Polypeptide–polymer bioconjugates
Luiz A.
Canalle
,
Dennis W. P. M.
Löwik
and
Jan C. M.
van Hest
*
Bio-Organic Chemistry, Institute for Molecules and Materials, Radboud University Nijmegen, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: j.vanhest@science.ru.nl; Fax: (+31) 24 3653393; Tel: (+31) 24 3653204
First published on 6th October 2009
Abstract
Creating bioconjugates by combining polymers with peptides and proteins is an emerging multidisciplinary field of research that has enjoyed increased attention within the scientific community. This critical review provides an overview of the strategies employed for the construction of these materials and will highlight the underlying synthetic methods used. This review is therefore relevant for chemists, material scientists and chemical biologists facing the challenge of constructing polypeptide–polymer bioconjugates in a controlled fashion (269 references).
 Dennis W. P. M. Löwik, Jan C. M. van Hest, Luiz A. Canalle | Luiz A. Canalle (right) was born in Caçador (Brazil) in 1981 and studied chemistry at the Radboud University Nijmegen. He received his MSc with honours in 1999 after traineeships at the Computational Genomics group of Prof. Martijn Huynen, the synthetic organic chemistry group of Prof. Floris Rutjes, and the natural products synthesis group of Prof. Emmanuel Theodorakis. He is currently a PhD student in the bio-organic group of Prof. Jan van Hest where he is working on the development of biofunctional coatings. |
Dennis W. P. M. Löwik (left) was born in 1969 in the Netherlands. After completing his MSc in organic chemistry in 1994, he received his PhD in 1998 under supervision of Prof. Rob Liskamp. This was followed by a post-doctoral position in the group of Prof. Chris Lowe until 2000. After a year’s stay in the group of Prof. Bert Meijer, he was appointed in 2001 at the Radboud University Nijmegen in the bio-organic chemistry group of Prof. Jan van Hest. As an assistant professor, he now has his focus on the emerging field of peptide-based materials, operating at the interface of chemistry, biology and physics. |
Jan C. M. van Hest (middle) conducted his doctoral research on molecular architectures based on dendrimers under supervision of Prof. Bert Meijer, for which the PhD title was granted in 1996. As a postdoctoral researcher, he investigated the possibilities of protein engineering for the preparation of materials under supervision of Prof. David Tirrell. In 1997 he then joined the chemical company DSM, where he worked as research scientist and later on as group leader on the development of innovative material concepts. In 2000 he was appointed as a full professor at the Radboud University Nijmegen to set up a new group in bio-organic chemistry, which focuses on bio-inspired hybrid materials and processes. |
1. Introduction
Traditionally, synthetic polymers have mainly been exploited for their structural properties and as such, have successfully replaced natural materials in a wide variety of applications. A reason for their success is that synthetic polymers can be obtained in a large range of compositions and architectures, which allows for a convenient fine tuning of their properties. However, even with the current synthetic techniques, there is still limited control over monomer composition and molecular weight. A synthetic polymer sample is therefore a mixture of many different macromolecules, which makes it difficult to translate information from the single polymer chain to the polymeric ensemble.On the other hand, naturally produced peptides or proteins can be regarded as highly refined polymers. They are monodisperse and have a precisely defined primary sequence, which allows them to hierarchically fold and organize into three-dimensional structures. This permits polypeptides to exhibit (bio)functional features like catalysis or receptor recognition. As a downside to this delicate folding behaviour, proteins are sensitive to temperature and pH changes. Furthermore, they have limited solubility in organic solvents, are susceptible to enzyme degradation, and their use in biomedical applications can be restricted by their possible toxicity and undesired elicited immune response.
In this review, we will focus on synthetic pathways that can be used to combine synthetic polymers with proteins or peptides. The resulting bioconjugates can synergistically combine the properties of the individual components and overcome their separate limitations. The protein or peptide element can impart (bio)functional properties to the bioconjugate, whereas the polymer component can improve protein stability, solubility and biocompatibility. The synthetic polymer can also introduce new properties such as self-assembly and phase behaviour, and even modulate protein activity. This emerging multidisciplinary field has opened the door to a wide range of applications which will be discussed throughout the article and which have already been extensively reviewed in recent literature.1–7 The main focus of this article, however, is the chemistry and synthetic strategies that are involved in the construction of these hybrid conjugates, and that have become more selective and specific in recent years, resulting in better defined polypeptide–polymer bioconjugates.
1.1 Specificity
Most traditional conjugation methods rely on a nucleophilic attack of a functional group of an amino acid side chain in the polypeptide on an appropriate electrophilic centre within the synthetic polymer. Due to the polyfunctional nature of proteins and peptides, however, such approaches ultimately lead to a mixture of products.As a result, the search for more specific modification methods has been the topic of significant academic research during the last decade. Site-specific modifications would first of all help with the purification and characterization of the obtained products, because mixtures are avoided. Furthermore, if the modification is directed to a specific location, the protein activity might be better preserved. For instance, the site for polymer conjugation can be located far away from the active site to avoid interference with the biological functioning of the protein. Alternatively, it can also be located nearby, or even within the active site, in order to control the biological activity of the protein.
No universal technique exists to date, but significant advances have been made in the development of more specific methods.8 When using naturally occurring functionalities in proteins or peptides, there has been a trend to modify amino acid targets that are less common. Alternatively, the conjugation step can be carried out under conditions directing the availability of the modification sites. Examples are the use of protective groups on peptides or the blockage of the active site of proteins with a substrate during the conjugation reaction. In some cases, biochemical approaches are used, such as mutagenesis to limit the amount of accessible, reactive amino acids, or the use of enzymes to enhance the regioselectivity of the conjugation reaction. In addition, unnatural functionalities can be introduced into proteins and peptides. Because this introduces a bigger pallet of potential chemical transformations, it is possible to establish bio-orthogonal coupling methodologies that tolerate a diverse array of natural functionalities. A less used method is to couple the protein and polymer via non-covalent interactions using, for example, natural cofactors.
1.2 Architecture
The architecture of the bioconjugate has to be adjusted to the application for which it is developed. The most common architecture is the head-to-tail conjugate (Fig. 1A). This set-up can be described as a linear AB block copolymer where the polymer serves as one block and the protein or peptide as the other. This architecture is often used for conjugates where polymers act as protective shields around the biomolecule. Alternatively, one can imagine a comb-shaped structure with peptides or proteins installed as side chain groups on a polymer backbone (Fig. 1B). The high loading capacity of this architecture makes it an interesting drug delivery system. It can furthermore be a favourable architecture in cases where the bioactivity is related to having multiple copies of a peptide in close proximity of each other. The density of the biomolecules can be increased even further by using dendrimers as the support structures (Fig. 1C).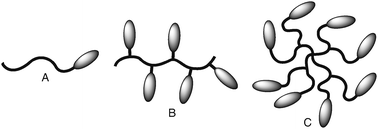 | ||
| Fig. 1 Cartoon representation of the most common architectures of bioconjugates. The curved lines represent the synthetic polymer component and the ellipses the peptide or protein: (A) head-to-tail conjugate, (B) comb-shaped structure, (C) dendritic architecture. | ||
1.3 Strategies
This review will be structured around the three different strategies used for the synthesis of polypeptide–polymer bioconjugates.The grafting to strategy is the most well-established and involves the coupling of a preformed polymer with a protein or peptide. Because this strategy tries to bring two macromolecules together, the coupling reaction has to be very efficient. This demanding coupling can sometimes be aided by an indirect functionalization. In this context, the protein is not reacted directly with a polymer, but is rather linked to a heterofunctional spacer first. In this way, a more reactive secondary functionality is introduced into the protein, which alleviates problems associated with the lack of reactivity when bringing two large molecules together.
The grafting from strategy is based on a polymer chain propagating (growing) from a protein or peptide surface. In this set-up, the protein or peptide acts as a macroinitiator. Consequently, an initiating moiety needs to be introduced into the biological molecule. This method has only become practical since the advance of controlled radical polymerization (CRP)9 techniques, which tolerate a wide variety of functional groups within the reaction mixture and can be performed under benign conditions. Alternatively, a peptide can be grown from a preformed synthetic polymer serving as the macroinitiator.
Finally, the grafting through strategy involves the polymerization of peptide-functionalized (macro)monomers. Peptides are functionalized with a polymerizable group, which serves as a handle to string the monomeric units together, thus resulting in a comb-shaped structure with a high density of peptides.
When compared to grafting to, grafting from and grafting through have the potential to facilitate an efficient synthesis of bioconjugates, because at first, only a small component needs to be coupled to the protein or peptide, after which the polymer component is grown in a stepwise fashion.
2. Grafting to using natural functionalities
2.1 Amine conjugation
The first-generation methods that were developed to create protein polymer hybrids under mild chemical conditions were based on reactions between the activated hydroxyl group of poly(ethylene glycol) (PEG) chains and primary amines from proteins.10 The latter groups are abundantly present on the surface of proteins in both lysine residues and the N-terminus. This facilitates easy functionalization by both alkylation, which maintains the positive charge of the protein at physiological pH, and acylation, which is accompanied by loss of charge at the conjugation site.Generally speaking, these first-generation methods are non-specific and result in the attachment of polymers at multiple sites on the protein. Nevertheless, this type of modification, also known as PEGylation, has been widely employed since the 70s to reduce the limitations of protein-based drugs, such as a short half-life and immunogenicity.3–5 PEGylation leads to an increased water solubility11 and stability,12 prevents clearance through the kidneys, and hence gives rise to a prolonged plasma lifetime.13 In one of the first examples, Abuchowski et al. showed that the covalent attachment of PEG chains to bovine liver catalase reduced the immunogenicity of this protein.14 Moreover, PEGylated catalase was shown to exhibit prolonged circulating times in the blood of mice. Since then, many different proteins have been modified such as hormones,11 antibodies15 and cytokines.16 Ultimately, these efforts have led to protein–polymer hybrid products on the market, of which PEG–bovine adenosine deaminase (ADAGEN) was the first to obtain FDA approval in 1990.3,17 Although PEGylation has been demonstrated to be an effective method of enhancing protein properties, one of the limitations is the accumulation of PEG in the liver, especially if high molecular weight PEG chains are employed.13
The oldest PEGylation method uses cyanuric chloride.14,18 Originally, cyanuric chloride was used in industrial applications to covalently couple dye molecules to fabrics. After being coupled to PEG, the second chloride of the PEG dichlorotriazine derivative (1) can easily react with amines from a protein at room temperature and at pH 9 to form a secondary amine linkage (2) (Scheme 1).10 A drawback of dichlorotriazine-activated PEG, however, is that it can react with other nucleophilic residues, such as cysteine, serine, tyrosine and histidine. Moreover, the third chloride, while less reactive, can still partially react with nucleophilic groups, which can result in crosslink formation. A way around this problem is to decorate cyanuric chloride with two PEG chains before conjugation, leaving only one chloride on the triazine ring available for coupling with the protein.19 A final important consideration for in vivo applications is still the potential toxicity of cyanuric chloride and its derivatives.
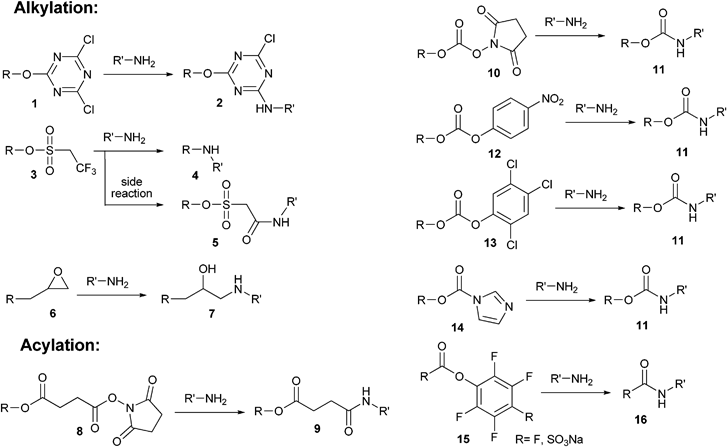 | ||
| Scheme 1 First-generation conjugation methods, which use activated hydroxyl groups for the functionalization of primary amines from peptides and proteins via alkylation and acylation. | ||
Another reagent that was designed to couple proteins to polymers via a secondary amine is tresylate (3) (Scheme 1).20 PEG tresylate has the advantage over PEG dichlorotriazine that it has a higher specificity towards amines. However, it was shown that during the conjugation with small amines, PEG tresylate does not always simply react by the familiar C–O cleavage process associated with sulfonic esters (4). Conjugation can also occur through a sulfoacetamide linkage (5).21,22 Because the resulting sulfoacetamide linkage is more susceptible to hydrolysis, the formation of such a heterogeneous mixture can hamper the applicability of this method.
Reacting the hydroxyl end-group of PEG with epichlorohydrin allows a terminal epoxy moiety to be introduced (Scheme 1). The epoxy-functionalized PEG (6) can subsequently be employed to modify the polymer chain with a protein by amine bond formation (7).23
Another widely used method for the activation of PEG towards amines uses N-hydroxysuccinimide (NHS) esters (Scheme 1). The NHS ester can be introduced into a PEG polymer chain by coupling it to the carboxylic acid of succinylated PEG.24 The succinimidyl succinate PEG (8) which is formed is highly reactive towards amines at physiological pH and forms amide bonds with proteins (9). The major drawback of this method is the potential hydrolysis of the ester bond of succinylated PEG. A way to avoid this problem is coupling of NHS to the PEG chain via a carbonate group (10).25,26 The carbamate bond that is formed after conjugation with a protein (11) is very stable against hydrolytic cleavage. Interestingly, the reactivity of the NHS ester appears to decrease when the distance between the activated ester and the PEG backbone is increased. This was illustrated when the spacer molecule was extended from a propionic acid to a butanoic acid.27 The reactivity was further decreased by introducing an α-branching methyl group into the spacer. This difference in reactivity was attributed to the steric hindrance of the extra branching moiety, demonstrating the subtlety of this coupling process.
In the abovementioned cases, the NHS group was introduced into the polymer chain in a post-polymerization modification process. In order to avoid possible problems that can arise from this approach, such as incomplete modification, NHS-functionalized initiators (17 and 18) have been employed to build up a functional polymer chain (Scheme 2). This approach was shown to be successful for the synthesis of polymethacrylates using Atom Transfer Radical Polymerization (ATRP).28,29 In accordance with the decreased reactivity of the propionic- and butanoic-based NHS ester after introducing a branching moiety,27 it was also found that when the NHS-functionalized initiator 17 was replaced by initiator 18, the resulting polymer was unable to couple to the protein.
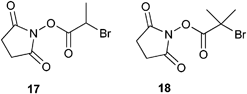 | ||
| Scheme 2 NHS-functionalized ATRP initiators. | ||
Other activated esters that have been used to couple polymers to proteins via acylation include p-nitrophenyl carbonate (12),30 trichlorophenyl carbonate (13),30 imidazole carbamate (14),31 pentafluorophenyl ester (15, R = F)32 and its more water soluble counterpart 4-sulfo-2,3,5,6-tetrafluorophenyl ester (15, R = SO3Na)33 (Scheme 1). All these derivatives are less reactive than the above mentioned succinimidyl carbonate. Generally speaking, however, a lower activity of the coupling moiety will result in a higher selectivity towards a specific amino acid residue within the protein.
Until now, we have seen strategies where polymers are first functionalized with a reactive group and then coupled to a peptide in a separate step. Instead, conjugation can also be accomplished directly by activating the polymer in situ with the use of a carbodiimide.34 In this manner, the carboxylic group of succinylated PEG24 was activated and coupled to various peptides.35 When using organic solvents, N,N′-diisopropyl carbodiimide (19, DIC) is the coupling reactant of choice. The water-soluble 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (20, EDC) is suitable for reactions in the aqueous phase (Scheme 3).
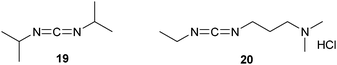 | ||
| Scheme 3 Structures of the carbodiimides DIC and EDC. | ||
A drawback of PEGylation, and of bioconjugation with polymers in general, is the polydisperse nature of the polymeric part of the conjugates, which could lead to populations of structures with different biological properties. This can of course be circumvented by making use of monodisperse polymers, as has been elegantly demonstrated by Kochendoerfer et al.36 A second, more prominent, limitation arises from the heterogeneous nature of the bioconjugates synthesized with the abovementioned first-generation PEGylation methods. Because the attachment of the PEG chains using these methods is non-specific, products generally suffer from a severe loss in bioactivity, ranging from 20% to 95%.37 This illustrates the requirement and importance of site-selective conjugation methods. Possibilities to gain the needed control over the macromolecular architecture of the bioconjugates will be discussed in the next sections.
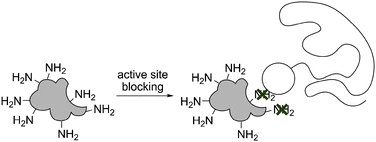 | ||
| Fig. 2 Active site blocking during bioconjugation. An inhibitor, substrate or ligand is added during the conjugation step, thereby shielding the reactive groups in the sensitive area (marked with a cross). | ||
A polymer can also be selectively coupled to the C-terminus of a peptide by first attaching a polymer to the solid support, followed by the subsequent assembly of the peptide from the polymer using standard solid phase chemistry.50 This approach can be seen as a first example of the grafting from strategy, which will be discussed in more detail in section 5. When a PEG chain is planned as the synthetic component, the commercially available Tentagel PAP resin can be used. This resin consists of a polystyrene matrix and a pre-attached PEG chain with a labile benzyl ether linkage. From this PEG chain, the peptide can directly be built up as the bioconjugate.51–56 By combining both strategies for the functionalization of the C- and N-terminus, an ABA triblock was synthesized with two outer polystyrene blocks.57
Besides modification of the peptide termini, amino acid side chains can also be conjugated with polymers during an SPPS procedure. Lu and Felix developed two routes for the site-selective PEGylation of lysine and aspartic acid residues.50 In the first route, the targeted amino acid was first coupled to a PEG chain and then built into the peptide during solid phase synthesis. In the second route, the peptide was first synthesized on the solid support with Nε-allyloxycarbonyl lysine or β-allyl aspartate at the conjugation site. These orthogonal protection groups were removed selectively, after which the peptide was site-selectively PEGylated, while still on the resin.
Mutter et al. used the concepts from SPPS and transformed them into a liquid-phase procedure.58 Peptides were assembled on a linear PEG support in a homogeneous solution. After each step, excess reagents and by-products were removed by membrane dialysis or precipitation of the growing peptide in diethyl ether. The advantage of this method is that intermediate products can be analyzed using standard techniques such as NMR. The method has, however, never found widespread application in the synthesis of peptide polymer conjugates, possibly because it can be difficult to find the optimal synthetic conditions.
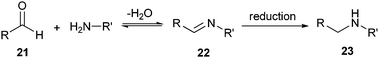 | ||
| Scheme 4 Mechanism of the reductive alkylation. | ||
Reductive alkylation proves to be quite efficient. When comparing immobilization methods for human serum albumin (HSA) to monoliths composed from methacrylate-based polymers,69 the reductive alkylation protocol resulted in a more efficient immobilization than the epoxy,23 succinimidyl carbonate25,26 and carbodiimide34 methods. These columns were subsequently used to separate both racemic warfarin and tryptophan, where the column prepared by reductive alkylation was found to give the best resolution.
However, reductive alkylation has as a drawback the use of sodium cyanoborohydride, which is both water-sensitive and has the potential for reducing disulfide bonds within proteins. A variation of this method avoids these problems by using a water-stable iridium catalyst for the reduction.70 However, this method is not as efficient as the classical reduction and no selectivity for the N-terminal amine has been shown yet. Another variation of the reductive alkylation procedure used an acetal moiety, which was converted to an aldehyde in situ before coupling. By storing the aldehyde functional polymer as its corresponding acetal derivative, a longer shelf life was guaranteed. The acetal moiety was either coupled to a pre-existing polymer71 or built into a polymer by synthesizing the polymer with an acetal-functional initiator.72 A similar chemical approach for surface immobilization was followed by Christman et al., who elegantly decorated a methacrylate surface with streptavidin by making use of an acetal-protected aldehyde functionality that can be deprotected by irradiation.73 Using this methodology together with photomasks, patterns were obtained with features as small as 500 nm.
2.2 Cysteine conjugation
The frequent occurrence of lysines on protein surfaces makes it hard to site-selectively target the amine groups of these residues. As mentioned above, reductive alkylation can target the N-terminus selectively, and active site blocking, mutagenesis and solid phase synthesis can be used to limit the amount of available lysines on the protein or peptide.Another, easier way to create well-defined polypeptide–polymer bioconjugates is to target a more specific functionality within the peptide or protein. In polypeptides there are only a few cysteines that do not participate in disulfide bonds. This low natural abundance makes it easier to functionalize proteins via the available cysteines at a specific location and thus minimize loss of biological activity. As an additional advantage, the overall charge of the polypeptide is maintained. In the absence of a free and accessible cysteine in the native structure, one can be added by site-directed mutagenesis74,75 or by introducing a cysteine-containing peptide connector in a fusion-protein.76 However, these strategies can be dangerous, as the addition of a single cysteine increases the possibility of incorrect disulfide formation and protein dimerization.
Generally speaking, conjugations that target cysteines can be divided into thioether- and disulfide-forming reactions, which will be discussed in the next sections.
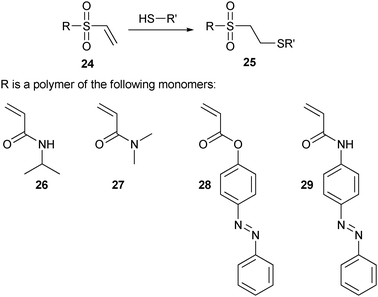 | ||
| Scheme 5 Cysteine conjugation using vinyl sulfone, together with some examples of the polymers employed. | ||
In one example of this strategy, chloroethyl sulfone was first introduced to a PEG chain by a four-step procedure, and then converted to vinyl sulfone in the presence of base.78 This gave the possibility to selectively attach the PEG derivative to the cysteine residues of reduced ribonuclease (RNase). In the area of tissue engineering, α,ω-divinyl sulfone PEG chains were crosslinked into hybrid hydrogel networks with protein–polymer-containing cell-binding sites.79 In another example, this method was furthermore used for the immobilization of the RGD protein on a surface.80
In another example of the application of cysteine conjugation using vinyl sulfone chemistry, polymers were prepared by polymerization of N-isopropylacrylamide (26, NIPAAm) via chain transfer free radical polymerization. The resulting polyNIPAAm is thermo-responsive due to its lower critical solution temperature (LCST). Above this critical temperature, water is expelled, turning the polymer into an insoluble hydrophobic state. The terminal hydroxyl group of polyNIPAAm was modified with divinyl sulfone, and the resulting thiol-reactive polymer was subsequently conjugated specifically to an unnaturally occurring cysteine in both streptavidin81–85 and endoglucanase 12A86,87 mutants. Likewise, copolymers of N,N-dimethyl acrylamide (27, DMA) and two different light-sensitive co-monomers, 4-phenylazophenyl acrylate (28, AZAA) and N-4-phenylazophenyl acrylamide (29, AZAAm), were attached to streptavidin using the vinyl sulfone moiety.82,83 Conjugation of these thermal- and light-sensitive polymers to proteins results in stimuli-responsive materials of which the thermal- and photo-induced changes are used to regulate substrate access to the protein’s active site. Such switchable systems might ultimately be employed in diagnostic assays and demonstrate the possibilities of specific conjugation methods that can introduce polymer chain close to the active site, without destroying the protein’s activity. These protein–polymer bioconjugates are commonly referred to as ‘smart’ materials, and besides light and temperature, examples are also known where their properties alter upon changes in pH and ionic strength.7
A more widely used Michael acceptor is the maleimide group (30) (Scheme 6). It reacts faster than vinyl sulfone and the conjugation can also be performed in slightly acidic conditions (pH 6–7). However, the resulting product (31) is less stable in water. The maleimide group can be introduced in various ways, for instance by first reacting an amine-terminated PEG (32) with maleic anhydride (33), followed by dehydratation and ring-closure of the intermediate maleamic acid by acetic anhydride and sodium acetate.88 In an alternative procedure, α-amino polyNIPAAm (35) was first coupled to an N-hydroxysuccinimide (NHS) activated maleimide (36) and then conjugated to a cysteine, which was introduced into cytochrome b5 by site-directed mutagenesis of a surface accessible threonine.89 The same strategy was used to conjugate the multi-subunit DNA restriction-modification enzyme EcoR124I endonuclease (REase) to α-functional maleimide polyNIPAAm.90
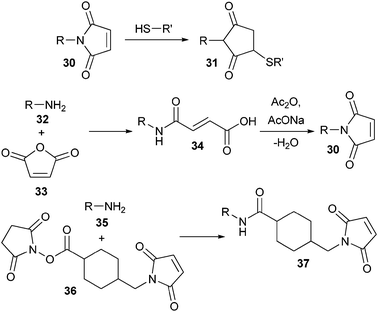 | ||
| Scheme 6 Use and synthesis of maleimide-functionalized polymers. | ||
An α-maleimide functional polymer can also be synthesized by performing an ATRP polymerization with a maleimide functional initiator. However, before polymerization, the maleimide initiator needs to be protected, for instance via a Diels–Alder reaction with dimethylfulvene (38)91 or furan (39) (Scheme 7).92 After polymerization, the protecting group can be liberated by refluxing the polymer at elevated temperature.
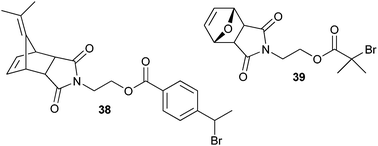 | ||
| Scheme 7 Maleimide-functionalized ATRP initiators. | ||
In one example of the maleimide-based coupling, the terminal carboxylic acid of polystyrene was converted into an acid chloride and reacted with maleimide under basic conditions.93 The single disulfide bond of Candida antarctica lipase B (Cal B) was reduced and the conjugation to the free cysteines was carried out in a mixture of tetrahydrofuran and water or on the air–water interface. This conjugation was employed to induce self-assembly of the protein and these structures have been coined as giant amphiphiles by the Nolte group.93 It was found that in aqueous media these amphiphilic structures form a variety of aggregates. Unfortunately, the aggregate formation is often accompanied by a severe loss in activity of the enzymes involved. A similar conjugation approach was used for the immobilization of a Cal B mutant onto a glass surface in which one cysteine of the disulfide bridge was mutated to an alanine.94 In a separate report, polyamine dendrimers with a maleimide-functionalized core were conjugated to the single cysteine of Bovine Serum Albumin (BSA) or a genetically engineered cysteine mutant of Class II hydrophobin (HFBI).95
On the other hand, it was proven possible to conjugate PEG to a cysteine buried inside a hydrophobic pocket of HSA using a two-step procedure.96 First, a glycoside bearing a dodecyl tail was functionalized with an NHS-activated maleimide. This hydrophobic spacer was used to facilitate conjugation to the buried cysteine. In a second step, the sugars were selectively oxidized using sodium m-periodate and the thus introduced aldehydes were coupled to PEG–hydrazine (see section 3.3 for hydrazone ligation). This two-step procedure was far more effective than direct conjugation of maleimide–PEG.
An alternative procedure, which is gaining interest, uses the century-old addition of thiols to alkenes through a photochemically or thermally induced radical mechanism (Scheme 8).97
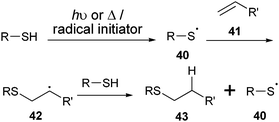 | ||
| Scheme 8 Mechanism of the thiol–ene coupling. | ||
This thiol–ene coupling (TEC) is bioorthogonal, efficient, and compatible with water and oxygen. Its potential for bioconjugation was first demonstrated by the conjugation of 1,2-polybutadiene (PBD) and PBD-b-PEG to short model peptides, resulting in peptide hybrid amphiphiles.98,99 More recently, this method has been used to pattern surfaces with proteins.100 To this end, polyamidoamine (PAMAM) dendrimers were first covalently coupled to a silicon oxide surface. Coupling of an aminocaproic acid spacer to the dendrimers, followed by functionalization with cystamine and subsequent reduction, resulted in the immobilization of the thiol-functionalized dendrimers on the surface. Treatment of the surface-bound thiol groups with alkene-functionalized biotin combined with irradiation through a photomask in the presence of a photoinitiator, led to a spatially selective coupling of biotin via thioester bond formation. Subsequent incubation with streptavidin finally produced a protein-patterned surface. In a separate report, the selective functionalization of a heterotelechelic polystyrene (PS) with both an alkene and an azide end-group furthermore demonstrated the orthogonality of the TEC with the copper-catalyzed azide–alkyne Huisgen 1,3-dipolar cycloaddition, which will be discussed in section 3.1.101
The most widely used activated disulfide is o-pyridyl disulfide (44) (Scheme 9). It reacts very efficiently towards the end product, because the o-pyridyl disulfide group is expelled from the substrate as a nonreactive compound (46), which can no longer participate in the thiol–disulfide exchange reaction. In line with this strategy, PEG was first functionalized using an NHS-activated o-pyridyl disulfide and then conjugated to mutagenically introduced cysteines on Pseudomonas Exotoxin A (PE).102 Alternatively, o-pyridyl disulfide was introduced to PEG by activating the terminal hydroxyl groups with p-nitrophenyl chloroformate, followed by coupling of 2-(2-pyridyldithio)ethylamine.103 These postpolymerization modifications can be circumvented by using pyridyl disulfide-functionalized initiators, which have been used successfully in the ATRP polymerization of 2-hydroxyethyl methacrylate (HEMA).104
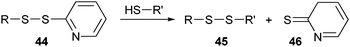 | ||
| Scheme 9 Thiol–disulfide exchange with o-pyridyl disulfide. | ||
In a demonstration of the usefulness of this conjugation chemistry, the heptameric protein pore α-hemolysin (αHL) was selectively PEGylated with a single polymer chain in its lumen.105 For its construction, single-cysteine αHL mutants were conjugated with an o-pyridyl disulfide PEG chain and then mixed with unmodified αHL unimers. After self-assembly of the heptamer, the desired product could be isolated. In another example, α-(o-pyridyl disulfide)-ω-(hydroxysuccinimide) PEG was first coupled to amine-functionalized atomic force microscopy (AFM) tips. In a second step, thiolated antibodies were conjugated to the cysteine-reactive end of the heterotelechelic PEG. The thus immobilized antibodies were then tested in single-molecule recognition experiments.106 Instead of only using the terminal end-group of a polymer for the immobilization of an antibody, side chain functionalization can be used to give high loading densities. With this idea in mind, polymer brushes of 2-methacryloyloxyethyl phosphorylcholine (MPC) and glycidyl methacrylate (GMA) were grown using ATRP.107,108 Using the epoxy groups of the GMA residues, o-pyridyl disulfide was introduced in the side chain of the polymer brush and used to immobilize antibody fragments.
Alternatives for the o-pyridyl disulfide activating group are the p-pyridyl disulfide,109 alkoxycarbonyl or o-nitrophenyl groups.110 The first activating group was used for the conjugation to the model protein papain and the last two showed good reactivity towards the tripeptide glutathione.
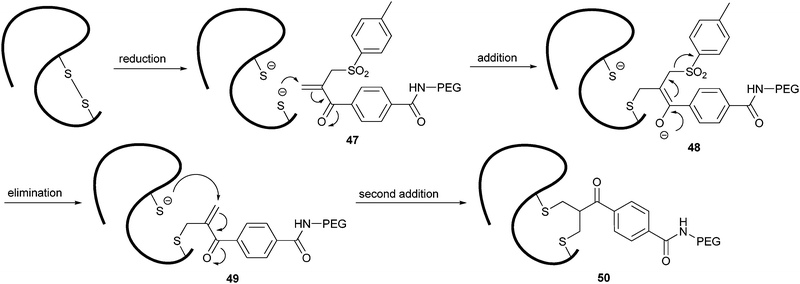 | ||
| Scheme 10 Mechanism of disulfide bridge formation using a bis-alkylating PEGylation agent. | ||
The addition of the first thiol (48) resulted in the elimination of a sulfinic acid derivative. The conjugated double bond that was formed (49) provided the addition site for the second thiol. With this method, PEG was conjugated via a three-carbon bridge that links the two original sulfur atoms (50). The sequential nature of the addition–elimination reactions ensures the efficient rebridging of the original disulfide bond, stabilizing the correct tertiary structure of the peptide. This strategy has been shown for the selective functionalization of interferon α-2, leptin, asparaginase and antibody fragments.112
2.3 Tyrosine conjugation
As a complement to the use of cysteines and lysines, tyrosine offers another possible conjugation site on proteins and peptides. An added advantage of tyrosine is that it can be introduced or removed genetically without changing the total charge of a protein (as with lysine) or its redox sensitivity (as with cysteine).It is known that tyrosines can react with diazonium salts (Scheme 11). In line with this method, poly(vinyl alcohol) was crosslinked into water-insoluble beads and then diazotized.113 Using these diazonium-functionalized beads (51), BSA and β-glucosidase were immobilized. However, it was noted that the diazonium moiety can also react with the electron-rich aromatic systems of the histidine side chain, which could result in uncontrolled side reactions.
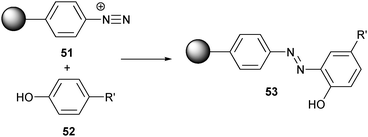 | ||
| Scheme 11 Reaction between tyrosines and a diazonium functionalized bead. | ||
Francis et al. developed two tyrosine conjugation methods that are more selective and could prove very useful in the synthesis of protein–polymer hybrids. The first one is based on a three-component Mannich-type reaction between tyrosine (52), an aliphatic aldehyde (54) and an aniline-derivatized fluorescent probe (55) (Scheme 12).114 This method was used to label chymotrypsinogen A, lysozyme and RNase A. However, this method requires a large excess of the aldehyde and aniline. Therefore, a second method was introduced, which only requires a small excess of the synthetic partners.115 There, an inert allylic acetate (57) was first activated by a palladium catalyst. The electrophilic π-allyl complex (58) was subsequently coupled to the tyrosine of chymotrypsinogen A (52), introducing either a fluorescent probe or hydrophobic tails on the protein (59).
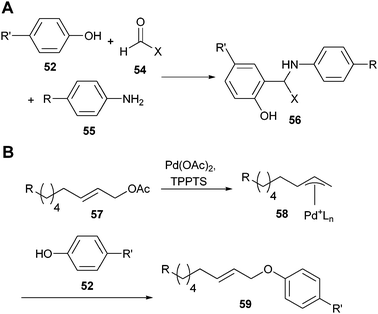 | ||
| Scheme 12 (A) Three component Mannich-type tyrosine conjugation; (B) palladium-assisted tyrosine conjugation. | ||
2.4 Native chemical ligation
Native chemical ligation (NCL) is based on a mechanism known from nature, which involves intein-associated protein splicing.116 It was first adopted in the preparation of synthetic peptides as a convenient technique that enables chemoselective coupling of two unprotected peptide segments.117 In the first step of the reaction, a peptide fragment with a C-terminal α-thioester (60) reacts with another polypeptide with an N-terminal cysteine (61). Initially, a thioester-linked species (62) is formed, which spontaneously rearranges via an intramolecular reaction to form a native peptide bond at the ligation site (63) (Scheme 13). This reaction proceeds with high yields at neutral pH. The requirement to have a cysteine at the ligation site limits the applicability of NCL, as cysteines are relatively rare amino acids in natural proteins. Several approaches, however, have recently been described that circumvent the use of cysteines.118,119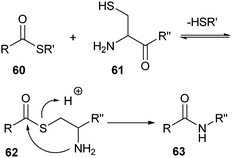 | ||
| Scheme 13 Mechanism of the native chemical ligation. | ||
NCL was first applied by Merkx and Meijer et al. as a bioconjugation tool for attaching oligopeptides and recombinant proteins to dendrimers.120,121 Hyperbranched polymers obtained from poly(propyleneimine) were reacted with succinimide-activated cysteine residues. This afforded dendrimers of the first, second and third generation with 4, 8 and 16 cysteine end-groups, respectively. Ligation reactions were performed with three oligopeptides (Leu–Tyr–Arg–Ala–Gly, Gly–Arg–Gly–Asp–Ser–Gly–Gly and Lys–Leu–Val–Phe–Phe–Gly–Gly) bearing mercaptopropionic acid–leucine (MPAL) thioesters at their C-terminus. For dendrimers of generations one and two, full conversion was observed. For the third-generation dendrimers, a mixture of products was found, probably due to steric crowding. After having shown the viability of this methodology for the coupling of peptides, it was extended for proteins by coupling green fluorescent protein (GFP) to a first-generation dendrimer. GFP was first expressed as an intein fusion protein (Scheme 14). Binding of this fusion protein on a resin (64) was followed by a nucleophilic attack with sodium 2-mercaptoethanesulfonate (MESNA) (66), which reacted with the transient thioester (65) at the intein fusion site. After coupling this GFP-based MESNA thioester (67) to the dendrimer, the remaining cysteines were further functionalized with peptide-based MPAL thioesters. A similar strategy was followed to immobilize peptides and proteins to a cysteine-functionalized dextran surface.122
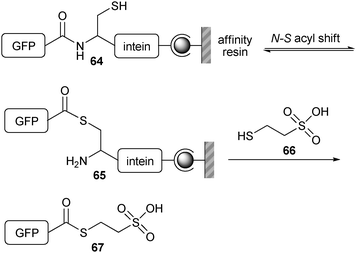 | ||
| Scheme 14 Functionalization of the N-terminus of GFP with MESNA (66) using an intein fusion protein. | ||
NCL was also used for the ligation of two nonsymmetric poly(lysine) dendritic wedges.123 One dendritic wedge with four cysteine residues along its periphery was first functionalized by NCL using the thioester-containing peptide Gly–Arg–Gly–Asp–Ser–Gly–Gly–MPAL, which can bind ανβ3 integrins. Then, the thioproline residue at the focal point of the dendritic wedge was converted to a cysteine residue by reacting it with methoxylamine. The second dendritic poly(lysine) wedge with sulfhydryl groups at its periphery was coupled to maleimide-functionalized diethylenetriamine pentaacetic acid (DTPA), a labeling agent in magnetic resonance imaging (MRI). The thioester at the focal point of this wedge could then be used in a native chemical ligation with the first peptide-functionalized wedge.
Becker et al. used discrete PEG-based oligomers carrying thioester moieties for the functionalization of an array of peptides and proteins using NCL.124 Here, it was shown that this procedure leads to the reversible attachment of polymer chains to any internal cysteine residues via thioester bonds. N-terminal cysteines on the other hand, are permanently modified through amide bond formation as shown in Scheme 13. This difference in bond formation between internal and N-terminal cysteines effectively allowed Rab GTPase, which contains three cysteines, to be selectively functionalized at the N-terminus.
2.5 Site-selective modification using enzymes
Site-selective PEGylation of proteins can be accomplished by using enzymes in the conjugation step. The first example of this biosynthetic approach used the enzyme transglutaminase (TGase).125 In its natural environment, TGase catalyses the post-translational modification of proteins by alkylating the amine group of glutamine with a primary alkylamine (Scheme 15).126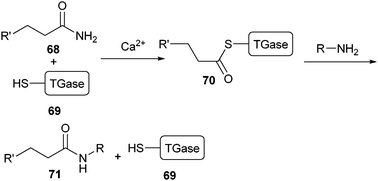 | ||
| Scheme 15 TGase is activated by Ca2+. In this conformation, thiol residue (69) is coupled to the glutamine side chain of its substrate (68). Transamidation of the intermediate structure 70 releases the alkylated product (71). | ||
Mammalian TGase has a stringent sequence requirement for the amine acceptor site around the glutamine residue. Therefore, in order to conjugate amine-functionalized PEG to the human protein interleukin-2 (hIL-2) using mammalian TGase, a short substrate sequence had to be cloned onto the N-terminus of hIL-2. The chimeric protein could be selectively functionalized at the appended substrate sequence and retained most of its bioactivity, while showing prolonged circulation in rat plasma. Alternatively, microbial TGase can be used for the conjugation reaction. This enzyme recognizes a wider variety of protein substrates as an amine acceptor. This allowed for one glutamine residue in native hIL-2 to be identified as a substrate for the microbial TGase. The other five glutamines in the hIL-2 sequence were left untouched and the well-defined bioconjugate that was formed retained its full bioactivity and possessed improved pharmacokinetics. Recently an excellent review was published covering the field of PEGylation mediated by transglutaminase.127 In another application of the enzyme, GFP and glutathione-S-transferase were immobilized on a surface using TGase.128 Both proteins were tagged with a glutamine-containing substrate sequence at their C-terminus and coupled to a β-casein coated surface, possessing reactive lysine residues.
For the site-specific conjugation of serine and threonine residues, a different enzyme has been used.129 In one example, three therapeutic human glycoproteins were expressed in Escherichia coli (E. coli) in their nonglycosylated form and PEGylated in a two-step procedure. First, their natural O-glycosylation site was functionalized with the sugar N-acetylgalactosamine (GalNac) using GalNac transferase. Subsequently, sialic acid-functionalized PEG was enzymatically coupled to the GalNac residue by sialyltransferase. The process provided a biologically active, chemically homogeneous bioconjugate with extended plasma half-life.
Another interesting pathway130 that is inspired by the natural selectivity of enzymes takes advantage of the ability of O6-alkylguanine-DNA alkyltransferase (AGT) to transfer the benzyl group of O6-benzylguanine (BG) to one of its cysteine residues.131 Originally, AGT was used in biochemistry for the labeling of proteins. To this end, the protein of interest is fused to an AGT tag and subsequently coupled covalently to a BG-functionalized fluorescent probe. Similarly, an AGT-fusion protein can react with a polymer in order to create a bioconjugate. This strategy was illustrated by reacting an AGT-fusion protein specifically with BG-functionalized polymer brushes, thereby immobilizing the fusion protein to the surface. Because the bioconjugation occurred exclusively via the reaction of the AGT tag, immobilization could be carried out directly from the crude cell lysates without the need for separate purification steps. The mild procedure left the fusion protein intact and free to react with its substrate. Furthermore, the density of protein on the surface was varied by changing the amount of BG that was anchored to the brush. Currently, three other tags are used for covalent labeling of proteins inside living cells: the CLIP tag,132 the tetracysteine tag133 and the HaloTag.134 One could envision that these tags could also be employed for bioconjugation using the same strategy.
3. Grafting to using unnatural functionalities
Peptides and proteins are both rich in structural complexity and diverse in their functional reactivity. We have seen that the high number of electrophilic and nucleophilic sites can make it difficult to selectively couple peptides and proteins to polymers using naturally occurring functional groups. Therefore, chemoselective ligation reactions have been developed in which two mutually and uniquely reactive functional groups react with one another. The chosen reactive groups are bio-orthogonal, tolerating a diverse array of other functionalities, and thus render protecting groups unnecessary. Because of their robust nature, these reactions are commonly grouped under the name ‘click’ reactions.3.1 Copper-catalyzed azide–alkyne Huisgen 1,3-dipolar cycloaddition
Thermally induced 1,3-dipolar cycloadditions have been intensively studied by Huisgen since the 1950s.135 Kolb and Sharpless,136 and Meldal and Tornoe137 showed that the 1,3-dipolar cycloaddition between azides (72) and alkynes (73) can be greatly accelerated by adding a copper catalyst, which allows the reaction to be performed at room temperature (Scheme 16).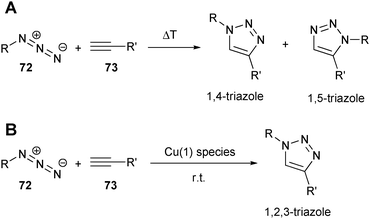 | ||
| Scheme 16 (A) Thermally induced cycloaddition between azides and alkynes; (B) regioselective copper-catalyzed azide–alkyne Huisgen 1,3-dipolar cycloaddition. | ||
This copper-catalyzed azide–alkyne Huisgen 1,3-dipolar cycloaddition (CuAAC) has recently enjoyed tremendous interest as a synthetic route for the synthesis of complex materials because of its high conversion and mild experimental conditions. Azides and alkynes can be easily introduced in both synthetic polymers and biomolecules. The reaction can be performed at room temperature and in water, making it compatible with biological systems. Because this method involves a cycloaddition rather than a nucleophilic substitution, proteins can be modified with extremely high selectivity, so little or no by-products are formed and no protective groups are required.
CuAAC was first used on proteins for the labeling of protein capsids of the cowpea mosaic virus (CPMV) with a fluorescent dye.138 The possibilities of the CuAAC for bioconjugation with polymers were first shown by the site-specific PEGylation of human superoxide dismutase-1 (SOD), a key enzyme in preventing the formation of reactive oxygen species in cells (Scheme 17).139 The alkyne was introduced into the PEG polymer by coupling propargylamine with an NHS ester-activated PEG (75). The azide was incorporated into the SOD protein (74) by introducing p-azidophenylalanine (77) as a 21st amino acid (Scheme 18). This was achieved by extending the repertoire of the cell’s translational machinery with an orthogonal tRNA and tRNA synthetase.140 This gives the possibility to site-selectively introduce an azide, while keeping the rest of the primary structure unchanged. The CuAAC ligation was performed in a phosphate buffer at pH 8, resulting in a PEGylated SOD (76) that had approximately the same bioactivity as the native enzyme.
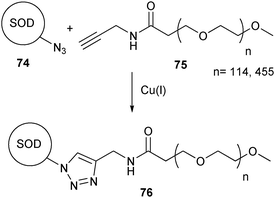 | ||
| Scheme 17 PEGylation of the SOD protein using CuAAc. | ||
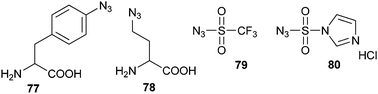
Alternatively, azide groups can be incorporated into recombinant proteins by expressing them in methionine-auxotrophic bacterial cultures growing in methionine-free medium which are supplemented with azido-homoalanine (78, Scheme 18).141 Because of the close resemblance of azido-homoalanine to methionine, azido-homoalanine is simply activated by methionyl-tRNA synthetase and thereby replaces each methionine in the proteins expressed. Although this means that several azido groups can be introduced into the protein, the low methionine abundance together with its hydrophobicity will limit the amount of surface accessible azides incorporated. Accordingly, this strategy was used to site-specifically PEGylate the enzyme Cal B.142
Instead of a biochemical approach, the azide group can also be introduced in a chemical manner into both peptides and proteins using diazo transfer agents (Scheme 18). Solid phase bound peptide amines can be easily converted into azides by a diazo transfer using triflyl azide (79) in the presence of divalent copper ions.143 The water-soluble diazo transfer agent 80144 can be used on proteins in an aqueous solution to substitute the amines of both lysine residues and the N-terminus by azide groups.145
In another example of CuAAC bioconjugation, the azide and alkyne functionalities switched places as an azide-terminated PS was coupled to either an alkyne-functionalized peptide or protein.146 The PS block was synthesized by ATRP and the terminal bromide was subsequently reacted with azidotrimethylsilane and tetrabutylammonium fluoride (TBAF) to introduce the azide group. The peptide with the sequence Gly–Gly–Arg was synthesized by SSPS and fluorescently labelled. While still on the solid support, the peptide was reacted with 3-butynylchloroformate, introducing the alkyne function onto the N-terminus. The subsequent CuAAC was performed in THF with the same copper catalyst used for the ATRP synthesis of the PS block. In order to demonstrate the scope of the reaction, the azide functional PS was also conjugated to an alkyne-functionalized BSA. Towards this goal, the available thiol from the cysteine-34 residue of BSA was first coupled to a maleimide bearing an alkyne. The alkynylated BSA was then reacted with the azide functional PS using the same reaction conditions as for the synthesis of the peptide–PS hybrid. Both peptide and protein bioconjugates displayed amphiphilic properties and their aggregation behaviour was investigated. In another example, the alcohol of α-hydroxy-ω-methoxy PEG was first tosylated and then substituted by sodium azide.147 The N-terminus of an oligopeptide was functionalized with pentynoic acid. The amphiphilic conjugates formed after CuAAC conjugation self-assembled into nanotubes, which formed soft hydrogels. In a final example, poly(oligo(ethylene glycol) acrylate) (POEGA) was prepared using ATRP and the terminal bromide group was replaced with an azide by reacting the polymer with sodium azide.148 The azido-POEGA was then reacted with the alkyne functional oligopeptide Gly–Gly–Arg–Gly–Asp–Gly.
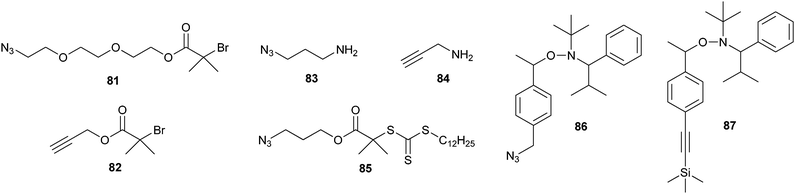
In the previous examples, the clickable groups on the polymer chain were introduced after the polymerization. An alternative strategy for obtaining clickable polymers is to use an alkyne or azido functional initiator. Scheme 19 shows some examples of azide- and alkyne-containing initiators for ATRP (81, 82),149,150 ring-opening polymerization (ROP) of amino acid N-carboxyanhydrides (NCA) (83, 84),150 RAFT (85)151 and nitroxide-mediated radical polymerization (86, 87).152 It has been shown that the azide and (protected) alkyne functionalities survive the polymerization conditions, effectively eliminating the need for postpolymerization modifications.
 | ||
| Scheme 19 Structures of azide- and alkyne-containing initiators. | ||
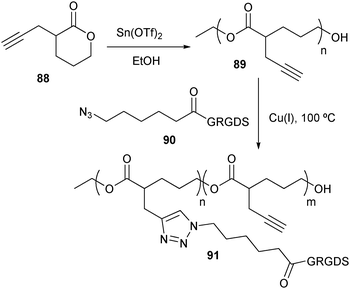
Besides the construction of linear bioconjugates, CuAAC has also been employed for the synthesis of polymers with a brush structure by introducing clickable groups in the monomer side chain (Scheme 20).153 A polyester with acetylene moieties was produced by ROP of α-propargyl-δ-valerolactone (88).154 Complementary to polyester 89, the amine terminus of a Gly–Arg–Gly–Asp–Ser oligopeptide sequence was first capped with 6-bromohexanoic acid, after which the bromide was substituted with an azide. Surprisingly the azide-terminated pentapeptide 90 could only be coupled to polyester 89 at 100 °C. Another branched structure was produced by coupling acetylene terminated dendrimers with azide-functional peptides via CuAAC.155 Because low reaction rates were observed, Liskamp et al. tried to accelerate the reaction by microwave irradiation. Varying yields were obtained, ranging from 14 to 97%, possibly due to steric hindrance.
 | ||
| Scheme 20 Side chain functionalization of a polyester with an oligopeptide using CuAAC. | ||
Even more complicated architectures are accessible by conjugating proteins to self-assembling block copolymers. An α,ω-diacetylene-functionalised PEG was first coupled to an azide-terminated PS via CuAAC to produce a PEG-b-PS polymer with an acetylene at its hydrophilic extremity. In water, these amphiphilic structures then assembled into polymersomes, placing the clickable groups at the surface of the aggregate. The accessibility of the acetylene was finally demonstrated by coupling either azido-functionalised horseradish peroxidase (HRP)145 or Cal B.156
In the area of surface immobilization, a heterotelechelic PEG linker carrying an alkyne and a cyclodiene terminal group was immobilized onto an N-(ε-maleimidocaproyl) functionalized glass slide via a Diels–Alder reaction.157 This resulted in an alkyne-terminated PEGylated surface, which could be used for the conjugation of azide-containing biomolecules via CuAAC in an aqueous solution.
The abovementioned examples impressively demonstrate the bio-orthogonality of the CuAAC. However, amino groups present in peptides and proteins can chelate the copper ions, thereby interfering with the cycloaddition reaction. Both the toxicity of the required copper, as well as the copper-induced denaturation of proteins158 furthermore indicate that this methodology needs to be improved. To this end, several copper-free ‘click’ reactions have been developed, which will be discussed in the next sections.
3.2 Staudinger ligation
In 1919 Staudinger and Meyer reported the reduction of azides using triphenylphosphine.159 This reaction involves the formation of an aza-ylide intermediate, which is hydrolyzed to generate an amine and triphenylphosphine oxide. When Saxon and Bertozzi incorporated a methoxy ester as an electrophilic trap into one of the phenyl rings (92), the nucleophilic aza-ylide 94 was captured before its hydrolysis (Scheme 21).160Via an intramolecular cyclization, the intermediate rearranged, and after hydrolysis ultimately coupled both reaction partners via an amide bond (96). Just like with the CuAAC, both reactive groups of this Staudinger ligation (i.e. the azide and the triphenylphosphine) are absent from peptides and proteins, and although they have a high intrinsic reactivity to one another, they do not react with any natural occurring functionalities. This property can be exploited in the synthesis of well-defined bioconjugates without the need for protecting groups.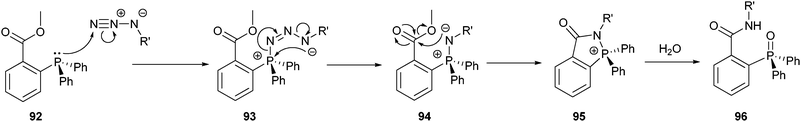 | ||
| Scheme 21 Mechanism of the Staudinger ligation. | ||
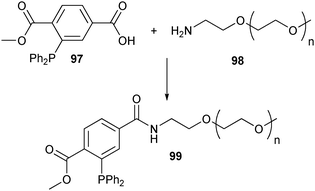
Accordingly, the Staudinger ligation was used for the site-specific PEGylation of azido-homoalanine containing trombomodulin.161 Functionalization of an amine-terminated PEG with phosphine 97, allowed PEG derivative 99 to be selectively conjugated to the C-terminus of the protein (Scheme 22).
 | ||
| Scheme 22 Functionalization of PEG with a phosphine moiety suited for the Staudinger ligation. | ||
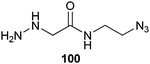
Alternatively, the C-terminus of RNase A was specifically functionalized with an azido group following the same strategy as for the synthesis of protein-based MESNA thioesters (Scheme 14). Here the target protein was expressed as an intein fusion protein and attacked by an azido-bearing α-hydrazine acetamide (100) (Scheme 23).162
Although the Staudinger ligation reaction works well, products retain a triaryl phosphine oxide moiety. In two independent reports163,164 traceless Staudinger ligations were reported in which the used phenylphosphine contained a cleavable linker (101), which was released once the aza-ylide attacked the carbonyl group (Scheme 24). The phosphonium species of the rearranged product 103 was subsequently liberated during the final hydrolysis step. Among the phosphines tested, 105 and 108 exhibited the best reactivity. One limitation of this ligation method was that it needed to be carried out in organic or mixed solvents. More recently however, a water soluble phenylphosphine (109) was developed, which should extend the applicability of this reaction.165 These traceless variants of the Staudinger ligation produce a clean amide bond after coupling, which might improve the biocompatibility of the prepared bioconjugates. However, the side-products formed might make purification of the product necessary.
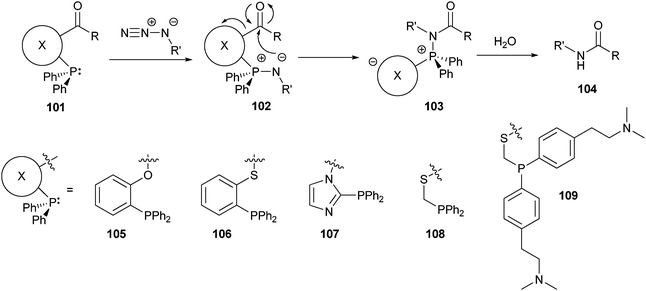 | ||
| Scheme 24 Mechanism of the traceless Staudinger ligation together with the structure of the cleavable linker incorporated in the phosphines tested. | ||
One of the first applications of the traceless Staudinger ligation was the assembly of peptide segments synthesized by SPPS into a final synthetic peptide. In contrast to NCL, the traceless Staudinger ligation does not need a cysteine residue at the ligation site. This freedom was first exploited in the total synthesis of RNase A.166 The full scope and the limitations of the Staudinger ligation in peptide chemistry still need to be explored, however. Questions about side chain dependence and whether or not protecting groups are required remain unanswered.
The use of the traceless Staudinger ligation has now also been extended to the immobilization of proteins via a surface-tethered PEG spacer.167,168 By having phosphine 106 on a PEG spacer, RNAse A was covalently coupled after which its immobilization was evaluated by a ribonucleolytic activity assay and immunostaining.
3.3 Hydrazone and oxime ligation
Instead of using an azide functionality for either a CuAAC or a Staudinger ligation, proteins and peptides can be site-selectively conjugated using ketones and aldehydes. These groups are not commonly present in natural proteins and can react with either hydrazide (110) or hydroxylamine (114) groups forming a hydrazone (112) or oxime (115) bond, respectively (Scheme 25).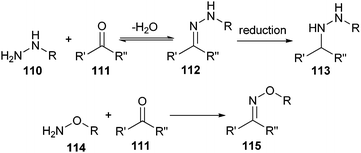
Hydrazone chemistry was originally developed to couple polypeptide fragments together into artificial proteins.169 Oxime chemistry was first employed to modify proteins with small organic substances,170 but was later also used for the ligation of polypeptide fragments.171 As a side reaction during these ligations, amino groups of proteins can form a Schiff base with the ketones or aldehydes, which can lead to crosslinked aggregates. However, this side reaction can be suppressed under acidic conditions, which protonates the amino groups. Because hydrazides and hydroxylamines are weaker bases than primary amines, the desired reaction can still occur. In this way, these coupling reactions can be performed regioselectively, making them good candidates for the well-defined formation of bioconjugates without the need for protecting groups during the conjugation step. Before elaborating on these conjugation methods, we will describe how to introduce ketone or aldehyde groups into proteins and peptides.
When working with glycoproteins, the incorporated carbohydrate provides a target that can be oxidized under mild conditions to afford the necessary ketones and aldehydes within the protein.172–174 Naturally occurring glycosylation sites are usually well removed from the protein active site and are readily accessible, making them excellent attachment sites for polymer chains. Using this method, multiple attachment sites are generated on the carbohydrate, but all modifications are confined to the glycosylation site. When no glycosylation sites are available, they can be engineered into the protein.175,176
Another way of introducing ketones or aldehydes into proteins is to oxidize the N-terminal serine or threonine with sodium periodate under mild conditions.177 A method with fewer restrictions on the nature of the N-terminal amino acid groups is the metal-catalyzed transamination.177 However, the conditions for this reaction are known to be potentially harmful to the protein. Also, both oxidation methods have only been used with varying success together with oxime coupling.
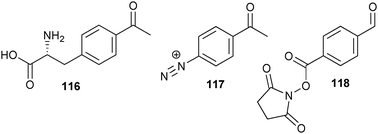
A more general strategy for the incorporation of ketones in larger proteins was first demonstrated by Tirrell et al., which is analogous to the earlier described multisite replacement approach of methionine for azido-homoalanine.178 Briefly, E. coli’s tRNA-synthetase for phenylalanine was mutated in such a way that it could also accept p-acetylphenylalanine (116) (Scheme 26). By feeding phenylalanine auxotrophic cell lines p-acetylphenylalanine instead of phenylalanine, the unnatural amino acid was incorporated at all phenylalanine positions. Schultz et al. created a cell line that introduced ketones site-specifically into proteins by using an orthogonal tRNA and tRNA synthetase couple, which made p-acetylphenylalanine amenable for ribosomal protein production incorporating it site-specifically as a 21st amino acid.179
 | ||
| Scheme 26 Structures of a ketone-containing amino acid and ketone-bearing tags. | ||
Another possibility is to react specific residues with ketone-bearing tags. In one such example, the tyrosine residues of the protein capsid of the tobacco mosaic virus (TMV) were reacted with p-acetylbenzenediazonium salt (117).180 The resulting surface-exposed ketones were coupled to hydroxylamine-functional PEG. In another example, NHS esters of p-formylbenzoic acid (118) were efficiently reacted with lysine residues in order to install aldehydes on the exterior of the MS2 capsid.181 Since this kind of chemistry is not site-specific, ketones can be introduced at multiple sites. So, even though the oxime conjugation itself is bio-orthogonal, constructing well-defined bioconjugates with this approach can be difficult.
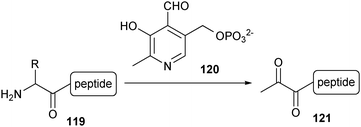
A more controlled way of introducing a ketone functionality into proteins was described by the group of Francis.182,183N-terminal amino acids can undergo a transamination reaction upon exposure to pyridoxal 5′-phosphate (120, PLP) at 37 °C in an aqueous solution (pH 6.5), resulting in the corresponding pyruvamide derivative (121) (Scheme 27). Because of the selectivity for the N-terminal amino group, the ketone is introduced at a single location, leaving the amines of the lysine side chains unchanged. This reaction was first demonstrated on proteins with an N-terminal glycine, valine, lysine and methionine,182 and more recently for N-terminal aspartic and glutamic acid.183 Apart from serine, threonine, cysteine, tryptophan and proline, it is expected that this reaction should also be applicable for the remaining natural amino acids at the N-terminus.
Hydrazide can be easily introduced into PEG184 and be reacted with ketones and aldehydes to produce a hydrazone linkage under acidic conditions. The hydrazone linkage is not very stable but may be reduced with sodium cyanoborohydride to a more stable alkyl hydrazide (113). In a proof-of-concept, it was shown that hydrazone formation is compatible with and orthogonal to CuAAC.185 Poly(norbornene)-based random copolymers possessing both ketone and azide functionalities were synthesized using ring-opening metathesis polymerization. A subsequent one-pot functionalization step with a library of small organic and biological molecules provided multifunctional polymers.
Alternatively, a ketone- or aldehyde-functionalized polypeptide can be attached to a hydroxylamine-bearing polymer. The oxime bond which is formed is more stable than the hydrazone linkage and does not need a successive reduction to produce a stable product. Using oxime chemistry, Kochendoerfer and co-workers site-selectively attached two branched polymer chains efficiently at two of the four glycosylation sites of synthetic erythropoiesis protein (SEP).36 The protein was synthesized as four individual peptide fragments by SPPS, where a non-coded, ketone-bearing amino acid, Nε-levulinyl lysine, was introduced at the desired positions. The PEG-based polymer was also synthesized on solid-phase resin and functionalized with hydroxylamine. The final construct was assembled by first coupling the PEG chains to the target sites with oxime-forming conjugation, followed by ligation of the four fragments using NCL. In a second study, this strategy was used to screen several SEP-polymer bioconjugates for their activity.186 Using the same strategy, the small anti-HIV protein CCL-5 was site-specifically PEGylated.187
Just like hydrazone formation, oxime ligation also has to be performed under acidic conditions. Since many proteins are unstable at low pH it would be desirable to perform the ligation under neutral conditions. This has recently been made possible by the addition of p-methoxyaniline, which acts as a nucleophilic catalyst and accelerates the reaction rate.188
Besides solid-phase chemistry, there have been other approaches developed for the convenient functionalization of polymers with a hydroxylamine moiety. One example is the solution-based displacement of the terminal alcohol group by N-hydroxyphthalimide under Mitsunobu conditions.180 Another example used tert-butyloxycarbonyl (Boc) protected hydroxylamine initiators for ATRP.189 Using such controlled radical initiators, HEMA, NIPAAm and poly(ethylene glycol) methacrylate (PEGMA) were polymerized with low polydispersities (PDI). Deprotection with trifluoroacetic acid liberated the hydroxylamine moiety, allowing a chemospecific coupling with Nε-levulinyl lysine-functionalized BSA.
In a further variation of the oxime coupling method, the two moieties used, i.e. the hydroxylamine and the ketone or aldehyde switch places. In this opposite approach, a PEG-based polyamide chain was functionalized with terminal serine residues, after which the alcohol functions were oxidized with periodate into glyoxylyl groups.190 Complementary to this, a hydroxylamine group was attached to the N-terminus of a peptide using Boc-aminooxyacetyl-N-hydroxysuccinimide during SPPS. In the final step, a well-defined bioconjugate was synthesized by coupling the synthetic polymer to the peptide through the formation of an oxime bond. In the same way, an aminooxyacetic acid was protected with acetone and incorporated into CCL-5 using SPPS.191 After deprotection, the peptide was conjugated to an aldehyde-functionalized PEG.
Using the same set-up, dendrimers were functionalized with a peptide.192 An amine-functional dendrimer was decorated with ketone groups by coupling levulinic acid using a carbodiimide. The twenty-mer peptide Glu–Tyr–Leu–Asn–Lys–Ile–Gln–Asn–Ser–Leu–Ser–Thr–Glu–Trp–Ser–Pro–Ala–Ser–Val–Thr was equipped with an N-terminal aminooxyacetic acid residue during SPPS and subsequently cleaved from the resin. Oxime ligation in an aqueous phosphate buffer afforded an average dendrimer loading of 63%. Attempts to fully load the dendrimer failed, probably due to steric crowding.
This methodology has also been translated to the chemoselective surface immobilization of peptides. Surface-bound hydroquinone was selectively oxidized to the ketone bearing benzoquinone, after which hydroxylamine-functional peptides were coupled via oxime ligation.193 Because of the incorporated redox activation step, the extent of immobilization could be modulated by changing the electrochemical potential of the surface. Alternatively, oxime ligation can be used together with lithography for surface patterning (Scheme 28). Surface-bound hydroxylamine groups were protected with nitroveratryloxycarbonyl (122, NVOC), which were selectively removed by shining ultraviolet light through a patterned photomask.194 A peptide bearing an N-terminal ketone (124) was subsequently immobilized according to the predetermined surface patterns.
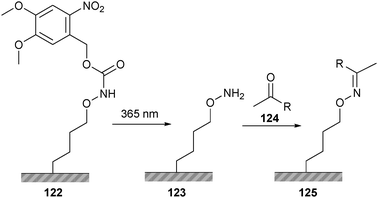 | ||
| Scheme 28 Surface patterning using oxime ligation and the photo-labile protection group NVOC. | ||
3.4 Miscellaneous copper-free click reactions
Several additional copper-free click reactions have been developed within chemical biology in recent years. Several methods have been developed to increase the reactivity of dipolarophiles towards azides in 1,3-dipolar cycloaddition. Prime examples are the oxanorbornadiene-based tandem cycloaddition–retro-Diels–Alder (crDA) reaction (126),195 the difluorinated cyclooctyne- (129)196 and dibenzocyclooctyne-based (132)197 strain-promoted azide–alkyne [3 + 2] cycloadditions (Scheme 29).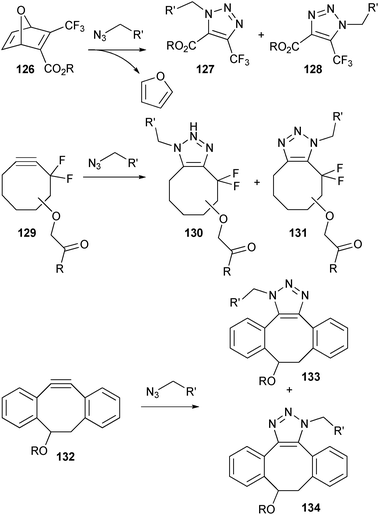 | ||
| Scheme 29 Examples of copper-free click reactions based on 1,3-dipolar cycloaddition with azides. | ||
All three reactions proceed with high yields and without the formation of interfering by-products. When comparing the extent of functionalization of an azide-containing coating, both the oxanorbornadiene-based crDA reaction and dibenzocyclooctyne-based cycloaddition gave better results than the CuAAC and Staudinger reactions.198 Solubility in aqueous solutions, however, remains an issue, with the oxanorbornadiene being the most hydrophilic. Examples of bioconjugations using these reactive groups are still limited,199 and most studies focus on the labeling of proteins with small organic probes. From these studies, however, it is apparent that these methods are good candidates for selective bioconjugation.
Lin and co-workers reported a method using tetrazoles (135), which generate nitrile imines (136) under UV light that can subsequently react with substituted alkene dipolarophiles (Scheme 30).200 This photoactivated 1,3-dipolar cycloaddition reaction was used to selectively label a genetically encoded alkene-containing protein (137). In another recent example of a 1,3-dipolar cycloaddition as a bioconjugation tool, norbornene-modified DNA (139) was smoothly reacted with several modified nitrile oxides (140).201
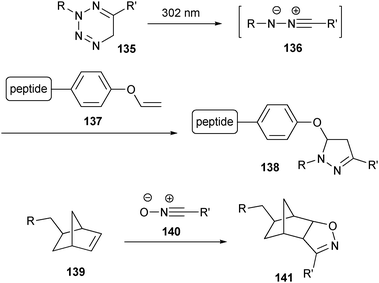 | ||
| Scheme 30 Examples of copper-free click reactions based on 1,3-dipolar cycloadditions with nitrile imines and nitrile oxides. | ||
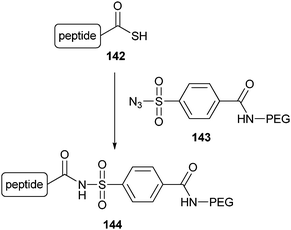
Another bioorthogonal coupling reaction, which was recently used for C-terminal PEGylation, is based on the thioacid/azide amidation reaction.202 In short, protein splicing of a recombinant protein yielded the small ubiquitin protein with a thioacid group at the C-terminus (142), which was subsequently selectively amidated by the electron-deficient sulfoazide 143 (Scheme 31). Only mono-PEGylated product was obtained, confirming the chemoselectivity of the reaction. However, the reaction was accompanied by some hydrolysis resulting in a yield of only about 65%.
Diels–Alder reactions are now also being explored as possible conjugation methods (Scheme 32). In one example, an inverse electron demanding Diels–Alder reaction between trans-cyclooctenes (145) and tetrazines (146) was followed by a retro-[4 + 2] cycloaddition, producing the conjugated product and nitrogen in quantitative yield.203,204
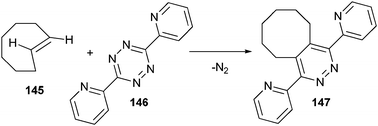 | ||
| Scheme 32 Conjugation reaction between trans-cyclooctene and tetrazine. | ||
Very fast conjugation of macromolecular blocks was shown to be possible between the thiocarbonate endgroup of a RAFT polymer (148) and a cyclopentadienyl-functionalized polymer (149).205 This retro-Diels–Alder reaction can be accelerated by using trifluoroacetic acid (TFA) as a catalyst, resulting in quantitative yields in just a few minutes (Scheme 33).
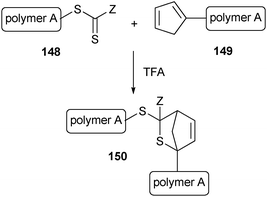 | ||
| Scheme 33 Conjugation of two macromolecular blocks though a Diels–Alder reaction. | ||
4. Grafting to using non-covalent interactions
Instead of covalently coupling polymers and proteins, bioconjugates can also be prepared by strongly coordinating polymers to proteins. This can be done by the use of cofactors. Cofactors are small organic molecules that are positioned in the active site of certain proteins and they are usually of great importance for the activity of these proteins. The cofactor can be removed from the protein and attached to a polymer. By simple mixing of the protein and the polymer-functionalized cofactor, a bioconjugate is created when the cofactor is reconstituted in the pocket of the protein. This strategy is efficient, provided that the modification of the cofactor does not hinder the complexation with the protein.This strategy was first explored using the well-studied biotin–(strept)avidin system. The cofactor biotin (vitamin B7) has an affinity for avidin (Ka ≈ 1015 M−1) or streptavidin (Ka ≈ 1013 M−1) which is so high that it can be regarded as irreversible.206 The naturally occurring carboxylic acid of biotin provides an easy way to couple it to pre-made polymers207,208, block copolymers,209,210 dendrimers,211 polymer brushes212 or for incorporation into crosslinked thin films.213,214 In order to circumvent the need for postpolymerization modifications, biotin can already be incorporated into the initiator that is used for the synthesis of the polymer. This approach has been carried out for azonitrile initiators,215 cyanoxyl-mediated polymerization,216,217 ATRP218–220 and RAFT.221,222 Because the acid group is not involved in the complexation with the protein, these modifications do not change the binding characteristics much. To facilitate a smooth incorporation of the cofactor into the protein, a hydrophilic207,211,218–220 or hydrophobic208,209,213,214 spacer can be included between biotin and the polymer chain.
Similarly, one of the carboxylic acids of the heme cofactor protoporphyrin IX was used for the attachment of a polystyrene chain. The heme-functionalized polymer was subsequently reconstituted into the HRP enzyme223 or the oxygen-binding protein myoglobin (Mb).224 The resulting giant amphiphiles were found to assemble into spherical aggregates, presumably vesicles. When this method was used to attach a PS-b-PEG copolymer to HRP and Mb, the obtained protein-containing ABC triblock formed a variety of more complicated aggregate structures.225 In all three examples, a hydrophilic spacer was built between the hydrophobic polystyrene and the cofactor to assure the compatibility between polymer and protein. Even so, the reconstitution had a negative effect on the biological activity of both heme proteins. In the case of HRP, the enzymatic activity decreased. For Mb, the stability of the oxy complex was reduced. This negative effect is probably due to a disturbed binding of the cofactor in the protein or a restricted access of substrates to the active site caused by the presence of the polymer chain. This illustrates that even when the position of polymer attachment is precisely known and controlled, it can still have a negative effect on protein activity when it is near the active site.
In the area of stimuli-responsive drug delivery, non-covalent interactions were used in the formation of hydrogels via the interaction of PEG-based heparin-functionalized star-polymers and the dimeric protein vascular endothelial growth factor (VEGF), which can bind heparin and acted as a crosslinker during gel formation.226 Stimuli-responsive erosion of the hydrogel was triggered after exposure to particles decorated with the VEGF receptors that bound the VEGF protein. When loaded with drugs, this hydrogel could therefore potentially be used for targeted delivery on the basis of ligand–receptor interactions.
5. Grafting from
We have seen that high requirements are put on the coupling chemistry when using the grafting to strategy, especially with regards to yield and specificity. Instead of conjugating a preformed polymer to a protein or peptide, a polymer can also be grown from these biological molecules. Alternatively, an amine terminal polymer can act as a macroinitiator of a polypeptide using ROP of amino acid N-carboxyanhydrides (NCA). The benefit of these strategies is that the need for a conjugation between macromolecules is averted, because only a small initiator moiety needs to be introduced to either the biological component or the polymer. The lower steric hindrance during the functionalization of the macromolecules simplifies the procedure. Furthermore, the product can be easily purified once the polymer is grown from the macroinitiator, as only the unreacted monomer has to be removed, as opposed to a preformed polymer.5.1 Peptide macroinitiators
One of the first examples of this strategy was reported by Börner et al. who used an oligopeptide as a macroinitiator.227 The pentapeptide Gly–Asp(t-Bu)–Gly–Phe–Asp(t-Bu) was synthesized by SPPS and the N-terminus was acylated with 2-bromopropionic acid, a standard initiator for ATRP. Next, the oligopeptide initiator was carefully cleaved from the solid support so as to avoid side group deprotection, and used for the ATRP of n-butyl acrylate. The resulting oligopeptide–poly(n-butyl acrylate) block copolymer had a number average molecular weight (Mn) of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and a PDI of approximately 1.19. However, interactions between the copper catalyst and the oligopeptide were observed, which caused a slow polymerization rate and a decrease of propagating radicals during the polymerization. Other reports described the same effects and slightly higher PDI’s (1.22–1.37) when using this strategy.228 This observation was ascribed to a low initiator efficiency of the amide-containing initiator. To overcome these drawbacks, oligopeptide-based initiators were synthesized for RAFT polymerizations. Using this copper-free controlled polymerization method, n-butyl acrylate (PDI ∼ 1.1)229,230 NIPAAm (PDI ∼ 1.2)230 and oligo(ethylene glycol) acrylate (OEGA) (PDI ∼ 1.2)230 were polymerized from peptide macroinitiators.
000 g mol−1 and a PDI of approximately 1.19. However, interactions between the copper catalyst and the oligopeptide were observed, which caused a slow polymerization rate and a decrease of propagating radicals during the polymerization. Other reports described the same effects and slightly higher PDI’s (1.22–1.37) when using this strategy.228 This observation was ascribed to a low initiator efficiency of the amide-containing initiator. To overcome these drawbacks, oligopeptide-based initiators were synthesized for RAFT polymerizations. Using this copper-free controlled polymerization method, n-butyl acrylate (PDI ∼ 1.1)229,230 NIPAAm (PDI ∼ 1.2)230 and oligo(ethylene glycol) acrylate (OEGA) (PDI ∼ 1.2)230 were polymerized from peptide macroinitiators.
Instead of functionalizing the N-terminus, amino acid side groups can be used as initiation sites. A peptide-based amphiphilic ABA triblock copolymer was prepared by the solid phase synthesis of a peptide with the sequence Ser–Ala–Gly–Ala–Gly–Glu–Gly–Ala–Gly–Ala–Gly–Ser–Gly.231 Before the peptide was cleaved from the solid support, the alcohol side groups of the two serines were functionalized with an α-bromo ester moiety to create a bifunctional ATRP initiator. Using living radical polymerization, methyl methacrylate (MMA) was polymerized in solution yielding a well-defined ABA triblock copolymer. Upon suspension of this amphiphilic triblock in a mixture of tetrahydrofuran and water, followed by the removal of tetrahydrofuran, polymersomes were formed. Alternatively, Fmoc-protected serine was first modified with an ARTP232 or nitroxide-mediated radical polymerization (NMP) initiator233 and then incorporated into a peptide by SPPS. This strategy allows for a polymer modification at a specific site without having to rely on a distinctly different chemistry of the targeted residue within the sequence.
Biesalski et al. prepared cyclic peptides with an alternating D- and L-amino acid sequence that self-assemble into hollow nanotubes.234,235 After the preparation and cyclization of the peptide on the solid support, the three available lysine residues were functionalized with an α-bromo ester group and the cyclic peptide initiator was cleaved from the resin. In solution, the ATRP initiating sites were exposed on the outer surface of the peptide nanotubes and polymerization yielded peptide assemblies decorated with polyNIPAAm234 or polybutylacrylate.235
Instead of synthesizing the peptide on the solid support and sequentially performing the polymerization in solution, Wooley and co-workers have shown that the entire bioconjugate can be synthesized on the resin. By functionalizing the N-terminus of a peptide with an alkoxyamine, a poly(acrylic acid)-b-poly(methyl acrylate) (PAA-b-PMA) copolymer was synthesized via NMP while keeping the macroinitiator on the resin.236 This method has also been extended to ATRP,237,238 showing the general applicability of this strategy.
5.2 Protein macroinitiators
Macroinitiations from complete proteins were initially conducted by simple free radical polymerization in the presence of proteins. The initiators are generally redox-,239 thermally-240 or radiation-induced.241 This creates radicals on the protein surface from which polymers were grown. In these examples, however, the grafting of the polymer occurs at completely random positions and there is no control over the amount of polymer chains attached.This approach was made more specific by Matyjaszewski et al. by attaching 2-bromoisobutyryl bromide to the lysine residues of α-chymotrypsin (αCT).242 Up to 8 initiating moieties were coupled from which PEGMA chains with a low PDI were grown via ATRP. However, the exact location of attachment could not be controlled.
Functionalization of cysteines gives better control over the location of the initiating site. For this purpose, BSA was first reduced. This freed three cysteines for functionalization with a pyridyl disulfide or maleimide ATRP initiator from which NIPAAm, PEGMA and dimethylaminoethyl methacrylate (DMAEMA) chains were grown.243,244 In the case of the pyridyl disulfide initiator, the disulfide bond between BSA and the polymer was reduced after polymerization. The detached polyNIPAAm was analysed by gel permeation chromatography (GPC) indicating a PDI of 1.34. This work was reproduced for the macroinitiation of PEGMA and NIPAAm using RAFT polymerization.245,246 This approach was also applied to a lysozyme mutant bearing a single cysteine, which was functionalized with an ATRP initiator.243
This macroinitiation strategy can be combined with a non-covalent coupling strategy. This was demonstrated using the abovementioned biotin–streptavidin couple. A biotinylated initiator was complexed in the streptavidin binding pocket. Subsequently macroinitiation from the protein complex was performed.247
5.3 Polymer macroinitiators
In the section concerned with SPPS, we saw that a peptide can be grown from a polymer using solid phase synthesis in a stepwise fashion.50–56 Using amine functionalized polymers (151), it is also possible to initiate the ROP of amino acid NCAs (152), forming poly(amino acids) (Scheme 34). Recent reviews have extensively discussed the synthesis of linear peptide–polymer block copolymers synthesized in this manner and the reader is referred to these papers for a more detailed overview.248,249 | ||
| Scheme 34 ROP of NCAs using a primary amine initiator and a nickel catalyst. bipy = 2,2′-bipyrydine; COD = 1,5-cyclooctadiene. | ||
The major drawback of the conventional amine-initiated NCA polymerization is that it is plagued by chain-breaking transfer and termination reactions, which prevent the poly(peptide) chain length from being controlled accurately. This problem can be overcome by using high vacuum techniques in order to create and maintain the conditions necessary for the living polymerization of NCAs with primary amines.250 A second way to get better control over the poly(peptide) synthesis is to replace the free primary amine initiator with its corresponding amine hydrochloride salt.251
Enhanced control of the NCA polymerization can also be obtained by using a zero-valent nickel complex (154) as the initiator.252 The transition metal mediates in the addition of monomers to the active polymer chain-end (155), suppressing chain-transfer and termination side reactions (Scheme 34).
More complex architectures are also accessible by using branched star-shaped macroinitiators. In one example, a star-shaped PEG with four terminal amines was used as a macroinitiator for the ROP of γ-benzyl-L-glutamate NCA.253 The outer poly(γ-benzyl-L-glutamate) blocks yielded a self-assembling star-shaped polymer–peptide copolymer. Similarly, a triarm, star-shaped PS was synthesized using ATRP.254 The three terminal bromides were first replaced by azides and then transformed via a Staudinger reduction. The resulting amine-terminated triarm was used to grow three poly(γ-benzyl-L-glutamate) arms onto the branched structure.
In another example of this macroinitiator strategy, a dual initiator was synthesized containing a primary amine and a nitroxide group.255 ROP yielded poly(γ-benzyl-L-glutamate) with high structural control. Macroinitiation of styrene by nitroxide-mediated controlled radical polymerization yielded a block copolymer with a polydispersity around 1.1. Both polymerizations were also successfully conducted in one pot without intermediate isolation owing to the high compatibility of both polymerization techniques.
6. Grafting through
An interesting peptide–polymer bioconjugate architecture places the peptide moieties as side chain functionalities on a polymer backbone. When aiming for these comb-shaped structures, the grafting to strategy can fall short as it is very difficult to achieve quantitative functionalization. To overcome this problem, another strategy has been developed in which the peptide is attached to the monomer unit. Therefore, after polymerization, every monomer is inherently functionalized. The disadvantage of this strategy is that synthesizing peptide-functionalized monomers is not trivial, and compatibility issues between the polymerization technique and peptide moiety have to be taken into account.As one of the first examples, the dipeptide Leu–Ala was introduced to both methacrylamide256 and methacrylate257 units, and polymerized by free radical polymerization. Extension of this procedure to an acrylamide with a Leu–Ala-based oligopeptide side chain, afforded an oligomer with a degree of polymerization between 3–8.258 The peptide moiety of the resulting product maintained its α-helical structure after polymerization in chlorobenzene, but was lost in DMF.
In another early example, norbornene-derived peptide-functionalized monomers with the cell adhesive sequences Gly–Arg–Gly–Asp and Ser–Arg–Asn (158) were polymerized via ring-opening metathesis polymerizations (ROMP) using a Grubbs’ catalyst (Scheme 35).259 Incorporation of a PEG-functionalized monomer (157) was found to prevent premature precipitation of the polymer during the course of polymerization and yielded a water soluble product. In a further study, a polymer substituted with a Gly–Arg–Gly–Asn–Ser peptide was shown to be significantly more active than the free peptide.260 This result demonstrates that the increased local concentration of peptides in comb-shaped bioconjugates can lead to multivalent interactions and thereby significantly enhance the observed biological activity. In a further example of the use of the Grubbs’ catalyst, dipeptide olefin monomers (160) were polycondensated via acyclic diene metathesis (ADMET) (Scheme 35).261
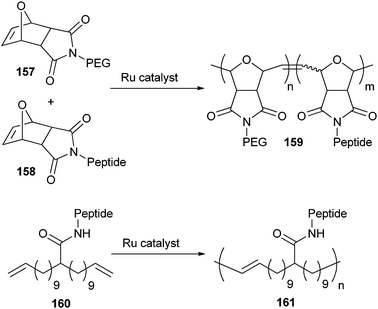 | ||
| Scheme 35 ROMP and ADMET of peptide-functionalized monomers. | ||
Isocyanide moieties have also been introduced as monomeric units on the N-terminus of oligopeptides.262,263 These monomers were subsequently polymerized using a nickel catalyst and formed rigid β-helical rods. This rigidity was caused by the hydrogen bonding network formed between the amide groups of the peptide side chains. Moreover, it was shown that the handedness of the helix could be steered by changing the enantiomeric configuration of the peptide side chain. Introduction of a hydrophobic PS block into these peptide-based materials furthermore resulted in the formation of polymersomes.264
The recent emergence of controlled radical polymerizations has provided a new momentum to the field of peptide-functionalized monomers by presenting better control over the polymerization reaction. The possibilities were first illustrated with the ATRP polymerization of a methacrylate coupled to Val–Pro–Gly–Val–Gly, a pentapeptide sequence derived from the structural protein tropoelastin, which exhibits LCST behaviour (162) (Scheme 36).265 The polymerization of this monomer with the initiator ethyl-2-bromo-2-methylpropionate (163, Ebib) proceeded in a controlled fashion and yielded polymers with a reasonably low PDI (1.25). Using a bifunctional PEG-based macroinitiator (165) it was even possible to synthesize triblock copolymers with fairly narrow molecular weight distribution (PDI 1.27). LCST of this ABA type block copolymer could be influenced by changing the degree of polymerization, polymer concentration, and pH.266 Using the same bifunctional initiator, monomers with an Ala–Gly–Ala–Gly side chain were also polymerized in a controlled manner (PDI 1.12).267 The living character of the ATRP process was furthermore illustrated by extending the block copolymer with a further MMA block via an in situ macroinitiation (PDI 1.17).
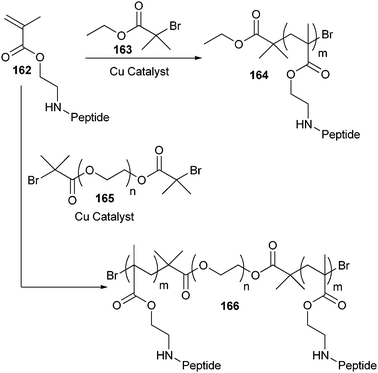 | ||
| Scheme 36 ATRP of peptide-functionalized monomers. | ||
It was possible to polymerize even bulkier peptide-functionalized monomers with ATRP. The cyclic β-sheet forming decapeptide gramicidin S was modified with a methacrylate handle and subsequently polymerised via ATRP.268 Although the polymerization was very slow, first-order kinetics were observed and a low PDI was obtained (1.09). The secondary structure of the peptide moiety was retained within the resulting polymer, as indicated by IR spectroscopy.
The study with the Val–Pro–Gly–Val–Gly-functionalized methacrylate monomer was repeated with RAFT.269 Using this technique, polymers with a higher degree of polymerization and lower PDI’s were isolated. In ATRP the copper catalyst might complex with the amide bonds in the peptide side chains and be deactivated. The absence of this peptide-induced deactivation in RAFT explains the improved polydispersities which were obtained.
7. Conclusions
In conclusion, the combination of synthetic polymers with polypeptides has opened the path towards a great new set of materials with unprecedented properties obtained in part because of the intrinsic (bio)functionalities of proteins and peptides. For many years, applications of polypeptide-polymer bioconjugates have been focussed on the field of drug delivery. New applications which are rapidly developing include diagnostics, tissue engineering and bioactive surfaces. The self-assembly properties of certain types of bioconjugates might make them useful components for nanotechnological applications, although this field is still on a more fundamental level.As these applications become ever more sophisticated, the requirement for selective and even specific coupling methods increases. This development has been illustrated in this review, where we have shown the advancement from early day coupling methods based on nucleophilic attacks of amines present in proteins and peptides, to the introduction of bio-orthogonal ‘click’ reactions. In the future, this trend will only continue into the expansion of more facile and widely applicable bio-orthogonal conjugation methods.
Acknowledgements
This work was supported by the Dutch technology foundation STW.References
- J. A. Opsteen and J. C. M. van Hest, in Macromolecular engineering. Precise Synthesis, Materials Properties, Application, ed. K. Matyjaszewski, Y. Gnanou and L. Leibler, Wiley, Weinheim, 2007, vol. 4, pp. 2645–2687 Search PubMed.
- H. A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1–17 CrossRef CAS.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459–476 CrossRef CAS.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS.
- A. S. Hoffman and P. S. Stayton, Macromol. Symp., 2004, 207, 139–151 CrossRef CAS.
- M. A. Gauthier and H. A. Klok, Chem. Commun., 2008,(23), 2591–2611 RSC.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, San Diego, 1996 Search PubMed.
- P. Wirth, J. Souppe, D. Tritsch and J. F. Biellmann, Bioorg. Chem., 1991, 19, 133–142 CrossRef CAS.
- O. B. Kinstler, D. N. Brems, S. L. Lauren, A. G. Paige, J. B. Hamburger and M. J. Treuheit, Pharm. Res., 1996, 13, 996–1002 CrossRef CAS.
- P. Caliceti and F. M. Veronese, Adv. Drug Delivery Rev., 2003, 55, 1261–1277 CrossRef CAS.
- A. Abuchowski, J. R. McCoy, N. C. Palczuk, T. Vanes and F. F. Davis, J. Biol. Chem., 1977, 252, 3582–3586 CAS.
- A. P. Chapman, Adv. Drug Delivery Rev., 2002, 54, 531–545 CrossRef CAS.
- A. Basu, K. Yang, M. L. Wang, S. Liu, R. Chintala, T. Palm, H. Zhao, P. Peng, D. C. Wu, Z. F. Zhang, J. Hua, M. C. Hsieh, J. Zhou, G. Petti, X. G. Li, A. Janjua, M. Mendez, J. Liu, C. Longley, Z. Zhang, M. Mehlig, V. Borowski, M. Viswanathan and D. Filpula, Bioconjugate Chem., 2006, 17, 618–630 CrossRef CAS.
- M. S. Hershfield, R. H. Buckley, M. L. Greenberg, A. L. Melton, R. Schiff, C. Hatem, J. Kurtzberg, M. L. Markert, R. H. Kobayashi, A. L. Kobayashi and A. Abuchowski, N. Engl. J. Med., 1987, 316, 589–596 CAS.
- A. Abuchowski, T. Vanes, N. C. Palczuk and F. F. Davis, J. Biol. Chem., 1977, 252, 3578–3581 CAS.
- Y. Gotoh, M. Tsukada, N. Minoura and Y. Imai, Biomaterials, 1997, 18, 267–271 CrossRef CAS.
- G. E. Francis, D. Fisher, C. Delgado, F. Malik, A. Gardiner and D. Neale, International Journal of Hematology, 1998, 68, 1–18 Search PubMed.
- H.-J. Gais and S. Ruppert, Tetrahedron Lett., 1995, 36, 3837–3838 CrossRef CAS.
- J. F. King and M. S. Gill, J. Org. Chem., 1996, 61, 7250–7255 CrossRef CAS.
- K. Bergström, K. Holmberg, A. Safranj, A. S. Hoffman, M. J. Edgell, A. Kozlowski, B. A. Hovanes and J. M. Harris, J. Biomed. Mater. Res., 1992, 26, 779–790 CrossRef CAS.
- A. Abuchowski, G. M. Kazo, C. R. Verhoest, T. Vanes, D. Kafkewitz, M. L. Nucci, A. T. Viau and F. F. Davis, Cancer Biochem.Biophys., 1984, 7, 175–186 Search PubMed.
- T. Miron and M. Wilchek, Bioconjugate Chem., 1993, 4, 568–569 CrossRef CAS.
- S. Zalipsky, R. Seltzer and S. Menonrudolph, Biotechnol. Appl. Biochem., 1992, 15, 100–114 CAS.
- US Pat., 5,672,662, 1997.
- V. Ladmiral, L. Monaghan, G. Mantovani and D. M. Haddleton, Polymer, 2005, 19, 8536–8545 CrossRef.
- F. Lecolley, L. Tao, G. Mantovani, I. Durkin, S. Lautru and D. M. Haddleton, Chem. Commun., 2004,(18), 2026–2027 RSC.
- C. O. Beauchamp, S. L. Gonias, D. P. Menapace and S. V. Pizzo, Anal. Biochem., 1983, 131, 25–33 CrossRef CAS.
- F. M. Veronese, R. Largajolli, E. Boccu, C. A. Benassi and O. Schiavon, Appl. Biochem. Biotechnol., 1985, 11, 141–152 CrossRef CAS.
- K. T. Wiss, O. D. Krishna, P. J. Roth, K. L. Kiick and P. Theato, Macromolecules, 2009, 42, 3860–3863 CrossRef CAS.
- M. Zambianchi, F. D. Maria, A. Cazzato, G. Gigli, M. Piacenza, F. D. Sala and G. Barbarella, J. Am. Chem. Soc., 2009, 131, 10892–10900 CrossRef CAS.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- M. Z. Atassi and T. Manshouri, J. Protein Chem., 1991, 10, 623–627 CrossRef CAS.
- G. G. Kochendoerfer, S. Y. Chen, F. Mao, S. Cressman, S. Traviglia, H. Y. Shao, C. L. Hunter, D. W. Low, E. N. Cagle, M. Carnevali, V. Gueriguian, P. J. Keogh, H. Porter, S. M. Stratton, M. C. Wiedeke, J. Wilken, J. Tang, J. J. Levy, L. P. Miranda, M. M. Crnogorac, S. Kalbag, P. Botti, J. Schindler-Horvat, L. Savatski, J. W. Adamson, A. Kung, S. B. H. Kent and J. A. Bradburne, Science, 2003, 299, 884–887 CrossRef CAS.
- G. W. M. Vandermeulen and H. A. Klok, Macromol. Biosci., 2004, 4, 383–398 CrossRef CAS.
- P. Caliceti, O. Schiavon, L. Sartore, C. Monfardini and F. M. Veronese, J. Bioact. Compat. Polym., 1993, 8, 41–50 CrossRef CAS.
- S. Salmaso, A. Semenzato, S. Bersania, M. Chinol, G. Paganelli and P. Caliceti, Biochim. Biophys. Acta, Gen. Subj., 2005, 1726, 57–66 Search PubMed.
- Y. Yamamoto, Y. Tsutsumi, Y. Yoshioka, T. Nishibata, K. Kobayashi, T. Okamoto, Y. Mukai, T. Shimizu, S. Nakagawa, S. Nagata and T. Mayumi, Nat. Biotechnol., 2003, 21, 546–552 CrossRef CAS.
- Y. Yoshioka, Y. Tsutsumi, S. Ikemizu, Y. Yamamoto, H. Shibata, T. Nishibata, Y. Mukai, T. Okamoto, M. Taniai, M. Kawamura, Y. Abe, S. Nakagawa, S. Nagata, Y. Yamagata and T. Mayumi, Biochem. Biophys. Res. Commun., 2004, 315, 808–814 CrossRef CAS.
- H. Shibata, Y. Yoshioka, S. Ikemizu, K. Kobayashi, Y. Yamamoto, Y. Mukai, T. Okamoto, M. Taniai, M. Kawamura, Y. Abe, S. Nakagawa, T. Hayakawa, S. Nagata, Y. Yamagata, T. Mayumi, H. Kamada and Y. Tsutsumi, Clin. Cancer Res., 2004, 10, 8293–8300 CrossRef CAS.
- M. Bach, P. Holig, E. Schlosser, T. Volkel, A. Graser, R. Muller and R. E. Kontermann, Protein Eng., Des. Sel., 2003, 16, 1107–1113 CrossRef CAS.
- G. B. Fields and R. L. Noble, Int. J. Pept. Protein Res., 1990, 35, 161–214 CAS.
- M. Bodansky and A. Bodansky, The Practice of Peptide Synthesis, Springer Verlag, Berlin, 2nd edn, 1995 Search PubMed.
- Y. A. Lu and A. M. Felix, Pept. Res., 1993, 6, 140–146 CAS.
- M. Pechar, P. Kopečková, L. Joss and J. Kopeček, Macromol. Biosci., 2002, 2, 199–206 CrossRef CAS.
- Y. A. Lu and A. M. Felix, React. Polym., 1994, 22, 221–229 CrossRef CAS.
- G. W. M. Vandermeulen, C. Tziatzios, R. Duncan and H. A. Klok, Macromolecules, 2005, 38, 761–769 CrossRef CAS.
- Y. A. Lu and A. M. Felix, Int. J. Pept. Protein Res., 1994, 43, 127–138 CAS.
- B. Kleine, W. Rapp, K. H. Wiesmüller, M. Edinger, W. Beck, J. Metzger, R. Ataulakhanov, G. Jung and W. G. Bessler, Immunobiology, 1994, 190, 53–66 Search PubMed.
- J. T. Meijer, M. J. A. G. Henckens, I. J. Minten, D. W. P. M. Löwik and J. C. M. van Hest, Soft Matter, 2007, 3, 1135–1137 RSC.
- T. S. Burkoth, T. L. S. Benzinger, D. N. M. Jones, K. Hallenga, S. C. Meredith and D. G. Lynn, J. Am. Chem. Soc., 1998, 120, 7655–7656 CrossRef CAS.
- J. H. Collier and P. B. Messersmith, Adv. Mater., 2004, 16, 907–910 CrossRef CAS.
- A. Rösler, H. A. Klok, I. W. Hamley, V. Castelletto and O. O. Mykhaylyk, Biomacromolecules, 2003, 4, 859–863 CrossRef.
- D. Eckhardt, M. Groenewolt, E. Krause and H. G. Börner, Chem. Commun., 2005,(22), 2814–2816 RSC.
- I. C. Reynhout, D. W. P. M. Löwik, J. C. M. van Hest, J. J. L. M. Cornelissen and R. J. M. Nolte, Chem. Commun., 2005,(5), 602–604 RSC.
- V. N. R. Pillai, M. Mutter, E. Bayer and I. Gatfield, J. Org. Chem., 1980, 45, 5364–5370 CrossRef CAS.
- S. S. Wong, Chemistry of Protein Conjugation and Cross-linking, CRC Press, Boca Raton, Florida, 1991 Search PubMed.
- R. Wetzel, R. Halualani, J. T. Stults and C. Quan, Bioconjugate Chem., 1990, 1, 114–122 CrossRef CAS.
- D. P. Baker, E. Y. Lin, K. Lin, M. Pellegrini, R. C. Petter, L. L. Chen, R. M. Arduini, M. Brickelmaier, D. Y. Wen, D. M. Hess, L. Q. Chen, D. Grant, A. Whitty, A. Gill, D. J. Lindner and R. B. Pepinsky, Bioconjugate Chem., 2006, 17, 179–188 CrossRef CAS.
- J. M. Harris, E. C. Struck, M. G. Case, M. S. Paley, J. M. Vanalstine and D. E. Brooks, J. Polym. Sci., Part A: Polym. Chem., 1984, 22, 341–352 Search PubMed.
- S. M. Chamow, T. P. Kogan, M. Venuti, T. Gadek, R. J. Harris, D. H. Peers, J. Mordenti, S. Shak and A. Ashkenazi, Bioconjugate Chem., 1994, 5, 133–140 CrossRef CAS.
- J. M. Goddard, J. N. Talbert and J. H. Hotchkiss, J. Food Sci., 2007, 72, E36–E41 CrossRef CAS.
- A. B. Jóźwiak, C. M. Kielty and R. A. Black, J. Mater. Chem., 2008, 18, 2240–2248 RSC.
- J. Holland, L. Hersh, M. Bryhan, E. Onyiriuka and L. Ziegler, Biomaterials, 1996, 17, 2147–2156 CrossRef CAS.
- O. Kinstler, G. Molineux, M. Treuheit, D. Ladd and C. Gegg, Adv. Drug Delivery Rev., 2002, 54, 477–485 CrossRef CAS.
- H. Lee, I. H. Jang, S. H. Ryu and T. G. Park, Pharm. Res., 2003, 20, 818–825 CrossRef CAS.
- R. Mallik, J. Tao and D. S. Hage, Anal. Chem., 2004, 76, 7013–7022 CrossRef CAS.
- J. M. McFarland and M. B. Francis, J. Am. Chem. Soc., 2005, 127, 13490–13491 CrossRef CAS.
- M. D. Bentley, M. J. Roberts and J. M. Harris, J. Pharm. Sci., 1998, 87, 1446–1449 CrossRef CAS.
- L. Tao, G. Mantovani, F. Lecolley and D. M. Haddleton, J. Am. Chem. Soc., 2004, 126, 13220–13221 CrossRef CAS.
- K. L. Christman, M. V. Requa, V. D. Enriquez-Rioz, S. C. Ward, K. A. Bradley, K. L. Turner and H. D. Maynard, Langmuir, 2006, 22, 7444–7450 CrossRef.
- R. J. Goodson and N. V. Katre, Bio-Technology, 1990, 8, 343–346 CrossRef CAS.
- S. Vanwetswinkel, S. Plaisance, Z. Y. Zhang, I. Vanlinthout, K. Brepoels, I. Lasters, D. Collen and L. Jespers, Blood, 2000, 95, 936–942 CAS.
- Y. Tsutsumi, M. Onda, S. Nagata, B. Lee, R. J. Kreitman and I. Pastan, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8548–8553 CrossRef CAS.
- M. P. Lutolf, N. Tirelli, S. Cerritelli, L. Cavalli and J. A. Hubbell, Bioconjugate Chem., 2001, 12, 1051–1056 CrossRef CAS.
- M. Morpurgo, F. M. Veronese, D. Kachensky and J. M. Harris, Bioconjugate Chem., 1996, 7, 363–368 CrossRef CAS.
- S. C. Rizzi and J. A. Hubbell, Biomacromolecules, 2005, 6, 1226–1238 CrossRef CAS.
- S. VandeVondele, J. Voros and J. A. Hubbell, Biotechnol. Bioeng., 2003, 82, 784–790 CrossRef CAS.
- Z. L. Ding, R. B. Fong, C. J. Long, P. S. Stayton and A. S. Hoffman, Nature, 2001, 411, 59–62 CrossRef CAS.
- V. Bulmus, Z. L. Ding, C. J. Long, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2000, 11, 78–83 CrossRef CAS.
- Z. L. Ding, C. J. Long, Y. Hayashi, E. V. Bulmus, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 1999, 10, 395–400 CrossRef CAS.
- P. S. Stayton, T. Shimoboji, C. Long, A. Chilkoti, G. H. Chen, J. M. Harris and A. S. Hoffman, Nature, 1995, 378, 472–474 CrossRef CAS.
- T. Shimoboji, Z. L. Ding, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2002, 13, 915–919 CrossRef CAS.
- T. Shimoboji, E. Larenas, T. Fowler, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2003, 14, 517–525 CrossRef CAS.
- T. Shimoboji, E. Larenas, T. Fowler, S. Kulkarni, A. S. Hoffman and P. S. Stayton, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16592–16596 CrossRef CAS.
- T. P. Kogan, Synth. Commun., 1992, 22, 2417–2424 CrossRef CAS.
- A. Chilkoti, G. H. Chen, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 1994, 5, 504–507 CrossRef CAS.
- S. S. Pennadam, M. D. Lavigne, C. F. Dutta, K. Firman, D. Mernagh, D. C. Gorecki and C. Alexander, J. Am. Chem. Soc., 2004, 126, 13208–13209 CrossRef CAS.
- Z. P. Tolstyka, J. T. Kopping and H. A. Maynard, Macromolecules, 2008, 41, 599–606 CrossRef CAS.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. J. L. M. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966–2973 CrossRef CAS.
- K. Velonia, A. E. Rowan and R. J. M. Nolte, J. Am. Chem. Soc., 2002, 124, 4224–4225 CrossRef CAS.
- K. Blank, J. Morfill and H. E. Gaub, ChemBioChem, 2006, 7, 1349–1351 CrossRef CAS.
- M. A. Kostiainen, G. R. Szilvay, J. Lehtinen, D. K. Smith, M. B. Linder, A. Urtti and O. Ikkala, ACS Nano, 2007, 1, 103–113 CrossRef CAS.
- S. Salmaso, A. Semenzato, S. Bersani, F. Mastrotto, A. Scomparin and P. Caliceti, Eur. Polym. J., 2008, 44, 1378–1389 CrossRef CAS.
- K. Griesbaum, Angew. Chem., Int. Ed. Engl., 1970, 9, 273–287 CAS.
- Y. Geng, D. E. Discher, J. Justynska and H. Schlaad, Angew. Chem., Int. Ed., 2006, 45, 7578–7581 CrossRef CAS.
- Z. Hordyjewicz-Baran, L. C. You, B. Smarsly, R. Sigel and H. Schlaad, Macromolecules, 2007, 40, 3901–3903 CrossRef CAS.
- P. Jonkheijm, D. Weinrich, M. Köhn, H. Engelkamp, P. C. M. Christianen, J. Kuhlmann, J. C. Maan, D. Nüsse, H. Schroeder, R. Wacker, R. Breinbauer, C. M. Niemeyer and H. Waldmann, Angew. Chem., Int. Ed., 2008, 47, 4421–4424 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- C. T. Kuan, Q. C. Wang and I. Pastan, J. Biol. Chem., 1994, 269, 7610–7616 CAS.
- J. T. Li, J. Carlsson, J. N. Lin and K. D. Caldwell, Bioconjugate Chem., 1996, 7, 592–599 CrossRef CAS.
- D. Bontempo, K. L. Heredia, B. A. Fish and H. D. Maynard, J. Am. Chem. Soc., 2004, 126, 15372–15373 CrossRef CAS.
- S. Howorka, L. Movileanu, X. F. Lu, M. Magnon, S. Cheley, O. Braha and H. Bayley, J. Am. Chem. Soc., 2000, 122, 2411–2416 CrossRef CAS.
- A. S. M. Kamruzzahan, A. Ebner, L. Wildling, F. Kienberger, C. K. Riener, C. D. Hahn, P. D. Pollheimer, P. Winklehner, M. Hölzl, B. Lackner, D. M. Schörkl, P. Hinterdorfer and H. J. Gruber, Bioconjugate Chem., 2006, 17, 1473–1481 CrossRef CAS.
- R. Iwata, R. Satoh, Y. Iwasaki and K. Akiyoshi, Colloids Surf., B, 2008, 62, 288–298 CrossRef CAS.
- Y. Iwasaki, Y. Omichi and R. Iwato, Langmuir, 2008, 24, 8427–8430 CrossRef CAS.
- C. Woghiren, B. Sharma and S. Stein, Bioconjugate Chem., 1993, 4, 314–318 CrossRef CAS.
- S. Herman, J. Loccufier and E. Schacht, Macromol. Chem. Phys., 1994, 195, 203–209 CrossRef CAS.
- S. Shaunak, A. Godwin, J. W. Choi, S. Balan, E. Pedone, D. Vijayarangam, S. Heidelberger, I. Teo, M. Zloh and S. Brocchini, Nat. Chem. Biol., 2006, 2, 312–313 CrossRef CAS.
- S. Brocchini, A. Godwin, S. Balan, J. W. Choi, M. Zloh and S. Shaunak, Adv. Drug Delivery Rev., 2008, 60, 3–12 CrossRef CAS.
- N. Curreli, S. Oliva, A. Rescigno, A. C. Rinaldi, F. Sollai and E. Sanjust, J. Appl. Polym. Sci., 1997, 66, 1433–1438 CrossRef.
- N. S. Joshi, L. R. Whitaker and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 15942–15943 CrossRef CAS.
- S. D. Tilley and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 1080–1081 CrossRef CAS.
- T. C. Evans and M. Q. Xu, Biopolymers, 1999, 51, 333–342 CrossRef CAS.
- P. E. Dawson, T. W. Muir, I. Clarklewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef CAS.
- C. Haase and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 1553–1556 CrossRef CAS.
- C. Haase, H. Rohde and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 6807–6810 CrossRef CAS.
- I. van Baal, H. Malda, S. A. Synowsky, J. L. J. van Dongen, T. M. Hackeng, M. Merkx and E. W. Meijer, Angew. Chem., Int. Ed., 2005, 44, 5052–5057 CrossRef.
- S. M. Chafekar, H. Malda, M. Merkx, E. W. Meijer, D. Viertl, H. A. Lashuel, F. Baas and W. Scheper, ChemBioChem, 2007, 8, 1857–1864 CrossRef CAS.
- B. Helms, I. van Baal, M. Merkx and E. W. Meijer, ChemBioChem, 2007, 8, 1790–1794 CrossRef CAS.
- A. Dirksen, E. W. Meijer, W. Adriaens and T. M. Hackeng, Chem. Commun., 2006,(15), 1667–1669 RSC.
- Y. Marsac, J. Cramer, D. Olschewski, K. Alexandrov and C. F. W. Becker, Bioconjugate Chem., 2006, 17, 1492–1498 CrossRef CAS.
- H. Sato, Adv. Drug Delivery Rev., 2002, 54, 487–504 CrossRef CAS.
- M. Griffin, R. Casadio and C. M. Bergamini, Biochem. J., 2002, 368, 377–396 CrossRef CAS.
- A. Fontana, B. Spolaore, A. Mero and F. M. Veronese, Adv. Drug Delivery Rev., 2008, 60, 13–28 CrossRef CAS.
- Y. Tanaka, Y. Tsuruda, M. Nishi, N. Kamiya and M. Goto, Org. Biomol. Chem., 2007, 5, 1764–1770 RSC.
- S. DeFrees, Z. G. Wang, R. Xing, A. E. Scott, J. Wang, D. Zopf, D. L. Gouty, E. R. Sjoberg, K. Panneerselvam, E. C. M. Brinkman Van Der Linden, R. J. Bayer, M. A. Tarp and H. Clausen, Glycobiology, 2006, 16, 833–843 CrossRef CAS.
- S. Tugulu, A. Arnold, I. Sielaff, K. Johnsson and H. A. Klok, Biomacromolecules, 2005, 6, 1602–1607 CrossRef CAS.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Correa, M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128–136 CrossRef CAS.
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269–272 CrossRef CAS.
- G. V. Los, C. Zimprich, M. G. McDougall, N. Karassina, R. Learish, D. H. Klaubert, A. Darzins, R. F. Bulleit and K. Wood, J. Neurochem., 2005, 94, 15–15.
- R. Huisgen, 1,3 Dipolar Cycloaddition Chemistry, Wiley, New York, 1984 Search PubMed.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192–3193 CrossRef CAS.
- A. Deiters, T. A. Cropp, D. Summerer, M. Mukherji and P. G. Schultz, Bioorg. Med. Chem. Lett., 2004, 14, 5743–5745 CrossRef CAS.
- L. Wang and P. G. Schultz, Angew. Chem., Int. Ed., 2005, 44, 34–66 CrossRef CAS.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS.
- S. Schoffelen, M. H. L. Lambermon, M. B. van Eldijk and J. C. M. van Hest, Bioconjugate Chem., 2008, 19, 1127–1131 CrossRef CAS.
- D. T. S. Rijkers, H. H. R. van Vugt, H. J. F. Jacobs and R. M. J. Liskamp, Tetrahedron Lett., 2002, 43, 3657–3660 CrossRef CAS.
- E. D. Goddard-Borger and R. V. Stick, Org. Lett., 2007, 9, 3797–3800 CrossRef CAS.
- S. F. M. van Dongen, R. L. M. Teeuwen, M. Nallani, S. S. van Berkel, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Bioconjugate Chem., 2009, 20, 20–23 CrossRef CAS.
- A. J. T. Dirks, S. S. van Berkel, N. S. Hatzakis, J. A. Opsteen, F. L. van Delft, J. J. L. M. Cornelissen, A. E. Rowan, J. C. M. van Hest, F. P. J. T. Rutjes and R. J. M. Nolte, Chem. Commun., 2005,(33), 4172–4174 RSC.
- N. Tzokova, C. M. Fernyhouph, P. D. Topham, N. Sandon, D. J. Adams, M. F. Butler, S. P. Armes and A. J. Ryan, Langmuir, 2009, 25, 2479–2485 CrossRef CAS.
- J. F. Lutz, H. G. Börner and K. Weichenhan, Macromolecules, 2006, 39, 6376–6383 CrossRef CAS.
- S. S. Gupta, K. S. Raja, E. Kaltgrad, E. Strable and M. G. Finn, Chem. Commun., 2005,(34), 4315–4317 RSC.
- W. Agut, D. Taton and S. Lecommandoux, Macromolecules, 2007, 40, 5653–5661 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172–1176 CrossRef CAS.
- S. Fleischmann, H. Kornber, D. Appelhans and B. I. Voit, Macromol. Chem. Phys., 2007, 208, 1050–1060 CrossRef CAS.
- B. S. Sumerlin, N. V. Tsarevsky, G. Louche, R. Y. Lee and K. Matyjaszewski, Macromolecules, 2005, 38, 7540–7545 CrossRef CAS.
- B. Parrish, R. B. Breitenkamp and T. Emrick, J. Am. Chem. Soc., 2005, 127, 7404–7410 CrossRef CAS.
- D. T. S. Rijkers, G. W. van Esse, R. Merkx, A. J. Brouwer, H. J. F. Jacobs, R. J. Pieters and R. M. J. Liskamp, Chem. Commun., 2005, 4581–4583 RSC.
- S. F. M. van Dongen, M. Nallani, S. Schoffelen, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Macromol. Rapid Commun., 2008, 29, 321–325 CrossRef CAS.
- X. L. Sun, C. L. Stabler, C. S. Cazalis and E. L. Chaikof, Bioconjugate Chem., 2006, 17, 52–57 CrossRef CAS.
- S. J. Stohs and D. Bagchi, Free Radical Biol. Med., 1995, 18, 321–336 CrossRef CAS.
- H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635–646 CrossRef CAS.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS.
- C. S. Cazalis, C. A. Haller, L. Sease-Cargo and E. L. Chaikof, Bioconjugate Chem., 2004, 15, 1005–1009 CrossRef CAS.
- J. Kalia and R. T. Raines, ChemBioChem, 2006, 7, 1375–1383 CrossRef CAS.
- E. Saxon, J. I. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141–2143 CrossRef CAS.
- B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2000, 2, 1939–1941 CrossRef CAS.
- A. Tam, M. B. Soellner and R. T. Raines, J. Am. Chem. Soc., 2007, 129, 11421–11430 CrossRef CAS.
- B. L. Nilsson, R. J. Hondal, M. B. Soellner and R. T. Raines, J. Am. Chem. Soc., 2003, 125, 5268–5269 CrossRef CAS.
- M. B. Soellner, K. A. Dickson, B. L. Nilsson and R. T. Raines, J. Am. Chem. Soc., 2003, 125, 11790–11791 CrossRef CAS.
- J. Kalia, N. L. Abbott and R. T. Raines, Bioconjugate Chem., 2007, 18, 1064–1069 CrossRef CAS.
- T. P. King, S. W. Zhao and T. Lam, Biochemistry, 1986, 25, 5774–5779 CrossRef CAS.
- R. R. Webb and T. Kaneko, Bioconjugate Chem., 1990, 1, 96–99 CrossRef CAS.
- K. Rose, J. Am. Chem. Soc., 1994, 116, 30–33 CrossRef.
- C. A. C. Wolfe and D. S. Hage, Anal. Biochem., 1995, 231, 123–130 CrossRef CAS.
- M. Wilchek and E. A. Bayer, Methods Enzymol., 1987, 138, 429–442 Search PubMed.
- Y. S. Youn, D. H. Na, S. D. Yoo, S. C. Song and K. C. Lee, Int. J. Biochem. Cell Biol., 2005, 37, 1525–1533 CrossRef CAS.
- S. O. Leung, M. J. Losman, S. V. Govindan, G. L. Griffiths, D. M. Goldenberg and H. J. Hansen, J. Immunol., 1995, 154, 5919–5926 CAS.
- M. L. Wang, L. S. Lee, A. Nepomich, J. D. Yang, C. Conover, M. Whitlow and D. Filpula, Protein Eng., Des. Sel., 1998, 11, 1277–1283 CrossRef CAS.
- H. F. Gaertner and R. E. Offord, Bioconjugate Chem., 1996, 7, 38–44 CrossRef CAS.
- D. Datta, P. Wang, I. S. Carrico, S. L. Mayo and D. A. Tirrell, J. Am. Chem. Soc., 2002, 124, 5652–5653 CrossRef CAS.
- Z. W. Zhang, B. A. C. Smith, L. Wang, A. Brock, C. Cho and P. G. Schultz, Biochemistry, 2003, 42, 6735–6746 CrossRef CAS.
- T. L. Schlick, Z. B. Ding, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2005, 127, 3718–3723 CrossRef CAS.
- E. W. Kovacs, J. M. Hooker, D. W. Romanini, P. G. Holder, K. E. Berry and M. B. Francis, Bioconjugate Chem., 2007, 18, 1140–1147 CrossRef CAS.
- J. M. Gilmore, R. A. Scheck, A. P. Esser-Kahn, N. S. Joshi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 5307–5311 CrossRef CAS.
- R. A. Scheck and M. B. Francis, ACS Chem. Biol., 2007, 2, 247–251 CrossRef CAS.
- A. Zalipsky and S. Menon-Rudolph, Poly(ethylene glycol) Chemistry and Biological Applications, ACS Books, Washington, DC, 1997 Search PubMed.
- S. K. Yang and M. Weck, Macromolecules, 2008, 41, 346–351 CrossRef CAS.
- S. Y. Chen, S. Cressman, F. Mao, H. Shao, D. W. Low, H. S. Beilan, E. N. Cagle, M. Carnevali, V. Gueriguian, P. J. Keogh, H. Porter, S. M. Stratton, M. C. Wiedeke, L. Savatski, J. W. Adamson, C. E. Bozzini, A. Kung, S. B. H. Kent, J. A. Bradburne and G. G. Kochendoerfer, Chem. Biol., 2005, 12, 371–383 CrossRef CAS.
- L. P. Miranda, H. Y. Shao, J. Williams, S. Y. Chen, T. Kong, R. Garcia, Y. Chinn, N. Fraud, B. O’Dwyer, J. Ye, J. Wilken, D. E. Low, E. N. Cagle, M. Carnevali, A. Lee, D. Song, A. Kung, J. A. Bradburne, X. Paliard and G. G. Kochendoerfer, J. Am. Chem. Soc., 2007, 129, 13153–13159 CrossRef CAS.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS.
- K. L. Heredia, Z. P. Tolstyka and H. D. Maynard, Macromolecules, 2007, 40, 4772–4779 CrossRef CAS.
- K. Rose and J. Vizzavona, J. Am. Chem. Soc., 1999, 121, 7034–7038 CrossRef CAS.
- H. Y. Shao, M. M. Crnogorac, T. Kong, S. Y. Chen, J. M. Williams, J. M. Tack, V. Gueriguian, E. N. Cagle, M. Carnevali, D. Tumelty, X. Paliard, L. P. Miranda, J. A. Bradburne and G. G. Kochendoerfer, J. Am. Chem. Soc., 2005, 127, 1350–1351 CrossRef CAS.
- J. P. Mitchell, K. D. Roberts, J. Langley, F. Koentgen and J. N. Lambert, Bioorg. Med. Chem. Lett., 1999, 9, 2785–2788 CrossRef CAS.
- E. W. L. Chan and M. N. Yousaf, J. Am. Chem. Soc., 2006, 128, 15542–15546 CrossRef CAS.
- S. Park and M. N. Yousaf, Langmuir, 2008, 24, 6201–6207 CrossRef CAS.
- S. S. van Berkel, A. J. Dirks, S. A. Meeuwissen, D. L. L. Pingen, O. C. Boerman, P. Laverman, F. L. van Delft, J. J. L. M. Cornelissen and F. P. J. T. Rutjes, ChemBioChem, 2008, 9, 1805–1815 CrossRef CAS.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzit, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS.
- X. H. Ning, J. Guo, M. A. Wolfert and G. J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS.
- L. A. Canalle, S. S. Van Berkel, L. T. de Haan and J. C. M. van Hest, Adv. Funct. Mater., 2009 DOI:10.1002/adfm.200900743.
- S. S. van Berkel, A. J. Dirks, M. F. Debets, F. L. van Delft, J. J. L. M. Cornelissen, R. J. M. Nolte and F. P. J. T. Rutjes, ChemBioChem, 2007, 8, 1504–1508 CrossRef CAS.
- W. Song, Y. Wang, J. Qu and Q. Lin, J. Am. Chem. Soc., 2008, 130, 9654–9655 CrossRef CAS.
- K. Gutsmiedl, C. T. Wirges, V. Ehmke and T. Carell, Org. Lett., 2009, 11, 2405–2408 CrossRef CAS.
- X. H. Zhang, F. P. Li, X. W. Lu and C. F. Liu, Bioconjugate Chem., 2009, 20, 197–200 CrossRef CAS.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS.
- N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411–2414 CAS.
- M. Wilchek and E. A. Bayer, Anal. Biochem., 1988, 171, 1–32 CrossRef.
- J. M. Hannink, J. J. L. M. Cornelissen, J. A. Farrera, P. Foubert, F. C. De Schryver, N. A. J. M. Sommerdijk and R. J. M. Nolte, Angew. Chem., Int. Ed., 2001, 40, 4732–4734 CrossRef CAS.
- S. Kulkarni, C. Schilli, A. H. E. Muller, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2004, 15, 747–753 CrossRef CAS.
- J. J. Lin, J. A. Silas, H. Bermudez, V. T. Milam, F. S. Bates and D. A. Hammer, Langmuir, 2004, 20, 5493–5500 CrossRef CAS.
- S. Kulkarni, C. Schilli, B. Grin, A. H. E. Muller, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2006, 7, 2736–2741 CrossRef CAS.
- D. S. Wilbur, P. M. Pathare, D. K. Hamlin, K. R. Buhler and R. L. Vessella, Bioconjugate Chem., 1998, 9, 813–825 CrossRef CAS.
- S. B. Jhaveri, M. Beinhoff, C. J. Hawker, K. R. Carter and D. Y. Sogah, ACS Nano, 2008, 2, 719–727 CrossRef.
- J. Anzai, Y. Kobayashi, N. Nakamura, M. Nishimura and T. Hoshi, Langmuir, 1999, 15, 221–226 CrossRef CAS.
- J. Anzai and M. Nishimura, J. Chem. Soc., Perkin Trans. 2, 1997,(10), 1887–1889 RSC.
- F. Ganachaud, A. Theretz, M. N. Erout, M. F. Llauro and C. Pichot, J. Appl. Polym. Sci., 1995, 58, 1811–1824 CrossRef CAS.
- S. J. Hou, X. L. Sun, C. M. Dong and E. L. Chaikof, Bioconjugate Chem., 2004, 15, 954–959 CrossRef CAS.
- X. L. Sun, K. M. Faucher, M. Houston, D. Grande and E. L. Chaikof, J. Am. Chem. Soc., 2002, 124, 7258–7259 CrossRef CAS.
- D. Bontempo, R. C. Li, T. Ly, C. E. Brubaker and H. D. Maynard, Chem. Commun., 2005, 4702–4704 RSC.
- K. Qi, Q. G. Ma, E. E. Remsen, C. G. Clark and K. L. Wooley, J. Am. Chem. Soc., 2004, 126, 6599–6607 CrossRef CAS.
- V. Vázquez-Dorbatt and H. D. Maynard, Biomacromolecules, 2006, 7, 2297–2302 CrossRef CAS.
- C. Y. Hong and C. Y. Pan, Macromolecules, 2006, 39, 3517–3524 CrossRef CAS.
- M. Bathfield, F. D’Agosto, R. Spitz, M. T. Charreyre and T. Delair, J. Am. Chem. Soc., 2006, 128, 2546–2547 CrossRef CAS.
- M. J. Boerakker, J. M. Hannink, P. H. H. Bomans, P. M. Frederik, R. J. M. Nolte, E. M. Meijer and N. Sommerdijk, Angew. Chem., Int. Ed., 2002, 41, 4239–4241 CrossRef CAS.
- M. J. Boerakker, N. E. Botterhuis, P. H. H. Bomans, P. M. Frederik, E. M. Meijer, R. J. M. Nolte and N. Sommerdijk, Chem.–Eur. J., 2006, 12, 6071–6080 CrossRef CAS.
- I. C. Reynhout, J. J. L. M. Cornelissen and R. J. M. Nolte, J. Am. Chem. Soc., 2007, 129, 2327–2332 CrossRef CAS.
- N. Yamaguchi, L. Zhang, B. S. Chae, C. S. Palla, E. M. Furst and K. L. Kiick, J. Am. Chem. Soc., 2007, 129, 3040–3041 CrossRef CAS.
- H. Rettig, E. Krause and H. G. Börner, Macromol. Rapid Commun., 2004, 25, 1251–1256 CrossRef CAS.
- D. J. Adams and I. Young, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6082–6090 CrossRef CAS.
- M. G. J. ten Cate, H. Rettig, K. Bernhardt and H. G. Börner, Macromolecules, 2005, 38, 10643–10649 CrossRef CAS.
- J. Hentschel, K. Bleek, O. Ernst, J. F. Lutz and H. G. Börner, Macromolecules, 2008, 41, 1073–1075 CrossRef CAS.
- L. Ayres, P. J. H. M. Adams, D. W. P. M. Löwik and J. C. M. van Hest, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6355–6366 CrossRef CAS.
- R. M. Broyer, G. M. Quaker and H. D. Maynard, J. Am. Chem. Soc., 2008, 130, 1041–1047 CrossRef CAS.
- K. Molawi and A. Studer, Chem. Commun., 2007,(48), 5173–5175 RSC.
- J. Couet and M. Biesalski, Macromolecules, 2006, 39, 7258–7268 CrossRef CAS.
- S. Loschonsky, J. Couet and M. Biesalski, Macromol. Rapid Commun., 2008, 29, 309–315 CrossRef CAS.
- M. L. Becker, J. Q. Liu and K. L. Wooley, Chem. Commun., 2003,(2), 180–181 RSC.
- M. L. Becker, J. Q. Liu and K. L. Wooley, Biomacromolecules, 2005, 6, 220–228 CrossRef CAS.
- Y. Mei, K. L. Beers, H. C. M. Byrd, D. L. Vanderhart and N. R. Washburn, J. Am. Chem. Soc., 2004, 126, 3472–3476 CrossRef CAS.
- A. George, G. Radhakrishnan and K. T. Joseph, J. Appl. Polym. Sci., 1984, 29, 703–707 CrossRef CAS.
- H. Keleş, M. Çelik, M. Saçak and L. Aksu, J. Appl. Polym. Sci., 1999, 74, 1547–1556 CrossRef CAS.
- I. Kaur, R. Barsola, A. Gupta and B. N. Misra, J. Appl. Polym. Sci., 1994, 54, 1131–1139 CrossRef CAS.
- B. S. Lele, H. Murata, K. Matyjaszewski and A. J. Russell, Biomacromolecules, 2005, 6, 3380–3387 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955–16960 CrossRef CAS.
- J. Nicolas, V. San Miguel, G. Mantovani and D. M. Haddleton, Chem. Commun., 2006,(45), 4697–4699 RSC.
- C. Boyer, V. Bulmus, J. Q. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS.
- J. Q. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. R. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099–3103 CrossRef CAS.
- D. Bontempo and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 6508–6509 CrossRef CAS.
- H. A. Klok and S. Lecommandoux, in Peptide Hybrid Polymers, 2006, vol. 202, pp. 75–111 Search PubMed.
- H. Schlaad and M. Antonietti, Eur. Phys. J. E, 2003, 10, 17–23 CrossRef CAS.
- T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2004, 5, 1653–1656 CrossRef CAS.
- I. Dimitrov and H. Schlaad, Chem. Commun., 2003,(23), 2944–2945 RSC.
- T. J. Deming, Nature, 1997, 390, 386–389 CrossRef CAS.
- C. S. Cho, Y. I. Jeong, S. H. Kim, J. W. Nah, M. Kubota and T. Komoto, Polymer, 2000, 41, 5185–5193 CrossRef CAS.
- S. Abraham, C. S. Ha and I. Kim, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2774–2783 CrossRef CAS.
- R. J. I. Knoop, G. J. M. Habraken, N. Gogibus, S. Steig, H. Menzel, C. E. Koning and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3068–3077 CrossRef CAS.
- H. Murata, F. Sanda and T. Endo, Macromolecules, 1996, 29, 5535–5538 CrossRef CAS.
- H. Murata, F. Sanda and T. Endo, Macromol. Chem. Phys., 1997, 198, 2519–2529 CrossRef.
- H. Murata, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1679–1682 CrossRef CAS.
- H. D. Maynard, S. Y. Okada and R. H. Grubbs, Macromolecules, 2000, 33, 6239–6248 CrossRef CAS.
- H. D. Maynard, S. Y. Okada and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 1275–1279 CrossRef CAS.
- T. E. Hopkins and K. B. Wagener, Macromolecules, 2004, 37, 1180–1189 CrossRef CAS.
- J. J. L. M. Cornelissen, J. J. J. M. Donners, R. de Gelder, W. S. Graswinckel, G. A. Metselaar, A. E. Rowan, N. A. J. M. Sommerdijk and R. J. M. Nolte, Science, 2001, 293, 676–680 CAS.
- J. J. L. M. Cornelissen, W. S. Graswinckel, P. J. H. M. Adams, G. H. Nachtegaal, A. P. M. Kentgens, N. A. J. M. Sommerdijk and R. J. M. Nolte, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 4255–4264 CrossRef CAS.
- D. M. Vriezema, J. Hoogboom, K. Velonia, K. Takazawa, P. C. M. Christianen, J. C. Maan, A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 2003, 42, 772–776 CrossRef CAS.
- L. Ayres, M. R. J. Vos, P. J. H. M. Adams, I. O. Shklyarevskiy and J. C. M. van Hest, Macromolecules, 2003, 36, 5967–5973 CrossRef CAS.
- L. Ayres, K. Koch, P. J. H. M. Adams and J. C. M. van Hest, Macromolecules, 2005, 38, 1699–1704 CrossRef CAS.
- L. Ayres, P. J. H. M. Adams, D. W. P. M. Löwik and J. C. M. van Hest, Biomacromolecules, 2005, 6, 825–831 CrossRef CAS.
- L. Ayres, G. M. Grotenbreg, G. A. van der Marel, H. S. Overkleeft, M. Overhand and J. C. M. van Hest, Macromol. Rapid Commun., 2005, 26, 1336–1340 CrossRef CAS.
- F. Fernández-Trillo, A. Dureault, J. P. M. Bayley, J. C. M. van Hest, J. C. Thies, T. Michon, R. Weberskirch and N. R. Cameron, Macromolecules, 2007, 40, 6094–6099 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |