruzewicz, A. P. Wong, M. Piel and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 975 CrossRef CAS.
- M. Mrksich, MRS Bull., 2005, 30, 180 CAS.
- (a) J. C. Love, D. B. Wolfe, R. Haasch, M. L. Chabinyc, K. E. Paul, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 2003, 125, 2597 CrossRef CAS; (b) A. Carvalho, M. Geissler, H. Schmid, B. Michel and E. Delamarche, Langmuir, 2002, 18, 2406 CrossRef; (c) X. Y. Jiang, D. A. Bruzewicz, M. M. Thant and G. M. Whitesides, Anal. Chem., 2004, 76, 6116 CrossRef CAS.
- F. Schreiber, Prog. Surf. Sci., 2000, 65, 151 CrossRef CAS.
- S. Onclin, B. J. Ravoo and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2005, 44, 6282 CrossRef CAS.
- (a) M. Mrksich, Curr. Opin. Colloid Interface Sci., 1997, 2, 83 CAS; (b) P. Kingshott and H. J. Griesser, Curr. Opin. Solid State Mater. Sci., 1999, 4, 403 CrossRef.
- For reviews on bioinert SAMs see: (a) M. Morra, J. Biomater. Sci., Polym. Ed., 2000, 11, 547 CrossRef CAS; (b) J. Genzer and K. Efimenko, Biofouling, 2006, 22, 339 CrossRef CAS; (c) N. Aldred and A. S. Clare, Biofouling, 2008, 24, 351 CrossRef CAS; (d) M. Schuler, D. Trentin, M. Textor and S. G. P. Tosatti, Nanomedicine, 2006, 1, 449 CrossRef CAS.
- (a) M. Veiseh and M. Q. Zhang, J. Am. Chem. Soc., 2006, 128, 1197 CrossRef CAS; (b) J. D. Cox, M. S. Curry, S. K. Skirboll, P. L. Gourley and D. Y. Sasaki, Biomaterials, 2002, 23, 929 CrossRef CAS.
- (a) L. Deng, M. Mrksich and G. M. Whitesides, J. Am. Chem. Soc., 1996, 118, 5136 CrossRef CAS; (b) Y.-Y. Luk, M. Kato and M. Mrksich, Langmuir, 2000, 16, 9604 CrossRef; (c) R. S. Kane, P. Deschatelets and G. M. Whitesides, Langmuir, 2003, 19, 2388 CrossRef CAS; (d) R. E. Holmlin, X. X. Chen, R. G. Chapman, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 2841 CrossRef CAS.
- (a) M. Caffrey and J. Wang, Annu. Rev. Biophys. Biomol. Struct., 1995, 24, 351 CAS; (b) B. J. Spargo, M. A. Testoff, T. B. Nielsen, D. A. Stenger, J. J. Hickman and A. S. Rudolph, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 11070 CAS; (c) D. A. Stenger, J. H. Georger, C. S. Dulcey, J. J. Hickman, A. S. Rudolph, T. B. Nielsen, S. M. McCort and J. M. Calvert, J. Am. Chem. Soc., 1992, 114, 8435 CrossRef CAS.
- S. F. Chen, L. Y. Lui and S. Y. Jiang, Langmuir, 2006, 22, 2418 CrossRef CAS.
- In this context a ligand is defined as a molecule that binds specifically to the integrin receptors of a cell.
- Review on cell adhesion to SAMs: M. Mrksich, Cell. Mol. Life Sci., 1998, 54, 653 Search PubMed.
- S.-K. Oh, M. Nakagawa and K. Ichimura, J. Mater. Chem., 2002, 12, 2262–2269 RSC.
- (a) B. Liedberg and P. Tengvall, Langmuir, 1995, 11, 3821 CrossRef CAS; (b) M. Lestelius, I. Engquist, P. Tengvall, M. K. Chaudhury and B. Liedberg, Colloids Surf., B, 1999, 15, 57 CrossRef CAS; (c) E. A. Burton, K. A. Simon, S. Hou, D. Ren and Y.-Y. Luk, Langmuir, 2009, 25, 1547 CrossRef CAS.
- B. M. Lamb, D. G. Barrett, N. P. Westcott and M. N. Yousaf, Langmuir, 2008, 24, 8885 CrossRef CAS.
- M. J. Humphries, Biochem. Soc. Trans., 2000, 28, 311 CrossRef CAS.
- O. Cherniavskaya, C. J. Chen, E. Heller, E. Sun, J. Provezano, L. Kam, J. Hone, M. P. Sheetz and S. J. Wind, J. Vac. Sci. Technol., B, 2005, 23, 2972 CrossRef CAS.
- (a) L. Hodgson, E. W. L. Chan, K. M. Hahn and M. N. Yousaf, J. Am. Chem. Soc., 2007, 129, 9264 CrossRef CAS; (b) Y. Arima and H. Iwata, Biomaterials, 2007, 28, 3074 CrossRef CAS; (c) Y. Arima and H. Iwata, J. Mater. Chem., 2007, 17, 4079 RSC; (d) H. Sato, Y. Miura, N. Saito, K. Kobayashi and O. Takai, Surf. Sci., 2007, 601, 3871 CrossRef CAS.
- (a) T. Zheng, D. Peelen and L. M. Smith, J. Am. Chem. Soc., 2005, 127, 9982 CrossRef CAS; (b) D. I. Rozkiewicz, Y. Kraan, M. W. T. Werten, F. A. de Wolf, V. Subramaniam, B. J. Ravoo and D. N. Reinhoudt, Chem.–Eur. J., 2006, 12, 6290 CrossRef CAS; (c) L. Liu, B. D. Ratner, E. H. Sage and S. Y. Jiang, Langmuir, 2007, 23, 11168 CrossRef CAS; (d) D. Peelen, V. Kodoyianni, J. Lee, T. Zheng, M. R. Shortreed and L. M. Smith, J. Proteome Res., 2006, 5, 1580 CrossRef CAS; (e) M. Nakajima, T. Ishimuro, K. Kato, I.-K. Ko, I. Hirata, Y. Arima and H. Iwata, Biomaterials, 2007, 28, 1048 CrossRef CAS; (f) W. L. Murphy, K. O. Mercurius, S. Koide and M. Mrksich, Langmuir, 2004, 20, 1026 CrossRef CAS.
- (a) T. Nakaji-Hirabayashi, K. Kato, Y. Arima and H. Iwata, Biomaterials, 2007, 28, 3517 CrossRef CAS; (b) K. Kato, H. Sato and H. Iwata, Langmuir, 2005, 21, 7071 CrossRef CAS.
- M. H. Lee, D. A. Brass, R. Morris, R. J. Composto and P. Ducheyne, Biomaterials, 2005, 26, 1721 CrossRef CAS.
- (a) E. V. Romanova, S. P. Oxley, S. S. Rubakhin, P. W. Bohn and J. V. Sweedler, Biomaterials, 2006, 27, 1665 CrossRef CAS; (b) O. Palyvoda, A. N. Bordenyuk, A. K. Yatawara, E. McCullen, C.-C. Chen, A. V. Benderskii and G. W. Auner, Langmuir, 2008, 24, 4097 CrossRef CAS; (c) O. Palyvoda, C.-C. Chen and G. W. Auner, Biosens. Bioelectron., 2007, 22, 2346 CrossRef CAS; (d) J. M. de la Fuente, A. Andar, N. Gadegaard, C. C. Berry, P. Kingshott and M. O. Riehle, Langmuir, 2006, 22, 5528 CrossRef CAS; (e) M. H. Lee, D. A. Brass, R. Morris, R. J. Composto and P. Ducheyne, Biomaterials, 2005, 26, 1721 CrossRef CAS.
- (a) R. T. Petty, H.-W. Li, J. H. Maduram, R. Ismagilov and M. Mrksich, J. Am. Chem. Soc., 2007, 129, 8966 CrossRef CAS; (b) M. Veiseh, O. Veiseh, M. C. Martin, F. Asphahani and M. Q. Zhang, Langmuir, 2007, 23, 4472 CrossRef CAS; (c) N. S. Sampson, M. Mrksich and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12870 CrossRef CAS; (d) T. Mori, K. Inamori, Y. Inoue, X. Han, G. Yamanouchi, T. Niidome and Y. Katayama, Anal. Biochem., 2008, 375, 223 CrossRef CAS; (e) S. Mandal, J. M. Rouillard, O. Srivannavit and E. Gulari, Biotechnol. Prog., 2007, 23, 972 CAS; (f) G. Zorn, I. Gotman, E. Y. Gutmanas, R. Adadi and C. N. Sukenik, J. Mater. Sci.: Mater. Med., 2007, 18, 1309 CrossRef CAS.
- E. Ruoslahti and M. D. Pierschbacher, Science, 1987, 238, 491 CrossRef CAS.
- W. Y. J. Kao and D. Lee, Biomaterials, 2001, 22, 2901 CrossRef CAS.
- Molecular switches, ed. B. L. Feringa, Wiley-VCH Verlag GmbH, Weinheim, 2001 Search PubMed.
- J. Nakanishi, T. Takarada, K. Yamaguchi and M. Maeda, Anal. Sci., 2008, 24, 67 CrossRef CAS.
- J. H. Collier and M. Mrksich, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2021 CrossRef CAS.
- J. L. Tan, J. Tien, D. M. Pirone, D. S. Gray, K. Bhadriraju and C. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1484 CrossRef CAS.
- X. Jiang, R. Ferrigno, M. Mrksich and G. M. Whitesides, J. Am. Chem. Soc., 2003, 125, 2366 CrossRef CAS.
- M. Mrksich and G. M. Whitesides, Trends Biotechnol., 1995, 13, 228 CrossRef CAS.
- G. M. Whitesides, E. Ostuni, S. Takayama, X. Y. Jiang and D. E. Ingber, Annu. Rev. Biomed. Eng., 2001, 3, 335 CrossRef CAS.
- (a) L. M. Tender, R. L. Worley, H. Y. Fan and G. P. Lopez, Langmuir, 1996, 12, 5515 CrossRef CAS; (b) L. M. Tender, K. A. Opperman, P. D. Hampton and G. P. Lopez, Adv. Mater., 1998, 10, 73 CrossRef CAS.
- D. A. Lauffenburger and A. F. Horwitz, Cell, 1996, 84, 359 CAS.
- A. B. Verkhovsky, T. M. Svitkina and G. G. Borisy, Curr. Biol., 1999, 9, 11 CrossRef CAS.
- H. Zhu, J. Yan and A. Revzin, Colloids Surf., B, 2008, 64, 260 CrossRef CAS.
- M. L. Turgeon, Clinical Hematology, Lippincott Williams & Wilkins, Boston, 3rd edn, 1999 Search PubMed.
- R. F. Siliciano, T. Lawton, C. Knall, R. W. Karr, P. Berman, T. Gregory and E. L. Reinherz, Cell, 1988, 54, 561 CrossRef CAS.
- R. S. Veazey, M. DeMaria, L. V. Chalifoux, D. E. Shvetz, D. R. Pauley, H. L. Knight, M. Rosenzweig, R. P. Johnson, R. C. Desrosiers and A. A. Lackner, Science, 1998, 280, 427 CrossRef CAS.
- A. Revzin, K. Sekine, A. Sin, R. G. Tompkins and M. Toner, Lab Chip, 2005, 5, 30 RSC.
- S. S. Shah, J. Y. Lee, S. Verkhoturov, N. Tuleuova, E. A. Schweikert, E. Ramanculov and A. Revzin, Langmuir, 2008, 24, 6837 CrossRef CAS.
- C. Zhao, I. Witte and G. Wittstock, Angew. Chem., Int. Ed., 2006, 45, 5469 CrossRef CAS.
- G. Wittstock, M. Burchardt, S. E. Pust, Y. Shen and C. Zhao, Angew. Chem., Int. Ed., 2007, 46, 1584 CrossRef CAS.
- H. Shiku, T. Takeda, H. Yamada, T. Matsue and I. Uchida, Anal. Chem., 1995, 67, 312 CrossRef CAS.
- C. Zhao, I. Zawisza, M. Nullmeier, M. Burchardt, M. Träuble, I. Witte and G. Wittstock, Langmuir, 2008, 24, 7605 CrossRef CAS.
- T. Buffeteau, B. Desbat and J. M. Turlet, Appl. Spectrosc., 1991, 45, 380 CAS.
- H. Kaji, M. Kanada, D. Oyamatsu, T. Matsue and M. Nishizawa, Langmuir, 2004, 20, 16 CrossRef CAS.
- H. Kaji, K. Tsukidate, T. Matsue and M. Nishizawa, J. Am. Chem. Soc., 2004, 126, 15026 CrossRef CAS.
- H. Kaji, K. Tsukidate, M. Hashimoto, T. Matsue and M. Nishizawa, Langmuir, 2005, 21, 6966 CrossRef CAS.
- H. Kaji, T. Kawashima and M. Nishizawa, Langmuir, 2006, 22, 10784 CrossRef CAS.
- H. Kaji, M. Hashimoto and M. Nishizawa, Anal. Chem., 2006, 78, 5469 CrossRef CAS.
- J. Nakanishi, Y. Kikuchi, T. Takarada, H. Nakayama, K. Yamaguchi and M. Maeda, J. Am. Chem. Soc., 2004, 126, 16314 CrossRef CAS.
- (a) T. Matsuda and T. Sugawara, Langmuir, 1995, 11, 2267 CrossRef; (b) B. Zhao, J. S. Moore and D. J. Beebe, Science, 2001, 291, 1023 CrossRef CAS; (c) Y. Luo and M. S. Shoichet, Nat. Mater., 2004, 3, 249 CrossRef CAS.
- K. Yamaguchi, T. Kitabatake, M. Izawa, T. Fujiwara, H. Nishimura and T. Futami, Chem. Lett., 2000, 228 CrossRef CAS.
- R. Michel, J. W. Lussi, G. Csucs, I. Reviakine, G. Danuser, B. Ketterer, J. A. Hubbell, M. Textor and N. D. Spencer, Langmuir, 2002, 18, 3281 CrossRef CAS.
- B. Geiger, A. Bershadsky, R. Pankov and K. M. Yamada, Nat. Rev. Mol. Cell Biol., 2001, 2, 793 CrossRef CAS.
- J. Nakanishi, Y. Kikuchi, T. Takarada, H. Nakayama, K. Yamaguchi and M. Maeda, Anal. Chim. Acta, 2006, 578, 100 CrossRef.
- J. Nakanishi, Y. Kikuchi, S. Inoue, K. Yamaguchi, T. Takarada and M. Maeda, J. Am. Chem. Soc., 2007, 129, 6694 CrossRef CAS.
- J. V. Small, T. Stradal, E. Vignal and K. Rottner, Trends Cell Biol., 2002, 12, 112 CrossRef CAS.
- W. Wood and P. Martin, Int. J. Biochem. Cell Biol., 2002, 34, 726 CrossRef CAS.
- S. Petersen, J. M. Alonso, A. Specht, P. Duodu, M. Goeldner and A. del Campo, Angew. Chem., Int. Ed., 2008, 47, 3192 CrossRef CAS.
- D. Kirchhofer, J. Gailit, E. Ruoslahti, J. Grzesiak and M. D. Pierschbacher, J. Biol. Chem., 1990, 265, 18525 CAS.
- R. Haubner, R. Gratias, B. Diefenbach, S. L. Goodman, A. Jonczyk and H. Kessler, J. Am. Chem. Soc., 1996, 118, 7461 CrossRef CAS.
- D. Liu, Y. Xie, H. Shao and X. Jiang, Angew. Chem., Int. Ed., 2009, 48, 4406 CrossRef CAS.
- J. Auernheimer, C. Dahmen, U. Hersel, A. Bausch and H. Kessler, J. Am. Chem. Soc., 2005, 127, 16107 CrossRef CAS.
- H. Rau, in Studies in Organic Chemistry: Photochromism, Molecules and Systems, ed. H. Dürr and H. Bonas-Laurent, Elsevier, Amsterdam, 1990, pp. 165–192 Search PubMed.
- N. Katsonis, M. Lubomska, M. M. Pollard, B. L. Feringa and P. Rudolf, Prog. Surf. Sci., 2007, 82, 407 CrossRef CAS.
- (a) M. N. Yousaf, B. T. Houseman and M. Mrksich, Angew. Chem., Int. Ed., 2001, 40, 1093 CrossRef CAS; (b) M. N. Yousaf, B. T. Houseman and M. Mrksich, Angew. Chem., 2001, 113, 1127 CrossRef.
- B. T. Houseman and M. Mrksich, Biomaterials, 2001, 22, 943 CrossRef CAS.
- M. Kato and M. Mrksich, Biochemistry, 2004, 43, 2699 CrossRef CAS.
- Y. Feng and M. Mrksich, Biochemistry, 2004, 43, 15811 CrossRef CAS.
- E. W. L. Chan and M. N. Yousaf, J. Am. Chem. Soc., 2006, 128, 15542 CrossRef CAS.
- N. P. Westcott and M. N. Yousaf, Langmuir, 2008, 24, 2261 CrossRef CAS.
- E. W. L. Chan and M. N. Yousaf, ChemPhysChem, 2007, 8, 1469 CrossRef CAS.
- W. S. Dillmore, M. N. Yousaf and M. Mrksich, Langmuir, 2004, 20, 7223 CrossRef CAS.
- W.-S. Yeo, C. D. Hodneland and M. Mrksich, ChemBioChem, 2001, 2, 590 CrossRef CAS.
- C. D. Hodneland and M. Mrksich, J. Am. Chem. Soc., 2000, 122, 4235 CrossRef CAS.
- (a) C. Roberts, C. S. Chen, M. Mrksich, V. Martichonok, D. E. Ingber and G. M. Whitesides, J. Am. Chem. Soc., 1998, 120, 6548 CrossRef CAS; (b) B. T. Houseman and M. Mrksich, J. Org. Chem., 1998, 63, 7552 CrossRef CAS.
- W.-S. Yeo, M. N. Yousaf and M. Mrksich, J. Am. Chem. Soc., 2003, 125, 14994 CrossRef CAS.
- W.-S. Yeo and M. Mrksich, Langmuir, 2006, 22, 10816 CrossRef CAS.
- E. W. L. Chan, S. Park and M. N. Yousaf, Angew. Chem., Int. Ed., 2008, 47, 6267 CrossRef CAS.
- W.-S. Yeo and M. Mrksich, Angew. Chem., Int. Ed., 2003, 42, 3121 CrossRef CAS.
- (a) M. Kantlehner, D. Finsinger, J. Meyer, P. Schaffner, A. Jonczyk, B. Diefenbach, B. Nies and H. Kessler, Angew. Chem., Int. Ed., 1999, 38, 560 CrossRef CAS; (b) M. Kantlehner, P. Schaffner, D. Finsinger, J. Meyer, A. Jonczyk, B. Diefenbach, B. Nies, G. Hölzemann, S. L. Goodman and H. Kessler, ChemBioChem, 2000, 1, 107 CrossRef CAS.
- H. Rau, in Studies in Organic Chemistry: Photochromism, Molecules and Systems, ed. H. Dürr and H. Bonas-Laurent, Elsevier, Amsterdam, 1990, pp. 165–192 Search PubMed.
- A. Higuchi, A. Hamamura, Y. Shindo, H. Kitamura, B. O. Yoon, T. Mori, T. Uyama and A. Umezawa, Biomacromolecules, 2004, 5, 1770 CrossRef CAS.
- J.-I. Edahiro, K. Sumaru, Y. Tada, K. Ohi, T. Takagi, M. Kameda, T. Shinbo, T. Kanamori and Y. Yoshimi, Biomacromolecules, 2005, 6, 970 CrossRef CAS.
- Y. Tada, K. Sumaru, M. Kameda, K. Ohi, T. Takagi, T. Kanamori and Y. Yoshimi, J. Appl. Polym. Sci., 2006, 100, 495 CrossRef CAS.
- Y. Ohmuro-Matsuyama and Y. Tatsu, Angew. Chem., Int. Ed., 2008, 47, 7527 CrossRef CAS.
- M. M. Stevens and J. H. George, Science, 2005, 310, 1135 CrossRef CAS.
- S. J. Todd, D. Farrar, J. E. Gough and R. V. Ulijn, Soft Matter, 2007, 3, 547 RSC.
- (a) I. Schechter and A. Berger, Biochem. Biophys. Res. Commun., 1967, 27, 157 CAS; (b) H. Matsubara, R. Sasaki, A. Singer and T. H. Jukes, Arch. Biochem. Biophys., 1966, 115, 324 CrossRef CAS; (c) E. Kraus and U. Femfert, Hoppe–Seyler Z. Physiol. Chem., 1976, 357, 937 CAS.
- R. M. Burch, M. Weitzberg, N. Blok, R. Muhlhauser, D. Martin, S. G. Farmer, J. M. Bator, J. R. Connor, C. Ko, W. Kuhn, B. A. McMillan, M. Raynor, B. G. Shearer, C. Tiffany and D. E. Wilkins, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 355 CAS.
| This journal is © The Royal Society of Chemistry 2010 |