Hydrogen generation from formic acid and alcohols using homogeneous catalysts
Tarn C. Johnson, David J. Morris and Martin Wills*
The Department of Chemistry, The University of Warwick, Coventry, UK CV4 7AL. E-mail: m.wills@warwick.ac.uk; Fax: +44 (0)24 7652 4112; Tel: +44 (0)24 7652 3260
First published on 2nd September 2009
Abstract
This tutorial review describes recent progress in the development of homogeneous catalytic methodology for the direct generation of hydrogen gas from formic acid and alcohols.
 David J. Morris, Martin Wills and Tarn C. Johnson | Tarn Johnson was born in Kettering in 1986. In 2008 he obtained his MChem degree from the University of Leicester. He is currently working with Professor Martin Wills at the University of Warwick for his PhD, investigating the use of organometallic catalysts for the generation of hydrogen from alcohols. |
David Morris was born in Kitwe (Zambia) in 1974 but grew up in English villages within North Hampshire (UK). He obtained a BSc from Bath in 1996 and joined Martin Wills at Warwick gaining an MSc and PhD. After a year in New Zealand David returned to Warwick and continues to postdoc with Martin. His research interests are asymmetric catalysis and sustainable energy vectors. |
Martin Wills started life some time before David Morris and a significant time before Tarn Johnson, in Swansea (Wales) but grew up in Reading (England). He obtained a BSc from Imperial College in 1985, then a DPhil from Oxford University, where he was supervised by Professor Steve Davies. After one year as a postdoc with Prof. W. Oppolzer in Geneva he took up a lectureship at Bath in 1989 then moved to Warwick in 1995, where he was promoted to a Chair in 2000. His research interests are in synthetic organic chemistry and asymmetric catalysis. |
Why hydrogen?
As fossil fuel reserves are progressively depleted, the development of a clean energy supply that is sustainable and can meet the rising global demand for energy is rapidly becoming one of the greatest challenges of the 21st century. One of the more promising solutions to this problem is that of a hydrogen economy,1 which has the advantage of a significant reduction in greenhouse gas emissions as hydrogen can be either combusted or converted directly into electricity via fuel cells, liberating water as the sole by-product. For these reasons the sustainable production of hydrogen is desirable. For a hydrogen economy to become a reality, improved techniques for the generation of hydrogen, its storage and its conversion into electrical energy need to be developed, ideally taking place under ambient conditions.The storage of hydrogen in a manner which is safe and reversible at mild temperatures is an important goal. Hydrogen stored in its elemental form as a gas or a liquid has safety implications due to its flammable nature and the need to keep it under pressure. The production of hydrogen directly from organic molecules, such as alcohols and formic acid, provides a potential method for the use of waste materials including glycerol, starch and cellulose-derived products as sources of the gas.
Currently, the majority of commercially produced hydrogen is generated by steam reforming, which is carried out at high temperatures (700–1200 K) and generates CO and CO2 as waste products. A recent review2 details many reforming, gasification and other notable reactions for producing hydrogen from fossil fuels and biomass-derived feedstocks. Another methodology for hydrogen production is biological production from bacteria.3 The focus of this tutorial review, however, will be on the catalytic dehydrogenation of primary and secondary alcohols, and formic acid, to yield molecular hydrogen. There are examples of both homogeneous and heterogeneous catalysts capable of dehydrogenating alcohols but only the former will be discussed here. Due to space requirements, the catalysts discussed do not represent an exhaustive list.
Homogeneous catalysts for the dehydrogenation of alcohols
Several classes of homogeneous catalyst have been used for alcohol dehydrogenation: i.e. the specific conversion of alcohols to oxidised derivatives through the direct generation of hydrogen.4–13 In one of the earliest examples of what can be termed an ‘acid-promoted’ reaction, Dobson and Robinson4 demonstrated the application of the complex [Ru(OCOCF3)2(CO)(PPh3)2] 1 in the dehydrogenation of a range of primary and secondary alcohols (Scheme 1). An excess of a fluorinated carboxylic acid is required. The proposed mechanism (Scheme 2) involves addition of the alcohol (to give 2) and loss of an acid (to give 3), followed by a β-elimination step to form a metal hydride species 4. Re-addition of acid liberates dihydrogen and regenerates the catalyst. With catalyst loadings in the order of ∼0.03 mol% and 12 mol of trifluoroacetic acid per mole of catalyst to promote the reaction, turnover frequencies (TOFs) of 2952 h−1 for heptan-1-ol and 1620 h−1 for cyclooctanol were achieved. Rates for low molecular weight primary and secondary alcohols were low (EtOH, propan-1-ol and propan-2-ol gave TOFs lower than 100 h−1). Notably, the highest TOF was achieved for benzyl alcohol with a value of 8172 h−1. | ||
| Scheme 1 Hydrogen generation using acid-promoted dehydrogenation of alcohols. | ||
 | ||
| Scheme 2 Proposed mechanism of acid-promoted dehydrogenation. | ||
Jung and Garrou5 reported a series of analogous catalysts making use of bidentate diphosphine ligands and reported on mechanistic aspects of the reaction, including deactivation mechanisms and the loss of the volatile trifluoroacetic acid ligands from the system. Hulshof et al.6,12 investigated this reaction further and found that the dehydrogenation of primary alcohols was complicated by decarbonylation (leading to catalyst deactivation) and aldol condensations under the reactions employed.6
In recent work, Hulshof et al.7 extended their studies to the use of preformed complexes 5 containing bidentate derivatives of the fluorinated acids as ligands for the dehydrogenation. The dimeric complex 5 (PPh2XXPPh2 = 2 × PPh3 or ligands 6–8), which was isolated and characterised by X-ray crystallography proved to be a very effective catalyst in 1-phenylethanol dehydrogenation (Scheme 3). A tentative catalytic cycle, based on that shown in Scheme 2, for the dehydrogenation was proposed. The reactions were typically carried out in refluxing toluene or xylene. Of a series of phosphines investigated, best results were achieved using derivatives of 5 containing the bidentate diphenylphosphinoferrocene (dppf 6) which gave TOFs of up to 383 h−1 although good results were also obtained with complexes of dppp 7 and dppb 8.

The use of base-promoted dehydrogenation by Morton and Cole-Hamilton catalysed by a series of ruthenium and rhodium complexes in 19878 and 19889 for the first time produced reasonable rates of hydrogen production (>100 h−1) for the lower molecular weight alcohols ethanol, propanol and isopropanol (Scheme 4, Table 1).9 These researchers recognised that, thermodynamically, the process of hydrogen formation from alcohols is generally unfavourable (the ΔG° for formation of hydrogen plus acetal from ethanol, for example, is +41.2 kJ mol−1).8,9 Whilst high temperatures and removal of the hydrogen gas (i.e. to prevent the reversible rehydrogenation of the generated carbonyl group) can provide a driving force, the conversion of alcohol to a mixture of methane, hydrogen and carbon dioxide converts the reaction to a thermodynamically favourable one (for ethanol; ΔG° = −33.3 kJ mol−1). This transformation was employed for the [Rh(bipy)2]Cl complex, which is capable of catalysing all the dehydrogenation steps in the latter process.8 Some activity was also reported with methanol as the substrate, which was not the case for the Robinson catalyst. The reported catalysts, [RuH2(N2)(PPh3)3] and [RuH2(PPh3)4], showed significant activity in their own right but it was also observed that rates could be increased under illumination with visible light, in some cases quite significantly. The most remarkable increase was seen for [RuH2(N2)(PPh3)3] with (CH2OH)2 as the substrate with an increase from 515.9 h−1 to 1185.3 h−1 on illumination. The reactions used 1–5 mol dm−3 of catalyst and 1 mol dm−3 NaOH was required. All catalytic reactions were performed at 150 °C, with the exception of [Rh(bipy)2Cl] which was used at 120 °C.
 | ||
| Scheme 4 Hydrogen generation using base-promoted dehydrogenation of alcohols. | ||
The use of related rhodium catalysts,10,13 including [RhCl(PPh3)3] and [RhH(PPri3)3], and extended results (Table 2) were reported later.10 In this study, the rates of formation of other gases, including methane and carbon dioxide, arising from subsequent reactions were also determined.
| Catalyst | Concentration/10−4 M | T/°C | TOF/h−1 | ||
|---|---|---|---|---|---|
| H2 | CH4 | CO2 | |||
| a Not measured.b Detected but not quantified. | |||||
| [RhCl(PPh3)3] | 2.82 | 150 | 7.5 | 0.17 | — |
| [RhCl(PPh3)3] + hν | 4.56 | 150 | 11.4 | b | — |
| [RhH(PiPr3)3] | 5.16 | 150 | 5.6 | 0.55 | — |
| [RhH(PiPr3)3] + hν | 4.14 | 150 | 23.1 | 1.85 | — |
| [Rh(bipy)2]Cl | 10.0 | 120 | 95.5 | 10.5 | 9.5 |
| [RuH2(N2)(PPh3)3] | 2.60 | 150 | 148.1 | a | — |
| [RuH2(N2)(PPh3)3] + hν | 3.48 | 150 | 210.2 | b | — |
| [RuH2(CO)(PPh3)3] | 2.82 | 150 | 62 | a | — |
| [RuH2(PPh3)4] | 2.98 | 150 | 23.8 | 0.40 | — |
| [RuH2(PPh3)4] + hν | 2.10 | 150 | 138.4 | 0.90 | — |
The role of base in these systems is quite pronounced with very little activity observed in its absence. The mechanism of the reaction is speculated to involve addition of alkoxide to a ruthenium hydride, followed by ketone or aldehyde elimination. Decarbonylation of aldehyde products is observed as a side reaction and this can result in catalyst poisoning. Irradiation with visible light is believed to accelerate the reaction by promoting release of CO from the metal and thus regenerating the catalyst.10 The effect of illumination may also be to increase the rate of the elimination of dihydrogen. There is already a precedent14,15 for photocatalytic systems for the dehydrogenation of alcohols to liberate dihydrogen with a good example being that of Griggs and Smith16 whose catalyst, [RhCl((P(OPh)3)3] (1.5 ppm), achieved a TOF of 6410 h−1 after 2 h at 21 °C with isopropanol as the substrate. Photocatalytic dehydrogenation of 2-propanol with carbonyl(halogeno)phosphine–rhodium complexes has also been reported.17 Isopropanol dehydrogenation using Wilkinson’s catalyst (RhCl(PPh3)3) was reported to give a TON of 980 in one example that was followed by monitoring generation of hydrogen. This required irradiation, and hydrogen generation essentially stopped when it was switched off.18 Although Wilkinson’s catalyst (i.e. the chloride form) is not active for iPrOH dehydrogenation, the corresponding hydride complex is.19 Addition of triethylamine to Wilkinson’s catalysts also results in its activation towards hydrogen generation from alcohols.
Methanol dehydrogenation, whilst challenging, has been reported in a number of papers.20–22 Liquid phase methanol dehydrogenation with Ru complexes has been reported,23 and ruthenium catalysed conversion of methanol into methyl formate has been described.24 The mechanism of formation of acetic acid from methanol by dehydrogenation has been studied,25 and photocatalytic methanol dehydrogenation using [IrH(SnCl3)5] has been described.26
It was recently demonstrated that a series of Ru arene, cyclopentadienyl and carbene complexes, including [CpRuCl(PPh3)2], [(indenyl)RuCl(PPh3)2], [(benzene)RuCl2]2, [(p-cymene)RuCl2]2, [PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ru(PCy3)2Cl2] and [Ru(IMes)(PPh3)2CO(H)2], are effective at dehydrogenation of 1-phenylethanol under basic conditions (typically involving reflux in toluene).27
Ru(PCy3)2Cl2] and [Ru(IMes)(PPh3)2CO(H)2], are effective at dehydrogenation of 1-phenylethanol under basic conditions (typically involving reflux in toluene).27
In recent years further advances have been reported, particularly, through the use of alternative ligands (Tables 3 and 4, Fig. 1).28,29 Beller et al. screened various ruthenium catalysts to identify suitable precursors for the in situ generation of active alcohol dehydrogenation catalysts. Two precursors, [RuCl3·xH2O] and [RuCl2(p-cymene)]2, were found to show a higher TOF for the dehydrogenation of isopropanol in the presence of two equivalents of PCy3 than the other catalysts tested (Table 3). Using 315 ppm of catalyst and 0.8 mol dm−3 NaOH at 90 °C, rates of 78 h−1 and 94 h−1 were obtained after 2 h which fell to 54 h−1 and 43 h−1 after 6 h for [RuCl3·xH2O] and [RuCl2(p-cymene)]2, respectively.
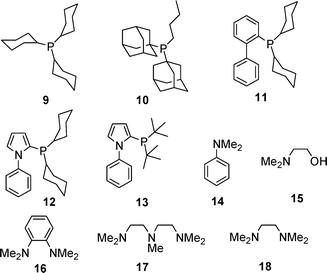 | ||
| Fig. 1 Ligands used in base-promoted hydrogen generation from alcohols (Table 4). | ||
| Catalyst | Ligandb | Base | Time/h | H2 volume/mL | TOF/h−1 |
|---|---|---|---|---|---|
| a 315 ppm [Ru], 0.8 M base, 90 °C, 6 h.b Where a phosphine is added, the P : Ru ratio is 2 : 1. | |||||
| [RuH4(PPh3)3] | — | NaOH | 2 | 45 | 49 |
| 6 | 74 | 27 | |||
| [RuH4(PPh3)3] | — | Na | 2 | 50 | 54 |
| 6 | 89 | 32 | |||
| [RuCl2(COD)] | PCy3 | NaOH | 2 | 33 | 36 |
| 6 | 54 | 20 | |||
| [RuCl3·xH2O] | — | NaOH | 2 | 22 | 24 |
| 6 | 54 | 20 | |||
| [RuCl3·xH2O] | PCy3 | NaOH | 2 | 72 | 78 |
| 6 | 148 | 54 | |||
| [RuBr3·xH2O] | PCy3 | NaOH | 2 | 20 | 22 |
| 6 | 39 | 14 | |||
| [Ru(acac)3·xH2O] | PCy3 | NaOH | 2 | 15 | 16 |
| 6 | 30 | 11 | |||
| [RuCl2(p-cymene)]2 | PCy3 | NaOH | 2 | 86 | 94 |
| 6 | 120 | 43 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
A series of phosphine ligands were tested28 using [RuCl3·xH2O] as the precursor with the best result being a TOF of 155 h−1 and 78 h−1 after 2 h and 6 h, respectively, with 315 ppm of catalyst, 0.8 mol dm−3 Na as the base and a ligand : catalyst ratio of 2 : 1 at 90 °C.
In subsequent work29 with isopropanol using a range of nitrogen ligands (Table 4), the [RuCl2(p-cymene)]2 precursor showed a significantly higher TOF of 192 h−1 after 2 h and 120 h−1 after 6 h at 90 °C when using a lower catalyst concentration of 16 ppm and 0.8 mol dm−3 sodium isopropoxide as the base. A range of nitrogen ligands were then tested using the [RuCl2(p-cymene)]2 precursor with a ligand : catalyst ratio of 1 : 1. The majority of the ligands tested displayed turnovers >200 h−1 and >100 h−1 after 2 h and 6 h, respectively. It was observed that the trialkylamines generally performed better than primary and secondary amines, however, ligand 15 achieved the best result of 373 h−1 after 2 h which dropped to 236 h−1 after 6 h, in initial tests.
Decreasing the catalyst concentration to 4 ppm and increasing the ligand : catalyst ratio to 10 : 1 gave turnovers of 313 h−1, 233 h−1 and 137 h−1 after 2 h, 6 h and 24 h, respectively, for ligand 15. TMEDA produced better results under these conditions: 519 h−1, 317 h−1 and 189 h−1 for 2 h, 6 h and 24 h, respectively. The TMEDA complex showed unprecedented stability with a TOF of 64 h−1 after 268 h with a total TON of 17215. The results clearly indicate that the activity of ruthenium catalysts can be influenced by the nature of coordinating ligands, although the levels of catalytic activity are fairly equal for a range of ligands of any particular category. Phosphines gave TOFs in a range of 101–115 h−1 in the first 2 h whilst polyamines performed somewhat better, giving TOFs in a higher range of 295–398 h−1 in the same time period. It would appear that di- and triamines offer significant potential as ligands for alcohol dehydrogenation, and are worthy of further investigation.
A 2-hydroxypyridine ligand proved to be crucial for the success of complex 19 in the dehydrogenation of 1-phenylethanol derivatives (Scheme 5).30 In the proposed mechanism, the hydroxyl group of the pyridine forms a cyclic complex with the iridium, thereby promoting hydrogen release (Scheme 6). Although conversion of 1-phenylethanol to acetophenone was incomplete even after refluxing for 20 hours in toluene, TONs of up to 2120 were achieved, and the catalyst was successfully applied to a wide range of alcohol substrates (yields of 75–97% for ca. 15 alcohols studied). Closely related complexes based on rhodium and iridium have been reported for this application recently.31
 | ||
| Scheme 5 Hydrogen generation using Ir(III) complexes of a 2-hydroxypyridine ligand. | ||
 | ||
| Scheme 6 Mechanism of hydrogen generation using complex 19. | ||
A number of pyridine-based, tridentate ligands, chelating in a pincer-type manner within ruthenium complexes such as 20 and 21, have proven to be highly active dehydrogenation catalysts for alcohols (Scheme 7).32,33 Hydrogen is produced from alcohols even at catalyst loadings as low as 0.2 mol% with the hydride 21, or 0.4 mol% of the nitrogen-bridged dimer 20 from which it is derived (Table 5).32 The complexes, and other derivatives thereof, were characterised in detail, including X-ray crystallographic analysis. The mechanism of the reaction is believed to involve a stepwise, base-activated mechanism similar to that previously discussed, via the intermediacy of a ruthenium dihydride complex. A related catalyst containing a pyridine-based, ‘PNN’ donor system has also given excellent results in hydrogen generation.33
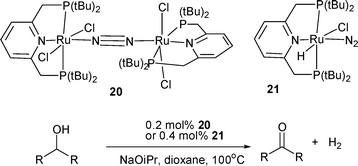
| Catalyst | Substrate | Time/h | Conversion (%) | Yield (%) | TON |
|---|---|---|---|---|---|
| 20 | Isopropanol | 70 | 60 | 57 | 144 |
| 21 | Isopropanol | 70 | 94 | 91 | 228 |
| 21 | 1-Phenylethanol | 100 | 64 | 64 | 161 |
| 21 | Cyclohexanol | 100 | 45 | 45 | 113 |
| 21 | 2-Butanol | 100 | 86 | 86 | 215 |
The conversion of alcohols to carboxylic acid derivatives potentially provides a thermodynamic driving force because the products benefit from a higher level of stabilisation through conjugation.34–41 The use of RuH2(PPh3)4 has been reported for formation of esters and lactones from alcohols and aldehydes,34 as has a ruthenium tetracyclone catalyst formed in situ from Ru3(CO)12 and acetylenes.35 The lactonisation of diols to 5 and 6 membered lactones catalysed by transition-metal hydrides under mild conditions has been described.36 The ruthenium hydride complex 22, containing a ‘PNN’ pincer ligand, catalyses the conversion of primary alcohols into both esters37 and amides38 with generation of hydrogen at each oxidation step (Scheme 8).

Using preformed complex 22, no base is required in the reaction. The chloride precursor, i.e.23, can also be used, but the addition of a base is required. Related complexes, containing bis(di(isopropylphosphine) donors, have been employed to convert alcohols into acetals in a selective manner.39 Amides may also be formed from primary alcohols and amines using a ruthenium complex of a carbene (typically 5 mol% catalyst required).40 Ruthenium hydride complexes have been used for the dehydrogenative conversion of 1,4-diols into lactones.41 Again this provides a thermodynamic driving force which counteracts the otherwise unfavourable thermodynamics associated with alcohol dehydrogenation reactions.8,9
Formic acid as a hydrogen storage material
Formic acid (FA), containing 4.4 wt% of hydrogen, represents a potentially attractive hydrogen storage material42,43 due to its formation via the reversible binding of carbon dioxide with dihydrogen44,45 and relative low toxicity (Scheme 9). | ||
| Scheme 9 Reversible formation of hydrogen and carbon dioxide from formic acid. | ||
The decomposition of formic acid using homogeneous catalysts has remained relatively underdeveloped despite several notable examples in the literature.46–49 The decomposition of FA to hydrogen and carbon dioxide is thermodynamically favoured due to the formation of two molecules of a gas.47 As such, FA conversion to hydrogen can be catalysed in an open system under relatively mild conditions. However the alternative decomposition—i.e. to CO and water— can lead to the formation of contaminants in the gas which cause complications for use in a CO-sensitive fuel cell.
As early as 1967,46 Coffey described the use of soluble metal complexes for formic acid decomposition, with predominant formation of carbon dioxide rather than carbon monoxide in the process. Of a series of Pt, Ru and Ir phosphine complexes tested, IrH2Cl(PPh3)3 gave the highest rate of decomposition. The use of the complex Rh(C6H4PPh2)(PPh3)2, containing a Rh–C σ-bond, resulted in formation of a formate complex that subsequently catalysed the formation of hydrogen from formic acid.47 Paonessa and Trogler, in 1982,48 found that a platinum dihydride complex catalysed the reversible formation of carbon dioxide and hydrogen from formic acid in a process that was somewhat dependent on the choice of solvent, and promoted by the addition of a small amount of sodium formate. The initial fast rate of the reaction was observed to slow as the gas pressures increase, but rise again when these gases were removed, an observation which has obvious implications for large scale hydrogen generation processes.48 Formic acid decomposition to hydrogen and carbon dioxide has been observed in the course of transition-metal catalysed reactions such as asymmetric transfer hydrogenation.50 In aqueous solution, nitrate ions have been found to be capable of promoting rhodium(III) catalysed formic acid decomposition to hydrogen.49 A molybdenum hydride complex for formic acid decomposition has been reported; the use of the hydride was important, as the equivalent halide complexes were inactive in this application.51
Puddephatt et al. reported extensive studies on the use of a binuclear, diphosphine-bridged, diruthenium catalyst 24 for the conversion of formic acid to H2 and CO2.52,53 The proposed mechanism (Scheme 10) was studied in detail and several intermediates were characterised by X-ray crystallography. The mechanism involves the initial formation of hydride dimer 25 from 24 through a series of protonation, formate addition, and subsequent reactions. Complex 25 then reacts with formic acid to generate hydrogen gas and complex 26, which next transfers a hydride from formate ligand to ruthenium to give 27, from which carbon dioxide is lost to regenerate 25 and complete the catalytic cycle. The reversibility of the reaction was studied, and the same catalyst was found to be competent in the formation of formic acid by hydrogenation of carbon dioxide.
 | ||
| Scheme 10 Catalytic cycle for hydrogen generation from a dimeric diruthenium complex. | ||
In recent research on the decomposition of formic acid in the presence of tertiary amine ligands,54–56 promising results were reported by Beller et al.54 Although the study focussed primarily on the use of triethylamine as the base, slightly higher volumes of hydrogen were produced in the presence of N,N-dimethylhexylamine and N,N-dimethylaminoethanol. The system was active for an extended period of time, and highly stable to impurities; added water or ethanol did not adversely influence the activity of the catalysts. Although an amine was required, it was not decomposed or consumed, hence aliquots of formic acid could be added to the reaction when required, giving full regeneration of catalytic activity. TOFs of 445.5 h−1 and 446.5 h−1 were measured after 2 h and 3 h, respectively, with 5.95 μmol of [RuCl2(PPh3)3] (pretreated in 1 mL DMF at 80 °C for 2 h) in 5HCO2H–2NEt3 (FA–TEA) at 40 °C. The TOF was 2688 h−1 after the first 20 minutes. Without pretreatment or the presence of DMF the rates were 238.5 h−1 after 2 h and 245.0 h−1 after 3 h. Interestingly, the rates were not dissimilar at 26.5 °C, measuring 181 h−1 and 204 h−1 after 2 h and 6 h, respectively. The pretreated system was demonstrated to run a PEM fuel cell with a maximum power of 47 mW at a voltage of 374 mV for more than 29 h.
During the course of an extensive study on the conversion of formic acid to hydrogen using the dimeric [RuCl2(p-cymene)]2 complex, the effects of 21 nitrogen bases including Et3N, nHexNMe2, amino alcohol 15 and others shown in Fig. 2 were studied.55 In the majority of cases, the ratio of FA to additive was 5 : 2, reflecting that in the original FA–TEA azeotrope. Table 6 summarises some of the results which were obtained. In all the examples shown, the level of CO in the gases produced in the reaction was shown to be less than 10 ppm, which is necessary for long term use in a fuel cell. The effect of additives such as KBr, KI and Mg salts was also investigated. A key finding was that amines and halide additives had a major influence on the activity of the catalysts, and that amidines, in particular, serve to increase hydrogen production sharply. When optimised (use of dppe was essential), a level of >330 mL hydrogen gas per hour was generated from small scale (5 mL) FA–amine mixtures.
 | ||
| Fig. 2 Amine additives used in hydrogen generation from formic acid (Table 6). | ||
| Additive | Vol gas 2 hb | Vol gas 3 hb | TON 2 h | TON 3 h | TOF 3 h |
|---|---|---|---|---|---|
| a Ratio FA : additive is 5 : 2, 5 mL scale at 40 °C for 3 h.b Gas is 1 : 1 CO2 : H2.c FA : additive = 5 : 4. | |||||
| Et3N | 41 | 61 | 15 | 21 | 7 |
| nHexNMe2 | 59 | 88 | 20 | 30 | 10 |
| 15 | 59 | 88 | 20 | 30 | 10 |
| 28c | 1.8 | 2.3 | 0.62 | 0.79 | 0.26 |
| 29 | 0.5 | 0.8 | 0.17 | 0.27 | 0.09 |
| 30 | 3.3 | 5.6 | 1.1 | 1.9 | 0.63 |
| 31 | 1.4 | 3.0 | 0.48 | 1.0 | 0.33 |
| 32 | 2.0 | 2.5 | 0.68 | 0.85 | 0.28 |
| 33 | 32 | 44 | 11 | 15 | 5 |
| 34 | 18 | 26 | 6.3 | 8.9 | 3 |
| 35c | 18 | 26 | 6.3 | 8.9 | 3 |
| 36 | 35 | 46 | 12 | 16 | 5.3 |
| 37 | 86 | 127 | 29 | 44 | 15 |
| 38 | 116 | 168 | 40 | 58 | 19 |
| 39 | 65 | 97 | 22 | 33 | 11 |
| 40 | 104 | 155 | 36 | 53 | 18 |
| 41 | 88 | 130 | 30 | 44 | 15 |
| 42 | 2.5 | 3.4 | 0.85 | 1.2 | 04 |
| 43 | 18 | 28 | 6.3 | 9.5 | 3.2 |
| 44 | 23 | 32 | 7.9 | 11 | 3.7 |
| 45 | 95 | 151 | 33 | 52 | 17 |
In the extensive study on nitrogen bases (Fig. 2, Table 6), an attempt was made to correlate the relationship of TON to base pKa. This resulted in the identification of three broad groupings. In the lowest pKa/low TON were the heterocyclic aromatic bases and urea. The aliphatic amines made up a large group which had pKas from 8 to 14 and TONs in the range of 8–40. The highest TONs (40–55) were exhibited by nitrogen bases with the highest pKas, notably the strong guanidine bases such as DBN, DBU, TBD and MTBD. These results show a strong correlation between base strength and activity, which may be useful in future catalyst design. In the context of other methods, the TONs and TOFs obtained with this system were competitive with those obtained with other Ru–nitrogen base systems. More significant however is the observation of very low levels of CO gas produced in the formic acid breakdown, typically less than 10 ppm. This makes the method highly valuable for use in CO-sensitive polymer electrolyte membrane (PEM) fuel cells.55
In another publication in 2008 by Beller et al.56 attempts to identify other suitable catalysts for formic acid decomposition linked to direct fuel cell utilisation yielded TOFs of 688 h−1 and 492 h−1. These were recorded after 2 h and 3 h, respectively, for 17.1 μmol [RuBr3·xH2O] with 3 equivalents of PPh3 in 5FA–2TEA at 40 °C. The catalyst was pretreated in 1 mL DMF at 80 °C for 2 h. The TOF was 3630 h−1 after the first 20 minutes. Unfortunately the catalyst was shown to deactivate as the reaction progressed. Other promising results involved 9.55 μmol [RuCl2(benzene)]2 in the presence of dppp (Ru : P, 1 : 6) which had turnovers of 127.0 h−1 and 458.7 h−1 after 2 h and 3 h, respectively, at 40 °C with catalyst pretreatment as described above. 29.75 μmol [RuCl2(benzene)]2 with 6 equivalents of PPh3 in 20 mL of 5HCO2H–4HexNMe2 was used to run a PEM fuel cell at 26.5 °C, generating 26 mW of power at 370 mV for 42 h (after an initial phase at 48 mW).
In a recent paper, Fellay et al.57 demonstrated a continuous, aqueous-based system containing hydrophilic ruthenium-based catalysts. In this system, formic acid is fed into a reactor containing a solution of [Ru(H2O)6(OTs)2] with meta-trisulfonated triphenylphosphine (TPPTS) in water with a small quantity of sodium formate to activate the catalyst. The evolved gases were released at a rate to maintain constant pressure within the reactor. Turnovers of 230 h−1 at 100 °C and 460 h−1 at 120 °C were achieved using 125 mmol Ru(H2O)6(TsO)2] and 250 mmol TPPTS. The hydrogen produced in this process was shown to be of high quality suitable for use in all types of fuel cells. Using this method, high pressures of gas could be generated, typically up to 220 bar, which reflects the thermodynamic favourability of the reaction even at elevated pressures. Indeed pressure of up to 750 bar was reported to be generated. The authors were able to create a continuous-flow method in which formic acid was continually added to a Parr autoclave reactor from which gases were simultaneously released as the formic acid was decomposed, thus providing an uninterrupted flow of hydrogen gas for power-supply. The mechanism of the formic acid decomposition was discussed in a recent full paper.58 Another water soluble catalyst, in this case [rhodium(pentamethylcyclopentadienyl)-(2,2-bipyridine)(H2O)]+, has been described by Suenobu et al. to be a very efficient one for formic acid decomposition at 298 K.59 There was no evidence of CO generation during the decomposition reaction, which reached a maximum rate at a pH of 3.8. Repeated additions of doses of formic acid could be carried out without catalyst decomposition. A series of mechanistic investigations revealed that the reaction in D2O gave rise to extensive formation of D2, which was believed to arise from an exchange reaction of the intermediate rhodium-hydride intermediate.
Whilst much effort has focussed on the use of formic acid as the hydrogen source, hydrogen production from aqueous formate salts is also a viable method.60 By coupling this to the reaction between water and CO, a method exists for hydrogen production from formic acid without the need to isolate it during the process.
Conclusions
The homogeneous dehydrogenation of alcohols, particularly the low molecular weight alcohols methanol, ethanol and isopropanol and the decomposition of formic acid, provide a viable method for the production of molecular hydrogen as a sustainable fuel source. Early work in the area served to establish the thermodynamics and mechanisms for dehydrogenation processes. Later research work has led to catalyst optimisation, particularly through studies of the effects of additives such as nitrogen bases and phosphines on the reactions. Formic acid decomposition has seen something of a renaissance of interest in recent years; it appears to offer a viable method for safe hydrogen transport, i.e. by virtue of its reversible formation. The ability to generate high pressures of hydrogen through formic acid degeneration offers possibilities for high energy output applications. Substantial further work needs to be completed, however, to identify stable and effective catalysts that function under mild conditions, and which work on more complex substrates, for example glycerin and starch. In the longer term, the goal of producing a commercial process for hydrogen generation and storage based on transformations of organic molecules appears to be worthwhile and achievable.Notes and references
- Hydrogen as a Future Energy Carrier, ed. A. Zuttel, A. Borgschulte and L. Schlapbach, Wiley VCH, Weinheim, 2008 Search PubMed.
- R. M. Navarro, M. A. Peña and J. L. G. Fierro, Chem. Rev., 2007, 107, 3952–3991 CrossRef CAS.
- J. Bartacek, J. Zabranska and P. N. L. Lens, Biofuels, Bioprod. Biorefin., 2007, 1, 201–214 Search PubMed.
- A. Dobson and S. D. Robinson, Inorg. Chem., 1977, 16, 137–142 CrossRef CAS.
- C. W. Jung and P. E. Garrou, Organometallics, 1982, 1, 658–666 CrossRef CAS.
- G. B. W. L. Ligthart, R. H. Meijer, M. P. J. Donners, J. Meuldijk, J. A. J. M. Vekemans and L. A. Hulshof, Tetrahedron Lett., 2003, 44, 1507–1509 CrossRef CAS.
- J. van Buijtenen, J. Meuldijk, J. A. J. M. Vekemans, L. A. Hulshof, H. Koojiman and A. L. Spek, Organometallics, 2006, 25, 873–881 CrossRef CAS.
- D. Morton and D. J. Cole-Hamilton, J. Chem. Soc., Chem. Commun., 1987, 248–249 RSC.
- D. Morton and D. J. Cole-Hamilton, J. Chem. Soc., Chem. Commun., 1988, 1154–1156 RSC.
- D. Morton, D. J. Cole-Hamilton, I. D. Utuk, M. Paneque-Sosa and M. Lopez-Poveda, J. Chem. Soc., Dalton Trans., 1989, 489–495 RSC.
- H. B. Charman, J. Chem. Soc. B, 1970, 584–587 RSC.
- R. H. Meijer, G. B. W. L. Lighart, J. Meuldijk, J. A. J. M. Vekemans, L. A. Hulsof, A. M. Mills, H. Kooijman and A. L. Spek, Tetrahedron, 2004, 60, 1065–1072 CrossRef CAS.
- E. Delgado-Lieta, M. A. Luke, R. F. Jones and D. J. Cole-Hamilton, Polyhedron, 1982, 1, 836–838 CrossRef.
- T. Yamakawa, T. Katsurao, S. Shinoda and Y. Saito, J. Mol. Catal., 1987, 42, 183–186 CrossRef CAS.
- K. Nomura and Y. Saito, J. Mol. Catal., 1989, 50, 303–313 CrossRef CAS.
- C. G. Griggs and D. J. H. Smith, J. Organomet. Chem., 1984, 273, 105–109 CrossRef CAS.
- K. Nomura, Y. Saito and S. Shinoda, J. Mol. Catal., 1989, 52, 99–111 CrossRef CAS.
- H. Arakawa and Y. Sugi, Chem. Lett., 1981, 1323–1326 CrossRef CAS.
- T. Matsubara and Y. Saito, J. Mol. Catal., 1994, 92, 1–8 CrossRef CAS.
- S. Shinoda, H. Itagaki and Y. Saito, J. Chem. Soc., Chem. Commun., 1985, 860–861 RSC.
- T. Fujii and Y. Saito, J. Mol. Catal., 1991, 67, 185–190 CrossRef CAS.
- L.-C. Yang, T. Ishida, T. Yamakawa and S. Shinoda, J. Mol. Catal. A: Chem., 1996, 108, 87–93 CrossRef CAS.
- H. Itagaki, S. Shinoda and Y. Saito, Bull. Chem. Soc. Jpn., 1988, 61, 2291–2294 CAS.
- T. A. Smith, R. P. Aplin and P. M. Maitlis, J. Organomet. Chem., 1985, 291, C13–C14 CrossRef CAS.
- T. Yamakawa, M. Hiroi and S. Shinoda, J. Chem. Soc., Dalton Trans., 1994, 2265–2269 RSC.
- K. Makita, K. Nomura and Y. Saito, J. Mol. Catal., 1994, 89, 143–150 CrossRef.
- G. R. A. Adair and J. M. J. Williams, Tetrahedron Lett., 2005, 46, 8233–8235 CrossRef CAS.
- H. Junge and M. Beller, Tetrahedron Lett., 2005, 46, 1031–1034 CrossRef CAS.
- H. Junge, B. Loges and M. Beller, Chem. Commun., 2007, 522–524 RSC.
- K.-I. Fujita, N. Tanino and R. Yamaguchi, Org. Lett., 2007, 9, 109–111 CrossRef CAS.
- A. M. Royer, T. B. Rauchfuss and S. R. Wilson, Inorg. Chem., 2008, 47, 395–397 CrossRef CAS.
- J. Zhang, M. Gandelman, L. J. W. Shimon, H. Rozenbery and D. Milstein, Organometallics, 2004, 23, 4026–4033 CrossRef CAS.
- J. Zhang, M. Gandelman, L. J. W. Shimon and D. Milstein, Dalton Trans., 2007, 107–113 RSC.
- S.-I. Murahashi, T. Naota, K. Ito, Y. Meeda and H. Taki, J. Org. Chem., 1987, 52, 4319–4327 CrossRef CAS.
- Y. Blum and Y. Shvo, J. Organomet. Chem., 1985, 282, C7–C10 CrossRef CAS.
- Y. Lin, X. Zhu and Y. Zhou, J. Organomet. Chem., 1992, 429, 269–274 CrossRef CAS.
- J. Zhang, G. leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS.
- C. Gunanathan, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2009, 131, 3146–3147 CrossRef CAS.
- L. U. Nordstrøm, H. Vogt and R. Madsen, J. Am. Chem. Soc., 2008, 130, 17672–17673 CrossRef CAS.
- J. Zhao and J. F. Hartwig, Organometallics, 2005, 24, 2441–2446 CrossRef CAS.
- S. Enthaler, ChemSusChem, 2008, 1, 801–804 CrossRef CAS.
- F. Joó, ChemSusChem, 2008, 1, 805–808 CrossRef CAS.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS.
- M. L. Man, Z. Zhou, S. M. Ng and C. P. Lau, Dalton Trans., 2003, 3727–3735 RSC.
- R. S. Coffey, Chem. Commun., 1967, 923–924 Search PubMed.
- S. H. Strauss, K. H. Whitmire and D. F. Shriver, J. Organomet. Chem., 1979, 174, C59–C62 CrossRef CAS.
- R. S. Paonessa and W. C. Trogler, J. Am. Chem. Soc., 1982, 104, 3529–3530 CrossRef CAS.
- R. B. King and N. K. Battacharyya, Inorg. Chim. Acta, 1995, 237, 65–69 CrossRef CAS.
- A. Fujii, S. Hashiguchi, N. Uematsu T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS.
- J. H. Shin, D. G. Churchill and G. Parkin, J. Organomet. Chem., 2002, 642, 9–15 CrossRef CAS.
- Y. Gao, J. Kuncheria, G. P. A. Yap and R. J. Puddephatt, Chem. Commun., 1998, 2365–2366 RSC.
- Y. Gao, J. K. Kuncheria, H. A. Jenkins, R. J. Puddephatt and G. P. A. Yap, J. Chem. Soc., Dalton Trans., 2000, 3212–3217 RSC.
- B. Loges, A. Boddien, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3962–3965 CrossRef CAS.
- H. Junge, A. Boddien, F. Capitta, B. Loges, J. R. Noyes, S. Gladiali and M. Beller, Tetrahedron Lett., 2009, 50, 1603–1606 CrossRef CAS.
- A. Boddien, B. Loges, H. Junge and M. Beller, ChemSusChem, 2008, 1, 751–758 CrossRef CAS.
- C. Fellay, P. J. Dyson and G. Laurenczy, Angew. Chem., Int. Ed., 2008, 47, 3966–3968 CrossRef CAS.
- C. Fellay, N. Yan, P. J. Dyson and G. Laurenczy, Chem.–Eur. J., 2009, 15, 3752–3760 CrossRef CAS.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, ChemSusChem, 2008, 1, 827–834 CrossRef CAS.
- H. Wiener, Y. Sasson and J. Blum, J. Mol. Catal., 1986, 35, 277–284 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |