Controlled polymer synthesis—from biomimicry towards synthetic biology
George
Pasparakis
*ab,
Natalio
Krasnogor
c,
Leroy
Cronin
d,
Benjamin G.
Davis
e and
Cameron
Alexander
*b
aInstitute of Electronic Structure and Laser (IESL), Foundation for Research and Technology Hellas (FORTH), P.O. Box 1527, 711 10, Heraklion, Crete, Greece. E-mail: gpasp@iesl.forth.gr
bSchool of Pharmacy, The University of Nottingham, University Park, Nottingham, UK NG7 2RD. E-mail: cameron.alexander@nottingham.ac.uk; Tel: +44 (0)115 846 7678
cSchool of Computer Science, University of Nottingham, Jubilee Campus, Wollaton Road, Nottingham, UK NG8 1BB
dWestCHEM, Department of Chemistry, University of Glasgow, UK G12 8QQ
eDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, UK OX1 3TA
First published on 6th October 2009
Abstract
The controlled assembly of synthetic polymer structures is now possible with an unprecedented range of functional groups and molecular architectures. In this critical review we consider how the ability to create artificial materials over lengthscales ranging from a few nm to several microns is generating systems that not only begin to mimic those in nature but also may lead to exciting applications in synthetic biology (139 references).
 George Pasparakis | George Pasparakis studied Materials Science at the University of Patras (BSc, 2005), before commencing graduate studies at the School of Pharmacy, University of Nottingham, under the supervision of Professor Cameron Alexander. His research project aimed at the construction of artificial cells based on smart glycopolymers that mediate specific cell–polymer interactions. Dr Pasparakis was awarded his PhD in July 2009. His research interests include cell–polymer interactions, drug delivery using smart polymers and soft materials for synthetic biology. He is currently a research associate at IESL-FORTH, Heraklion, Greece. |
 Natalio Krasnogor | Natalio Krasnogor is an Associate Professor and Reader in the School of Computer Science at the University of Nottingham, where he leads the Interdisciplinary Optimisation Laboratory (IOL). His research activities lie at the interface of Computer Science and the Natural Sciences. In particular, he develops competitive search methodologies and intelligent decision support systems for transdisciplinary optimisation, modelling of complex systems and very-large datasets processing. He has applied his expertise to bioinformatics, systems biology, synthetic biology, nanoscience and chemistry. Dr Krasnogor is a member of the editorial board for the journal Artificial Evolution and Applications. He co-chaired the 1st and 2nd European Conferences on Synthetic Biology. |
 Leroy Cronin | Leroy Cronin is a Professor of Chemistry at the University of Glasgow, where he is also an EPSRC Advanced Research Fellow. His current research interests include polyoxometalate clusters, non-equilibrium supramolecular assembly, complex and emergent chemical systems and self-growing nano and micron-scale structures and molecular evolution. He received a silver medal in the Young European Chemists Award (2006), Nexxus young scientists award (2006) and is also a recipient of a 2007 Leverhulme Prize and the 2008 Morino Foundation Lectures. In 2009 he was elected Fellow of the Royal Society of Edinburgh, Fellow of the Royal Society of Chemistry, and was being appointed to the Gardiner chair in Glasgow. |
 Benjamin G. Davis | Benjamin Davis is a Professor of Chemistry, and Fellow and Tutor in Organic Chemistry, Pembroke College. His group’s research centres on chemical biology with an emphasis on carbohydrates and proteins. These interests encompass synthetic protein and biocomponent design and use, as well as synthesis and methodology, protein engineering, molecular biology, and glycoscience with the goal of medicinal application. He has published >100 papers, >20 patents and has received the 1999 RSC Meldola medal, 2001 RSC Carbohydrate Award, an AstraZeneca Strategic Research Award, a DTI Smart Award, a Mitzutani Foundation for Glycoscience Award, the 2002 Philip Leverhulme Prize, the 2004 Royal Society's Mullard medal, the 2005 RSC Corday-Morgan medal and the 2008 ACS Isbell Award. |
 Cameron Alexander | Cameron Alexander is Professor of Polymer Therapeutics in the School of Pharmacy at the University of Nottingham. He is a Fellow of the Royal Society of Chemistry, a member of the editorial board of the Journal of Materials Chemistry, and has published more than 90 refereed articles. Research in his group centres on the synthesis of responsive/‘smart’ materials using controlled polymerisation techniques, the study of cell–polymer interactions at surfaces and interfaces, drug gene and cell delivery, and developing materials for synthetic biology. Professor Alexander was recently appointed to an EPSRC Leadership Fellowship (2009–2014) in the area of personalised medicines. |
Introduction
Life depends on polymers, and the adage ‘the whole is greater than the sum of the parts’ is entirely appropriate for natural macromolecules. The functions of many biopolymers, from information storage in nucleic acids to cell-binding by lectins, are characterised by an enhanced activity of the individual monomer components through incorporation in a polymer chain.1 Changes in the way the units are organised can completely alter the overall activity of the polymer, with the obvious examples being DNA strands—even a single mismatch in an otherwise complementary sequence disrupts binding. By contrast, most synthetic polymers have, until recently, exhibited properties that represent ‘averaged’ functionalities of ensembles of chains. This has primarily been due to difficulties in putting monomers of different functionalities together precisely in a synthetically tractable manner. However, advances in controlled polymer synthesis,2–7 especially those involving free radical polymerisations, are allowing large wholly synthetic molecules to be put together in highly directed ways. It is now possible to assemble monomer units with side-chains of very different functionalities into polymers, leading to materials which have truly unprecedented properties. In this review we highlight polymers with structures and functions complementary to those in nature, and illustrate how new materials, which may lead to applications beyond biomimicry and towards synthetic (and artificial) biology, can be produced.Structural precision in polymeric materials
The precise assembly of natural polymers underlies their selectivities in function, which have been tuned through successive cycles of evolution against an enormous diversity of fitness functions.8 The well-known sequences of nucleic acids, the primary structures of proteins and the varying constituents of carbohydrates and lipids have thus formed the basis of information storage and transfer, metabolic processing and cellular architecture and compartmentalisation. Both spatial and temporal control of these units in the cell relies on many interdependent synthesis and processing mechanisms and a plethora of correction strategies to ensure that the correct materials form.Synthetic polymerisation procedures also rely on multiple reaction stages, many of which can be connected, but in general there are limited error correction stages during a lab-based polymer synthesis. As a result, most artificial polymers have to be post-synthetically purified, and this usually involves precipitation or fractionation procedures that cannot select for properties more defined than molar mass ranges. With the exception of artificial peptides, oligonucleotides and dendrimers, which have been synthesised in precise but intensive step-wise procedures, synthetic polymers have thus not possessed the intricacies of structure or function of their natural counterparts. New polymerisation methods are changing this distinction, as practical routes to assemble complex molecular architectures from a whole host of accessible monomers are now available. The fidelity of the synthetic routes is enabling polymer chemists to assemble sequence-specified macromolecules, with all the associated extra functionality that this may afford (Fig. 1).
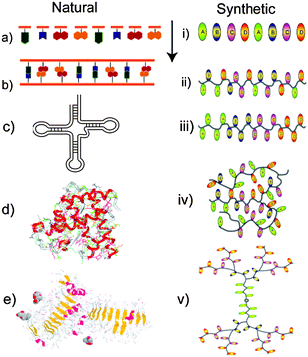 | ||
| Fig. 1 Structures of natural and synthetic polymers. Monomer units (a, i) can be assembled into defined (b) or random (ii) sequences. Further complexity is possible via regions of self-complementarity as in t-RNA (c), or in block copolymers (iii). Cross-linking of polymer chains occurs in both natural polymers via S–S links in proteins (d) or in network polymers (iv). Combinations of blocks with complex architectures can be found in glycoproteins (e) and dendrimers (v). | ||
Linear polymers in nature vary from those with very precisely defined sequences, such as RNA, DNA, peptides and proteins, to poly(saccharides) such as alginic acids and pectin where there is considerable flexibility in monomer sequence throughout the chain. In part these differences reflect the functions of the molecules: nucleic acids have evolved for information storage and thus require high fidelity of sequence, whereas many polysaccharides are structural materials and need to be flexible both in sequence and function. More complex structures such as block copolymers and supramolecular networks are found throughout biology, with typical examples being viral coat proteins, bacterial peptidoglycans, myosin filaments and microtubules. A key feature of many of these higher order architectures is that they derive from more simple linear polymers that are predisposed to assemble in particular ways to give the final objects. This in turn enables control over lengthscales ranging from several nm in small proteins to micron-sized structures in cells.
Synthetic counterparts of supramolecular assemblies are now becoming much more feasible owing to the increased control in the ‘primary sequences’ of artificial polymers. This is enabling structures and architectures such as micelles, vesicles, core–shell particles etc. to be prepared more readily than before and a degree of overall order analogous to (if simplified compared to) those seen in natural systems (Fig. 2).
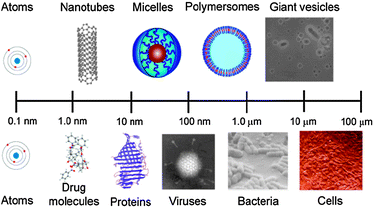 | ||
| Fig. 2 Lengthscales and structures/architectures in synthetic and natural systems of increasing complexity. | ||
Functional biomimicry in synthetic systems
The changes in architecture in these tri-blocks have potential to be used in a variety of biomedical settings, from sensing to controlled encapsulation and release, as, for example, hydrophobic drugs encapsulated in the micellar form of the polymers might be released during the transition to vesicular architecture.
However, PNIPA copolymers are perceived to exhibit significant cytotoxicity and biocompatibility issues (even though in fact there is little toxicology data for this material in the open literature24,25) while synthesis of PNIPA to controlled molar masses has only become readily accessible since the advent of radical addition fragmentation transfer (RAFT)6 polymerisation. In efforts to circumvent these problems the Lutz group proposed an alternative class of polymers that could potentially replace PNIPA. Oligo-ethylene glycol methacrylates were found to exhibit sharp LCST onsets over a wide range of temperatures simply by varying co-monomer ratios. The polymers were prepared by atom transfer radical polymerisation (ATRP)2 which provided well-defined materials of controlled molecular weight and low polydispersities,26 in contrast to polymerisation of NIPA, which by ATRP routes is difficult to control with high degrees of precision due to catalyst deactivation by the amide segment of the monomer. Controlled polymerisation of the oligo-ethylene glycol methacrylates by ATRP has enabled complex block architectures and enhanced functionalities to be generated.27–29 Recent work from this group has extended the potential applications of these biocompatible responsive polymers to controlled cell attachment.30 Related PEG-methacrylates have been polymerised under ‘bio-friendly’ synthesis routes to obtain well-defined polymers.31 These particular PEG-side-chain poly(methacrylates) were found to be responsive not only to temperature but also to salts of the Hofmeister series, which are also of relevance in protein stability. Hybrid block copolymers could be synthesised composed of statistical sequences of two structurally-similar monomers, poly(ethylene glycol)methacrylate ethyl ether of molar mass 246 (PEGMA-EE 246) and poly(ethylene glycol)methacrylate methyl ether of molar mass 475 (PEGMA-ME 475), from which was grown an outer block of PEGMA-ME 475. These polymers self-assembled in water due to the difference in the hydrophilic nature of the blocks, in a manner similar to those of the PNIPA-containing tri-blocks (Fig. 3) described by Sundararaman et al.23 Interestingly, the PEGMA-EE 246-stat-PEGMA-ME 475-graft PEGMA-ME 475 materials can be considered as “mono-hybrid” polymers as they consist solely of pendant PEG chains of different lengths. As such their sequences show similarities to carbohydrate polymers in nature, for example certain glycosaminoglycans which display pendant saccharide chains of varying length. The ability of these PEGMA-based polymers to assemble into supramolecular structures reversibly dependent on stimuli points to a variety of practical applications. From a pharmaceutical perspective, the “mono-hybrid” PEG-methacrylate polymers should give rise to fewer breakdown products, implying reduced complexity in future toxicological profiling.
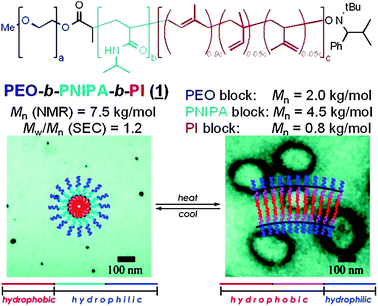 | ||
| Fig. 3 Structures of ABC triblock stimuli-responsive polymers. Increase in temperature above LCST of the middle block induced a change from micellar to vesicular structures. Published in ref. 23. Copyright © 2008 American Chemical Society. | ||
Another class of materials as potential candidates to replace PNIPA as switchable systems was explored by Hoogenboom et al. who studied the physicochemical properties of poly(oxazolines).32 The latter are novel polymers that can be produced by living cationic polymerisation of 2-oxazoline monomers. Various substituents can be used at the 2-position (i.e. ethyl, propyl etc.). It was found that the LCST of these polymers was dependent on the molecular weight, in a manner analogous to PNIPA. Since 2-propyl-oxazoline is an isomer form of NIPA (and indeed the amino acid leucine), these new polymers could be considered as NIPA alternatives particularly for applications where reversibility of the phase transition is disfavoured (for example as injectable scaffolds) as poly(oxazolines) are known to maintain their collapsed state if exposed above LCST for long periods of time. Similar work by Iwasaki et al. showed that novel biodegradable polyphosphoester polymers produced by ring opening polymerisation exhibit similar LCST behaviour to polyoxazolines that can also be fine tuned according to the ratio of the monomers used (i.e. 2-ethoxy-2-oxo-1,3,2-dioxaphospholane and 2-isopropoxy-2-oxo-1,3,2-dioxaphospholane).33
The induction of a change in LCST via a non-reversible pH-mediated reaction was exploited by Zou et al., who prepared a new polymer with tuneable LCST based on poly(N-[(2,2-dimethyl-1,3-dioxolane)methyl]acrylamide).35 The starting monomer, [(2,2-dimethyl-1,3-dioxolane)methyl]-acrylamide, was polymerised by ATRP to yield a material with a sharp LCST at 20 °C. Cleavage of the dioxolane groups left side-chains with hydrophilic diol moieties, raising the LCST. Controlled incorporation of the ratio of protected–cleaved dioxolanes in these polymers enables fine tuning of the LCST, while the diol groups can easily be converted to other groups to alter LCST, self-association or other properties as desired. Polymers of this type are likely to be of practical interest as most thermoresponsive polymers used to date are difficult to functionalise post-synthesis and thus are rather limited in function compared to biological counterparts (Fig. 4).
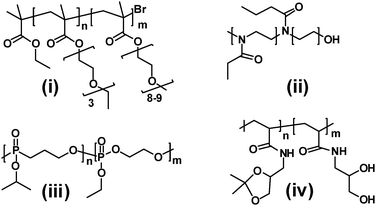 | ||
| Fig. 4 Structures of thermoresponsive polymers. Structures i–iv are described in ref. 27–33. | ||
It is important that systems are able to deal with a large number of stimuli that may vary spatially and temporally; as such combinations of responses are important in applications such as sensing and actuation. The Sumerlin group have recently reported a polyboronic acid block combined with a poly(NIPA) block which assembles/disassembles in response to biochemical stimuli, in this case temperature, pH and sugar (Fig. 5).36
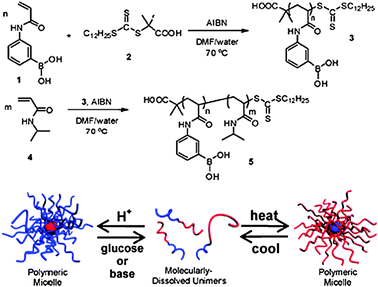 | ||
| Fig. 5 Triply responsive boronic acid-containing polymers. Supramolecular structures can be inverted through changes in temperature, pH and molecular recognition. Reproduced from ref. 36 with permission. | ||
These materials have clear parallels with natural polymers in that they are able to change conformation due to a ligand binding event. The possibilities for these polymers of encapsulating an active material, with an obvious example being insulin, then releasing it in response to glucose, suggest practical uses in therapies as well as in biosensing events if regulatory approval can be achieved.
Synthetic–natural hybrids
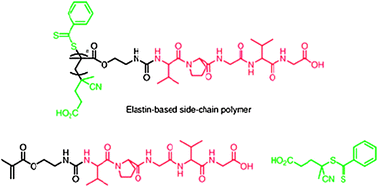 | ||
| Fig. 6 Polymer–peptide hybrid materials with side-chain elastin-like sequences (shown in red) pendant from a polymethacrylate backbone grown from RAFT agent (in green). Reproduced from ref. 37 with permission. | ||
RAFT techniques were used to prepare polymers of different molecular weights but with very low polydispersities, enabling fine control of the transition temperature of the polymers in aqueous solutions. Owing to the low variation in molar mass range due to the RAFT methodologies, it was found that simple mixing of two different batches of polymers, differing only in the weight average molecular weight, resulted in a single LCST value which was the average of the transition temperatures of the individual polymers. Thus it was possible to adjust the LCST of elastin-based polymers over a wide range of temperatures of physiological interest. The normal functions of elastin are to provide resilience and elasticity in connective tissue, arteries and ligaments, thus the ability to tailor the properties of elastin-hybrids with fine control over LCST, and in combination with a synthetic polymer backbone, offers much potential in tissue repair and regenerative medicine.
The Chilkoti41 and Ghandehari42 groups have performed systematic studies on other elastin-like polypeptides (Val-Pro-Gly-Xaa-Gly, X being any amino acid besides proline), also exploiting the inverse transition temperature behaviour. These materials have a number of advantages compared to more established responsive polymers which render them particularly attractive for the clinic: (i) they are potentially less cytotoxic as they are solely made of amino acids, (ii) solvation properties such as LCST can be fine tuned by incorporating any moiety at the X position, (iii) self-assembled superstructures can be controlled by varying the hydrophobic to hydrophilic ratio, and (iv) perhaps most importantly, these materials can exhibit multivalent interactions with biological hosts directly derived from their peptidic nature which can be varied by incorporation of any oligopeptide-targeting ligand. Hence, ELPs are promising candidates for targeted drug delivery. For example, tumour targeting can be achieved by temperature stimulation (via local hyperthermia) to induce localised self-assembly and accumulation of ELP nanocarriers that could target cancer cells by multivalent host–guest interactions.
In addition to hybrid blocks where the natural component is a low–medium molar mass compound, polymers grafted to, or grown from, proteins have been prepared. The benefits of polymer conjugation to proteins are well-known in the pharmaceutical sector, where attachment of poly(ethylene glycol) (PEG), known as PEGylation, has become an important industrial and therapeutic area. This is because proteins administered as drugs via systemic injection are rapidly degraded or eliminated. The attachment of a hydrophilic polymer such as PEG to the protein means that the resulting polymer–protein conjugate can circulate longer in the system and thus is more likely to reach its intended biological target. The efficiency of protein bioconjugation has dramatically improved recently with the introduction of the “growing from” approach facilitated by living polymerisations which provides superior control on the final bioconjugate topology and uniformity. In particular, site-specific conjugation can be obtained within minutes/hours of polymerisation by ATRP or RAFT and well-defined polymers can be grown from the protein molecules that act as macroinitiators (Fig. 7). Such conjugates are likely to exhibit better defined biological activity and less complex regulatory hurdles for biomedical applications.
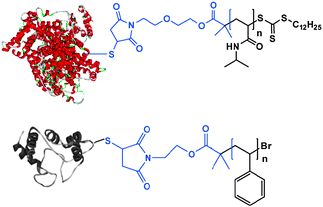
De et al. explored the “grafting from” method by using RAFT polymerisation to synthesise defined polymer–protein bioconjugates.43 A maleimido-functionalised RAFT agent was selectively reacted with the accessible sulfhydryl groups on bovine serum albumin which was then used as a macro-transfer agent to grow NIPA. Polymerisation was conducted in aqueous solution by using a water-soluble initiator at room temperature. Well-defined polymer–protein conjugates were produced as evidenced by SEC and PAGE analysis. Interestingly, the protein esterolytic activity could be retained after polymer growth and regulated by thermal oscillations, i.e. higher activity was observed at temperatures below LCST whereas lower protein activity was apparent when the NIPA segments were in the collapsed state above LCST presumably due to the hydrophobic interactions of the polymer chains with the protein active site. The exact mechanisms of the bioconjugates’ activities in respect to temperature remain elusive, but it is to be expected that these materials would behave as polymer amphiphiles giving rise to self-assembled superstructures at temperatures where the PNIPA chains become hydrophobic. To date, there have been limited data reported on transient supramolecular structures in these conjugates, but ‘switching’ of the functions of these conjugates by temperature-induced changes in attached thermoresponsive grafts/blocks has been demonstrated in a number of diverse systems.22,44
Another interesting approach towards self-assembled superstructures46 involving natural building blocks47,48 was shown by Adams et al. who synthesised novel block copolymers that can form vesicles or toroids. Oligopeptides of valine or phenylalanine were synthesised via conventional peptide synthesis and coupled to 2-aminoethyl methacrylate.49 The peptidic monomers could be polymerised by ATRP in hexafluoroisopropanol which was found to be a good solvent for polymer growth under controlled conditions. Modified PEG derivatives were used as macroinitiators that served as hydrophilic segments. Interestingly, the pendant oligopeptide segments were found to form beta sheets as shown by circular dichroism. Also, toroid-like structures were observed in some valine-rich batches: this is a rather uncommon supramolecular conformation. These materials are potentially biodegradable due to the methacrylate-based backbone, the biocompatible PEG segments and the peptide-based monomers. This combination of attributes is advantageous for utilisation in biological environments. Mesoscopic sacs and membranes formed by the interfacial combination of a peptide amphiphile with a high molecular weight polysaccharide hyaluronic acid have recently been presented by Stupp et al.50 These are robust self-sealing systems formed by osmotic pressure and self-assembly and represent a new type of polymer–protein aggregate.
As apparent from Fig. 8, a wide variety of structures and assembly types are possible, driven by synthetic polymer self-association as well as the DNA-pairing components. Strong solvophobic interactions of individual blocks were shown to generate micelles either in aqueous solvents where hydrophobic polymers formed the cores with a surface corona of DNA strands, or inverse micelles in dichloromethane when PEG-chains formed the corona with DNA-paired strands in the interior. The inverse design approach, exploiting the ability to direct DNA assembly from the bottom-up into geometrically well-defined shapes, such as catenanes, knots, and cubes, has effectively established the field of DNA-nanotechnology. These complex structures are now being developed for a variety of applications ranging from sensors and diagnostic agents to controlled release devices,54 wherein a perturbation that removes base-pairing or which cleaves the DNA strands from the synthetic block removes the driving force for self-association and supramolecular order.
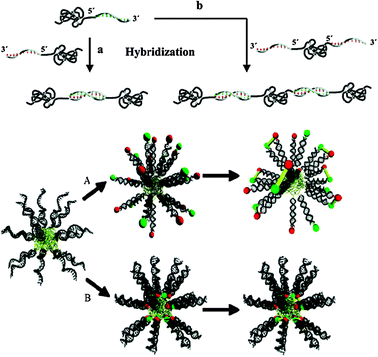 | ||
| Fig. 8 Polymer–nucleic acid hybrids. The structures are indicative of the diversity of architectures possible via different positioning of the base-pair hybrids. Reproduced from ref. 52 with permission. | ||
The combinations of biological functionality with those of synthetic polymers are extremely diverse, and the induction of ‘switching’ behaviour in these systems adds to the variety of functions these materials can perform. The syntheses and structure–function relationships of such ‘macromolecular chimera’ have also been recently reviewed.55–59
Self-assembled functional polymers
Complex polymer topologies and functional superstructures through self-assembly46,60,61 include systems that, while bio-inspired, are wholly synthetic. Structures such as functional vesicular/micellar systems that mimic cell compartments or organelles are much more accessible through controlled polymerisation techniques. The final architectures can be derived either from direct synthesis alone, programmed self-assembly of blocks as a consequence of synthesis, or through a combination of predisposed assembly followed by post-functionalisation, in a manner analogous to (though different in mechanism from) post-translational modification of proteins.Examples of complex polymer topologies formed by living polymerisation techniques alone include linear-dendritic (or branched) architectures obtained by consecutive living polymerisations of different monomers in the presence (or absence) of an inime.62,63 Despite the fact that these methods provide excellent control of the polymer topology, they are somewhat limited by the extra synthesis step required for the inimer.
In a recent study, Hong et al. interestingly demonstrated a facile method to control the polymer topology in Michael-addition polymerisations by simple temperature control.64 Polymerisation of disulfide bridged diacrylate with N-methyl ethylenediamine at low temperatures (T < 25 °C) produced linear polyamino esters whereas a slight increase in the polymerisation temperature (i.e. 48 °C) rendered the secondary amines formed active enough to induce branched topology as demonstrated by detailed 13C NMR analysis. Both linear and branched topologies could be degraded using dithiothreitol due to the redox-sensitive disulfide bonds of the diacrylate monomer. This study is an advance in the synthesis of functional/responsive materials with complex topology (i.e. branched, dendritic etc.) without the need to synthesise branching agents/crosslinkers as is often required. Additionally, the fact that this method could potentially be performed under solely aqueous conditions renders it even more attractive for the synthesis of polymeric nanocarriers for biomedical applications.
In order to obtain more elaborate functional supramolecular materials, it is sometimes necessary to carry out post-synthesis modification of self-assembled polymers. Many natural structures are post-synthetically modified, with post-translation glycosylation and phosphorylation being well-known examples. For synthetic systems, robust chemistries must be introduced that will allow for multiple functionalities to be achieved in labour/cost-effective reaction steps. Among others, the ‘click’ reaction methodology and in particular the Huisgen cycloaddition have been successfully applied in post-polymerisation functionalisation routes.3,65 Click reactions are particularly attractive due to their compatible reaction conditions with ATRP which is often the route of choice for polymer synthesis (see ref. 3 for example) and also can be applied under relatively mild synthesis conditions that are often required in presence of sensitive biomolecules (i.e. proteins).
An interesting approach was demonstrated by Li et al. who synthesised block copolymer vesicles of poly(butadiene)-block-poly(ethylene oxide) that were end-capped with azidoacetic acid to introduce the azide functionality.66 In order to determine the critical number of azide groups that can be further derivatised without disrupting the stability of the polymer bilayer, different ratios of azide-capped to non-modified polymers were tested for vesicle formation and were further reacted with an alkyne-containing dendron. Up to 40% of azide-containing polymers could be incorporated in the vesicle bilayer, however significant aggregation phenomena were observed above this ratio. The vesicles synthesised could be potentially used for biomedical applications where multivalency is necessary to achieve high affinity interactions with biological hosts such as lectin–carbohydrate interactions found in nature (see ref. 67 as an excellent recent example).
Opsteen et al. exploited click chemistry to derivatise vesicle-forming PS-b-PAA polymers.68 The latter were produced by consecutive ATRP reactions and further conversion of the –Br ends to azides with azidotrimethylsilane and tetrabutylammonium fluoride. The polymers could form vesicles in aqueous solutions that could be further decorated with alkyne-containing moieties. The principle was demonstrated by decoration of the vesicle shell with alkyne dansyl probes and other alkyne fluorescent derivatives (i.e. green fluorescent protein).
An et al. ingeniously employed concerted chemical reactions (including Huisgen addition reaction) to produce core–shell nanoparticles of well-defined heterofunctional polymers.69 Starting from an azide-containing chain transfer agent, polymers were produced via the RAFT process which could be further derivatised by Huisgen alkyne addition on the azide end and consecutive one-pot aminolysis/Michael addition reactions at the thioester end of the polymer chains. The principle was demonstrated by the production of core–shell PNIPA–PDMA nanoparticles by precipitation polymerisation. The core of the nanoparticles was decorated with fluorescein by using hydroxyethylamine as the aminolysis agent and subsequent Michael addition of an acrylate derivative of the dye whereas the shell was derivatised with a fluorescent dansyl label via a click reaction by using CuSO4/sodium ascorbate as catalyst. This study is significant in terms of functional polymers for biomedical/biosensing applications as it provides a facile route to well-defined polymers with chemically rich functionality.
Supramolecular polymer architectures and biomedical applications
Assemblies of polymers into supramolecular architectures and their subsequent derivatisation enable the generation of container-type systems analogous to natural organelles. These in turn can be considered for applications such as drug and gene delivery. Vesicles have been evaluated as nanocarriers for nucleic acids as they are topologically very similar to natural cellular compartments. The ability to include water-soluble cargo within the vesicles’ aqueous interiors along with the possibilities to fine-tune their response according to (bio)chemical stimuli70 is leading to intense interest in vesicles for therapeutic uses.An excellent example of this concept was demonstrated by the Hubbell group who synthesised novel polymer vesicles with disulfide linked blocks that could be disrupted under conditions analogous to those of lysosomal compartments in cells. The vesicles consisted of polypropylene sulfide produced by ring opening polymerisation which was linked via a disulfide bridge with a deprotected thioacetate PEG.71 The resulting polymer could form vesicles in aqueous solutions that could be disrupted/degraded under intracellular conditions due to the redox-sensitive nature of the disulfide bridges linking the two polymer blocks. The concept was demonstrated with cell uptake experiments by using calcein loaded vesicles and non-disulfide-rich vesicles as controls. The –SS-containing vesicles could release their calcein load at times relevant to the early endosome formation (that is approximately 10 minutes) indicative of vesicle disruption due to the accumulation of natural reductants such as cysteine and glutathione. On the contrary, the fluorescence intensity of calcein was found to be significantly lower in the control samples.
In another study the research groups of Armes, Battaglia and Ryan have employed pH-sensitive diblock copolymers of poly(2-(methacryloyloxy) ethyl-phosphorylcholine)-co-poly(2-(diisopropylamino)ethyl methacrylate) to encapsulate and deliver DNA molecules for gene therapy applications.72 The polymers formed vesicles at physiological pH, optimal for DNA encapsulation, whereas the vesicles were able to dissociate at pH 5–6 i.e. the pH found in acidifying endosomes in cell interiors. Plasmid-DNA loaded vesicles successfully transfected human dermal fibroblast cells and Chinese hamster ovary (CHO) cells. The results showed the high potential of these novel self-assembling and self-dissociating polymers as gene-delivery candidates. The transfection potency of these polymer delivery agents has yet to be demonstrated in more “difficult” cell lines (for example myoblasts, which have previously shown lower transgene expression than CHO cells in gene-delivery experiments). However, the advantage of the self-assembling block copolymer vesicle approach is that varying ‘doses’ of specific cell-targeting or membrane-disrupting groups can be encoded into the vesicle surfaces by simple mixing of end-capped copolymers. Intriguingly, the same research groups have recently shown enhanced cellular entry of mixed vesicles/polymersomes wherein the corona of the vesicles contained both poly(2-(methacryloyloxy) ethyl-phosphorylcholine) and PEG. Evidence for specific block and domain structures was obtained for the outer surfaces of these nanoparticles, indicating a further linkage of structure through self-assembly at the nanoscale and behaviour/function.
In contrast to the examples given above, there are certain cases where vesicle stability is desirable (for example in prolonged drug release systems) and too rapid a stimulus response constitutes a functional disadvantage. Yuting et al. contributed a novel means to “lock” block copolymer vesicles, composed of poly(N-isopropylacrylamide-co-3-aminopropylmethacrylamide), into superstructural order.73 Vesicle formation was induced by a temperature stimulus that resulted in the collapse of the NIPA segments, which drove the initial self-assembly process. Rapid mixing of the vesicles with the anionic polymer poly(sodium 2-acrylamido-2-methylpropanesulfonate) caused association with the cationic vesicular shell, so forming polyelectrolyte complexes that stabilised the vesicles into retention of structural integrity independent of the temperature. This strategy is advantageous in the sense that the double hydrophilic nature of the block copolymer does not require organic solvents to induce self-assembly, that is a simple temperature stimulus can formulate vesicles solely in aqueous solutions. In addition, the structural “locking” of the vesicles renders them particularly stable for applications where temperature variations are negligible (for example for systemic drug delivery applications). The anionic-co-cationic nature of these vesicles could also have potential for polyelectrolyte biopolymer condensation for protein and nucleic acid delivery applications.
Hydrogels and synthetic extracellular matrix mimics
The extracellular matrix (ECM) in eukaryotic organisms comprises a number of polymers with defined properties to regulate physical properties, maintain structural order and mediate cell–cell interactions and communication. Many ECM polymers also possess the attribute of structuring the aqueous environment around them, through complex 3-D architectures and exterior/surface display of high affinity water binders. These structural hydrogels have long been a focus for polymer scientists, and interest in this area has grown considerably in recent years as the demand for cell supports, tissue scaffolds and engineered/regenerated organs has developed.Hydrogels already constitute a large portion of functional biomaterials in the medical field where they find applications as contact lenses, implants, wound dressings, sustained drug release matrices and, more recently, sensors/actuators and cell delivery agents. For the more advanced applications in tissue engineering, which require a cell support role that mimics certain features of an ECM, very precise control of hydrogel structure and properties is needed. For example, in the case of biodegradable hydrogels that are used in vivo it is crucial to tailor the mechanical properties, the biodegradation profile and also to be able to predetermine the structure of the degraded fragments for the intended pharmacological outcome. In this context, controlled polymerisation and defined polymer networks with structural uniformity and controlled topology throughout the polymer mesh are highly desirable.74,75 Malkoch et al. demonstrated a significant advance in the generation of well-defined hydrogel networks by preparing diacetylene and tetraazide PEG derivatives that were chemically crosslinked under Huisgen addition reaction conditions.76 The resulting hydrogels were well-defined as the essentially stoichiometric nature of the coupling reaction enabled uniform distribution of the crosslinks in between the polymer mesh to be developed. The click reaction also allowed for fine tuning of the crosslinking degree by systematic variation of the azide/acetylene ratio. The hydrogels that were produced in this way exhibited superior mechanical properties as well as swelling behaviour when compared to conventional photocrosslinked hydrogels.
Biomimetic strategies can also be employed to synthesise hydrogel ECM analogues (Fig. 9). Ehrbar et al. synthesised novel hydrogels that are both crosslinked and degraded in a biomimetic manner.77 Eight-armed vinyl sulfone PEG hydrogel precursors were end-capped with the thiol-containing peptides Ac-FKGGGPQGIWGQ-ERCG-NH2 or H-NQEQVSPL-ERCGNH2 by using Michael addition reactions. These peptides were biochemically crosslinked by addition of the thrombin-activated factor XIIIa (transglutaminase crosslinking enzyme). Also a modified derivative of the peptide RGDSP—which is commonly used to mediate cell spreading and proliferation—was attached to the hydrogel by factor XIIIa. Finally, the peptide sequences used as crosslinks were designed to exhibit substrate specificity with metallo proteases produced by cells in order to mediate the biodegradation of the hydrogels during cell migration. Human dermal fibroblasts were cultured on the hydrogels and were found to spread more profoundly in presence of the RGD moieties within the polymer network. Hydrogel degradation also occurred due to concurrent formation of an interconnected cellular network during growth and migration.
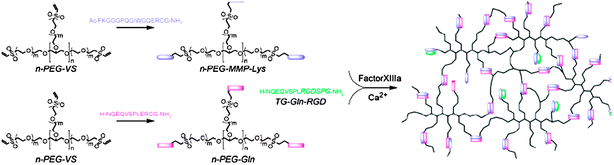 | ||
| Fig. 9 ECM mimics enzymatically-cleavable hydrogels. Peptides are linked via factor XIIIa to generate a cell-supportive scaffold, which is remodelled in situ as cells grow and release matrix metalloproteinases. Reproduced from ref. 77 with permission | ||
Kiick described a related approach to biomimetic ECM-type systems, utilising heparinized star-PEG derivatives crosslinked with heparin binding PEG-attached proteins such as antithrombin III.78 Due to diffusion, the hydrogels eroded slowly, releasing growth factors controllably into the assay media. The protein release rate could be determined by the mechanical properties of the hydrogels, that is by controlling the crosslinking density of the polymer network.
These examples demonstrate how biomimetic design of polymeric biomaterials can exhibit dynamic and adaptive behaviour similar to natural systems. Such materials might constitute a new platform of artificial extracellular matrices for 3D cell and tissue culture.
Other important classes of polymers in this category are microgels and nanogels. Microgels (including colloidal gels) find applications in drug delivery for smart drug release technologies, in medical imaging, and as injectable sol–gel scaffolds for regenerative medicine.79–81 The biomimetic approach has been extended by the Bae group into the synthesis of hydrogels with lengthscales and functionalities designed to resemble those of viruses (Fig. 10).82 These nanogel-type materials contained a core of poly(L-histidine-co-phenylalanine) which encoded structural changes due to protonation at the reduced pH values found in endo/lysosomes and solid tumours. The nanogel shells were composed of PEG units decorated with the protein BSA, thus giving the particles a steric shield against non-specific biological adsorption. The nanogel was additionally functionalised with folic acid residues to achieve tumour-targeting capability via the overexpressed folate receptor found in certain cancer cells. Nanogels loaded with the anti-cancer drug doxorubicin were found to enter cancer cells in consecutive cycles, in a similar manner to virus particles migrating to different cells and causing infection. Release of the encapsulated doxorubicin was observed to take place in cell endosomes where the nanogels swelled due to the decrease in pH that occurs in the natural cell processing of endocytosed materials. The swelling degree was high enough to allow for release of the nanogel into the cytosol with subsequent shrinking. Migration of the nanogel from the cell compartment resulted in triggering of new ‘infection’ cycles which were eventually terminated due to gradual drug depletion and loss of nanogel structural integrity.
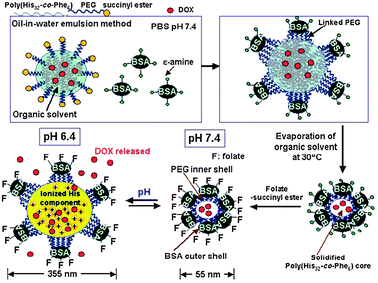 | ||
| Fig. 10 Viral mimetics, consisting of a hydrated PEG-based hydrogel, pH-swellable core and anti-cancer drug payload. Reproduced from ref. 82 with permission. | ||
A promising related approach to complex supramolecular conjugates has been developed by the Finn group83 who have explored the effects of surface modifying virus particles (which in themselves are self-assembled) on polyvalency and infection potency. Click chemistry along with ATRP was used to decorate the virus particles with glycopolymers and oligopeptides.84 These virus-polymer chimeras exhibited enhanced polyvalent properties and are of interest as carriers for therapeutic agents as they combine the already sophisticated capsids selected by evolution with desired ligands/polymers that can introduce (or even suppress, if desired) specific binding events for local or systemic drug administration.
Synthetic organelles, functional containers and cell mimics
The organisation of block copolymers into cell-like structures is a logical extension to the biomimetic self-assembly philosophy. In particular, block copolymers with structures that assemble to generate asymmetric functionality have been a major focus, with differential activities outside the self-assembled structure to those inside. The concept of ‘polymersomes’ i.e. polymer-analogues of liposomes has been elegantly expounded in a number of applications,85–88 and this field is now one of the most active in polymer science. Potential applications of polymersomes are, as with micellar systems, strongly biased towards encapsulation and drug delivery, but there have been a number of extremely elegant studies of potentially new functions. A recent example of ‘active’ polymersomes has been demonstrated by Hammer et al. who prepared block-copolymer-based vesicles that could mimic the adhesion and rolling capabilities of leukocytes.89 The vesicles were based on the commercially available block copolymer poly(butadiene-block-ethylene oxide). The hydroxyl ends of the polymer were used to attach biotin moieties which were further used to accommodate streptavidin-based segments. In an effort to mimic the selectin–integrin concerted mechanism of leukocyte adhesion and rolling onto the endothelium, the vesicle-forming polymers were decorated with (a) the sialyl LewisX carbohydrate that mediates rolling by moderate adhesion, and (b) the anti-ICAM-1 factor, known to facilitate rolling suppression developing strong adhesion forces with its ICAM-1 counterpart. The polymers were able to retain their vesicle-forming properties despite the relatively high degree of end group derivatisation. Vesicle tracking experiments showed that the vesicles exhibited rolling and adhesive behaviour under simulated flow conditions on P-selectin/ICAM-1 surfaces, and further, that these could be tuned by variation of the two ligands on the vesicles’ surfaces.Polymer–cell interactions through sugar–protein recognition events have also been demonstrated using vesicle-forming double hydrophilic block copolymers (Fig. 11). Pendant glucose units on the surface of synthetic glycopolymer vesicles specifically interacted with E. coli species that expressed carbohydrate recognition sites on their pili.90 The multivalent adhesion of the bacterial membrane to the vesicle bilayer induced molecular cargo (in this case a dye) to be transported from the vesicles to within the bacterial cytoplasm. This diffusion-driven molecular transport was most likely to have been due to confined bimolecular perturbation along the vesicle bilayer induced by the multivalent sugar–protein recognition events. If this is borne out by further experiments, then one might anticipate that such systems could exhibit non-linear or emergent properties as they appear to adopt behaviour that derives more strongly from their superstructural conformation rather than the physicochemical properties of their individual building parts.
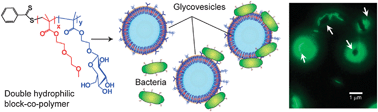 | ||
| Fig. 11 Self-assembly of double-hydrophilic block copolymers leads to vesicle formation, while specific recognition of green-fluorescent E. coli MG1655pGFP (fimH positive) at the surface of the vesicles is shown schematically and in fluorescence microscopy. | ||
Polymer superstructures and synthetic cells
The above examples of hybrid synthetic–natural polymers, viral conjugates, virus and cell analogues lead to the intriguing possibility that synthetic polymers might be used not just for biomimicry but perhaps for entirely new biologies. While such an idea might seem far-fetched, and indeed a fully synthetic biology is still a long way from being realised, nevertheless, some guiding principles from current biology are being used to extend synthetic systems across the boundaries of ‘artificial’ and ‘natural’ behaviour. For example, complex hierarchical structures from synthetic block copolymers have the possibility of embedded additive properties through their assembled blocks, leading to new properties by induction of emergent behaviour i.e. by symmetry breaking and asymmetric structural perturbation. This is widespread in biological ensembles, as organisation into compartments inherently leads to non-isotropic concentration gradients and reagent/product distributions. Self-assembling block copolymers are thus inherently useful as a generic materials platform to test emerging principles and concepts related to artificial cell applications.Steps on the route to synthetic biology necessitate development of confined/enclosed reaction systems that are gated in some way between the interior and exterior of the synthetic cell, e.g. by switchable pores or transport mechanisms (Fig. 12). Pore-formation in the lipid bilayers of natural cell membranes is facilitated by immobilised proteins that act as molecular gates allowing specific biomolecular permeability. An artificial analogue was constructed by Chiu et al., who synthesised novel functional polymer vesicles with pH-dependent permeability. The vesicles consisted of a copolymer produced by the partial transesterification of a polyacrylic acid precursor polymer with 1,2-distearoyl-rac-glycerol.91 The polymer was found to form stable multivesicular structures (small vesicles trapped within larger ones) when synthesised by the double emulsion dispersion method. The mechanism suggested was that the esterification process induced formation of multiblocks of distearoyl-glycerol acrylates within the polymer chain, which in turn facilitated the self-assembly properties of the polymer. It was therefore assumed that discrete pores of acrylic acid residues were able to be formed alongside the vesicular bilayer which could also be ionised and allow for molecular transport within the vesicle compartment at alkaline pH. Conversely the pores were “closed” in acidic environment where acrylic acid is not charged. The principle was demonstrated with small fluorescent probes such as calcein and but was also successful when larger probes were used (i.e. haemoglobin). These vesicles thus resembled the ion-gates found in mammalian cells where ion and protein transport is facilitated through specific transmembrane proteins. Also the multivesicular assemblies bore similarities to cellular compartments within the cytoplasm. In principle, this study is a pointer for synthetic biology applications such as the construction of artificial cells and advanced vehicles for targeted/smart delivery of therapeutics.
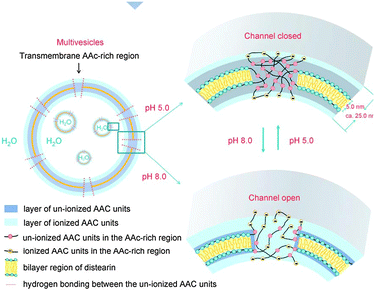 | ||
| Fig. 12 Transport into multivesicular structures via pH-switchable channels. Reproduced from ref. 91 with permission. | ||
A similar approach to selectively permeable vesicles by pH perturbations was contributed by the Kataoka group who have been very active in the field of polyion complex vesicles (PICsomes).92 PICsomes consist of block–copolymer pairs that share a block of opposite charge and a common PEG-segment. The self-assembly of these ensembles is mainly driven by electrostatic interactions which renders them pH-sensitive. Vesicles were formed by the mixing of the basic PEG-bl-poly[(5-aminopentyl)-α,β-aspartamide] (pKa 10.47) and the acidic PEG-bl-poly(α,β-aspartic acid) (pKa 4.88) which are both equally charged at physiological pH. Dynamic light scattering and confocal laser scanning microscopy revealed that the vesicles could be fragmented to smaller particles by a pH decrease to acidic levels. Remarkably, a portion of vesicles could be regenerated by a subsequent pH increase. This pH-responsive behaviour was explained by the difference of the protonation degree of the carboxylate moieties of the polymers and the mobility of the counterions. The principle was demonstrated by the pH-governed uptake and release of TRITC-dextran which was followed by confocal laser scanning microscopy. Membrane permeability could therefore be controlled by simple pH perturbations. The results from these studies suggest that wholly synthetic vesicles could act as selectively and sequentially compartmentalised reactors where input of reactants and product release can be controlled by chemical stimuli.
The study of polymersomes as microreactors has also been pursued very actively by the Nolte and van Hest groups, who have precisely positioned enzymes within polymer bilayers or in the aqueous compartment of vesicles.93 These groups have demonstrated the principle by incorporation of different enzymes i.e. Candida antarctica lipase (CAL), horseradish peroxidase, and glucose oxidase, either within the bilayer or in the outer (or inner) vesicular compartment of poly(styrene)-b-poly(isocyanoalanine(2-thiophen-3-yl-ethyl)amide). By carefully selecting appropriate substrates they were able to construct a continuous closed-loop tandem-like reaction concerted by all three enzymes. In another study from the same group, polymer vesicles were used as microreactors for lactone ring opening polymerisation.94 CAL enzyme was again either immobilised within the vesicle bilayer or in the aqueous compartment and polymerisation was observed to take place when lactone-monomers of certain hydrophobicity were used. Different polymer products were derived from the bilayer-bound enzyme and the encapsulated lipase. Of particular note was that CAL enzyme in the aqueous compartment produced similar products to non-encapsulated enzyme whereas the bilayer-bound CAL only produced low molecular weight polymer products perhaps due to the limited accessibility of the enzyme within the vesicular bilayer. More recently, a polymer vesicle system was reported based on a PEG-b-polyboronate block copolymer that could facilitate controlled permeability in response to the presence of sugar.95 The sugar-boronate binding perturbed the bilayer structural uniformity allowing permeability from discrete regions of the vesicular bilayer.
These studies clearly point to the significance of local physicochemical perturbations, close to the bilayer, in the functional properties of these systems that seems to be derived solely by their superstructural conformation rather than the individual properties of their building blocks. In this sense, there is a clear pointer to emergent behaviour96 as the reaction products change the reaction conditions and feedback loops become theoretically possible.
Another intriguing possibility is that synthetic polymer vesicles/polymersomes might behave not just as individual cell analogues, but as populations of artificial cells, perhaps leading towards tissue, organ and even organism mimics, however simplified. In order to do this, one has to consider communication between artificial cells, and the ‘language’ of this communication. At the simplest level, logic operations provide the basis for communication, and this of course bridges natural and computational systems. A discussion of the latter is beyond this review, but nevertheless, there already exist many examples of molecular logic systems.97–103 Replacement of natural information storage and processing materials is more difficult than for structural materials but polymers of controlled structure and function are now beginning to be used in more active roles, such as processing operations. While most molecular logic studies have been carried out by DNA-based materials,104–107 there is also an increasing focus on operations, and indeed artificial cells, that do not use nucleic acids as the basis for information storage and transfer. For example, Nguyen et al.108 explored aspects of artificial cellularity in a solely abiotic system by investigating emerging behaviour in a “soup” of dynamic block components that could be reversibly interconnected producing self-assembling amphiphiles in a dynamic combinatorial manner (Fig. 13).
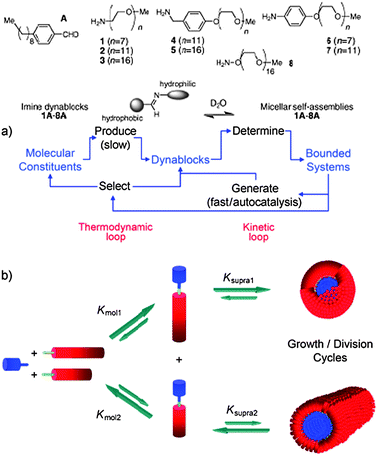 | ||
| Fig. 13 Dynamic combinatorial self-assembly of amphiphiles Schiff base formation from amine- and aldehyde-precursors leads to linked hydrophilic and hydrophobic blocks. These in turn self-assemble into different architectures based on the individual block lengths and packing parameters, each of which is dynamically linked to the synthesis. Reproduced from ref. 108 with permission. | ||
A dramatic amplification of the population of the self-assembled products was observed due to their intrinsic property to solubilise the hydrophobic dynamic blocks and stabilise hydrolysable linking imine bonds. The study ingeniously probed strong emergence in abiotic systems via an autocatalytic operation mode which is not uncommon in biological systems, however, these concepts were largely uninvestigated before the advent of synthetic biology.
The development of new tools and systems for synthetic biology implies a relationship with current biological building blocks. However, the development of totally abiotic or inorganic biology, could revolutionise our understanding of biology. The most promising starting point is with development of self-assembling superstructures based on inorganic polymers. Recently the transformation of metal-oxide crystals into dynamic self-growing tubular networks has been demonstrated.109 The assembly initiates when the crystals are immersed in an aqueous solution containing a low concentration of an organic cation, see Fig. 14. A membrane immediately forms around the crystal that then gives birth to micron-scale tubes and the growth is driven by an osmotic pressure within the membrane sack around the crystal which ruptures to release the pressure. Although this is a rudimentary system, the use of inorganic building blocks for new types of membranes, catalysts, information carrying and energy storing units offers considerable potential for future developments in synthetic biology.
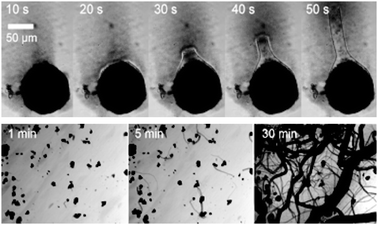 | ||
| Fig. 14 Self-assembly of inorganic superstructures from crystals of inorganic metal oxides. An organic counterion is used to coat the crystal which causes build up of osmotic pressure and the inorganic ‘monomers’ are flowed out of the crystal polymerising upon contact with the bulk solvent to growth tubes. The top view shows this process over 50 seconds for a single crystal and the bottom view shows this process on a large number of crystals. | ||
Self-assembling superstructures—towards synthetic and artificial biology
The generation of ‘life-like’ properties in synthetic systems is one that is now becoming experimentally tractable with an increasing level of molecular control and sophistication.110,111 The manipulation of biological components and principles using already demonstrated physical and computational methodologies forms one approach to synthetic biology, in a manner distinct from the quest for “artificial life” in a computational and philosophical context,112 although they are closlely related. This is primarily because synthetic cells, organs etc. might include new materials that are useful in biomedical applications.113 Further, the quest for artificial life could shed light on the events that led to the emergence of life since the underpinning process that allows the assembly of any artificial system will almost certainly share common features.The conceptual bases for synthetic biology and artificial life are in fact well-established,114–116 but the functional implementations of hypothesised ‘life-like’ behaviours in abiotic systems are only now beginning to be realised. One approach to artificial cells utilises a top-down approach (i.e. taking a biological system and reducing its components systematically until an organism no longer functions)117,118 while a second involves a bottom-up methodology (assembling components or information units until an aspect of ‘life’ appears).96,119 Key components of artificial cells include metabolism, (auto)catalysis and information transfer/replication with artificial molecules and cell-like compartments:120–124 in all cases, self-assembly and compartmentalisation of materials are central to function, and polymeric superstructures are implicit in the function. Also, the ability to encapsulate synthetic reaction networks within the compartments, and for the overall system to be dissipative, appear to be important additional requirements. Recently, a cellular mimic has been demonstrated125 with the ability to stimulate bacterial quorum sensing126 as a result of the sugar-forming formose reaction within a vesicle reaction-chamber, and the subsequent interaction of these sugars with the bacterium Vibrio harveyi. The measurable output of light by the bacteria in response to the products of this ‘proto-metabolism’ could be considered a form of chemical ‘interrogation’ of one by the other, and thus a step towards realising the ‘Turing test’ paradigm recently proposed for an imitation game involving real and artificial cells.127 The reaction in this case took place within phospholipid vesicles, rather than polymers, but it is possible to envisage similar reaction systems utilising amphiphilic polymers or other self-assembling units; indeed such systems may have advantages over phospholipids in terms of the breadth of conditions they can tolerate, and the ability to fine-tune the permeability of the vesicles to control the reactions within.
Ultimately, if it is possible to engineer or ‘emerge’ a functioning life-like artificial chemical cell, or an assembly of a ‘highly-functional’ polymer system, one key question arises: when might life-like behaviour arise, and how could it be tested? Rigorous mathematical formalisms that capture cell processes and computational methodologies such as dissipative particle dynamics (DPD)128 and P systems129 are increasingly being used for simulation purposes. DPD simulations are coarser grained than conventional molecular dynamics algorithms and hence can be used to capture longer processes. Moreover, the relative ease by which intermolecular interactions can be coded in DPD,130,131 the potential for parallelisation and the fact that DPD has a rigorous statistical mechanics interpretation,132 makes it an ideal method for simulating bilayers,133 micelles,134 vesicles135 and hence complex supramolecular objects. Whilst DPD is a suitable methodology for simulating self-assembled containers, the capture of processes such as gene transcription dynamics is more difficult due to the different time scales between membranes fusion and transcription rates. For modelling the mechanistic nature of complex gene and signalling networks P systems are used instead.128 These are an “executable” biology136 technique that capture compartments (e.g. nested vesicles) topologically, but through (variants of) Gillespie’s stochastic simulation algorithm137 P-systems can accurately follow the biochemical events associated with biological regulatory networks. Hybrid DPD–P system computational simulations have also been proposed to model leaky vesicles138 and computational liposomes.139 Such complex entities are still theoretical at this stage, but are becoming increasingly ‘life-like’ over multiple iterations. In turn the ever-increasing power of the synthetic (polymer) chemistry toolbox is enabling the structural imitation of multi-component natural systems as well as the confirmation of computationally simulated supramolecular architectures: the sophistication of function of these systems will begin to emerge as these methodologies mature.
Conclusions
In this review, we have provided selective examples from the recent literature to illustrate how the rational design of functional polymers with precise architecture and hierarchy directly impacts on the functional interactions of these materials with biological systems. The behaviour of these materials directly stems from the interplay of controlled synthesis leading to dynamic behaviour, self-assembly and application through a function such as a change in conformation. In addition it can be seen that controlled polymer synthesis—i.e. largely synthetic routes that allow for precise control of macromolecular/supramolecular functionalisation and micro-topology—is exercising an influence ranging beyond biomimicry. Precision polymers are now being developed for theoretical and practical considerations that extend beyond those currently found in nature and perhaps towards an entirely synthetic biology.Acknowledgements
We thank the Engineering and Physical Sciences and Biotechnology and Biological Sciences Research Councils (Grants EP/G042462/1, EP/D022347/1, D021847/1 and BB/F01855X/1) and the University of Nottingham, U.K., for funding.Notes and references
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef.
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS.
- J. Geng, J. Lindqvist, G. Mantovani and D. M. Haddleton, Angew. Chem., Int. Ed., 2008, 47, 4180–4183 CrossRef CAS.
- T. Pintauer and K. Matyjaszewski, Coord. Chem. Rev., 2005, 249, 1155–1184 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- R. Narain and S. P. Armes, Chem. Commun., 2002, 2776–2777 RSC.
- F. Ciciriello, G. Costanzo, S. Pino, C. Crestini, R. Saladino and E. Di Mauro, Biochemistry, 2008, 47, 2732–2742 CrossRef CAS.
- F. Carriere, C. Withers-Martinez, H. van Tilberugh, A. Roussel, C. Cambillau and R. Verger, Biochim. Biophys. Acta, 1998, 1376, 417–432 CAS.
- H. Y. Wang and G. Oster, Nature, 1998, 396, 279–282 CrossRef CAS.
- A. Gennerich and R. D. Vale, Curr. Opin. Cell Biol., 2009, 21, 59–67 CrossRef CAS.
- E. S. Gil and S. A. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- C. d. l. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- M. Hruby, J. Kucka, H. Mackova, O. Lebeda and K. Ulbrich, Chem. Listy, 2008, 102, 21–27 CAS.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS.
- I. Roy and M. N. Gupta, Chem. Biol., 2003, 10, 1161–1171 CrossRef CAS.
- A. S. Hoffman, P. S. Stayton, O. Press, N. Murthy, C. A. Lackey, C. Cheung, F. Black, J. Campbell, N. Fausto, T. R. Kyriakides and P. Bornstein, Polym. Adv. Technol., 2002, 13, 992–999 CrossRef CAS.
- A. Chilkoti, M. R. Dreher, D. E. Meyer and D. Raucher, Adv. Drug Delivery Rev., 2002, 54, 613–630 CrossRef CAS.
- B. Twaites, C. D. Alarcon and C. Alexander, J. Mater. Chem., 2005, 15, 441–455 RSC.
- Y. Tsuda, M. Yamato, A. Kikuchi, M. Watanabe, G. P. Chen, Y. Takahashi and T. Okano, Adv. Mater., 2007, 19, 3633–3636 CrossRef CAS.
- A. Kikuchi, J. Kobayashi, T. Okano, T. Iwasa and K. Sakai, J. Bioact. Compat. Polym., 2007, 22, 575–588 CrossRef CAS.
- S. S. Pennadam, M. D. Lavigne, C. F. Dutta, K. Firman, D. Mernagh, D. C. Gorecki and C. Alexander, J. Am. Chem. Soc., 2004, 126, 13208–13209 CrossRef CAS.
- A. Sundararaman, T. Stephan and R. B. Grubbs, J. Am. Chem. Soc., 2008, 130, 12264–12266 CrossRef CAS.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef CAS.
- I. Ankareddi, M. M. Bailey, C. S. Brazel, J. E. Rasco and R. D. Hood, Birth Defects Res., Part B: Dev. Reprod. Toxicol., 2008, 83, 112–116 Search PubMed.
- J. F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- K. Skrabania, J. Kristen, A. Laschewsky, O. Akdemir, A. Hoth and J. F. Lutz, Langmuir, 2007, 23, 84–93 CrossRef CAS.
- J. F. Lutz, S. Pfeifer and Z. Zarafshani, QSAR Comb. Sci., 2007, 26, 1151–1158 Search PubMed.
- J. F. Lutz, J. Andrieu, S. Uzgun, C. Rudolph and S. Agarwal, Macromolecules, 2007, 40, 8540–8543 CrossRef CAS.
- E. Wischerhoff, K. Uhlig, A. Lankenau, H. G. Borner, A. Laschewsky, C. Duschl and J. F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 5666–5668 CrossRef CAS.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS.
- R. Hoogenboom, H. M. L. Thijs, M. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760 RSC.
- Y. Iwasaki, C. Wachiralarpphaithoon and K. Akiyoshi, Macromolecules, 2007, 40, 8136–8138 CrossRef CAS.
- D. C. Wu, Y. Liu and C. B. He, Macromolecules, 2008, 41, 18–20 CrossRef CAS.
- Y. Q. Zou, D. E. Brooks and J. N. Kizhakkedathu, Macromolecules, 2008, 41, 5393–5405 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2009, 2106–2108 RSC.
- F. Fernandez-Trillo, J. C. M. van Hest, J. C. Thies, T. Michon, R. Weberskirch and N. R. Cameron, Chem. Commun., 2008, 2230–2232 RSC.
- F. Fernandez-Trillo, A. Dureault, J. P. M. Bayley, J. C. M. van Hest, J. C. Thies, T. Michon, R. Weberskirch and N. R. Cameron, Macromolecules, 2007, 40, 6094–6099 CrossRef CAS.
- J. Hyun, W. K. Lee, N. Nath, A. Chilkoti and S. Zauscher, J. Am. Chem. Soc., 2004, 126, 7330–7335 CrossRef CAS.
- D. W. Urry, A. Pattanaik, J. Xu, T. C. Woods, D. T. Mcpherson and T. M. Parker, J. Biomater. Sci., Polym. Ed., 1998, 9, 1015–1048 CrossRef CAS.
- M. R. Dreher, A. J. Simnick, K. Fischer, R. J. Smith, A. Patel, M. Schmidt and A. Chilkoti, J. Am. Chem. Soc., 2008, 130, 687–694 CrossRef CAS.
- Z. Megeed, J. Cappello and H. Ghandehari, Adv. Drug Delivery Rev., 2002, 54, 1075–1091 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS.
- A. S. Hoffman and P. S. Stayton, Prog. Polym. Sci., 2007, 32, 922–932 CrossRef CAS.
- B. Le Droumaguet and K. Velonia, Angew. Chem., Int. Ed., 2008, 47, 6263–6266 CrossRef CAS.
- J. Rodríguez-Hernández, F. Chécot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef CAS.
- H. Iatrou, H. Frielinghaus, S. Hanski, N. Ferderigos, J. Ruokolainen, O. Ikkala, D. Richter, J. Mays and N. Hadjichristidis, Biomacromolecules, 2007, 8, 2173–2181 CrossRef CAS.
- Y. Mei, K. L. Beers, H. C. M. Byrd, D. L. VanderHart and N. R. Washburn, J. Am. Chem. Soc., 2004, 126, 3472–3476 CrossRef CAS.
- D. J. Adams, D. Atkins, A. I. Cooper, S. Furzeland, A. Trewin and I. Young, Biomacromolecules, 2008, 9, 2997–3003 CrossRef CAS.
- R. M. Capito, H. S. Azevedo, Y. S. Velichko, A. Mata and S. I. Stupp, Science, 2008, 319, 1812–1816 CrossRef CAS.
- F. E. Alemdaroglu, N. C. Alemdaroglu, P. Langguth and A. Herrmann, Adv. Mater., 2008, 20, 899–902 CrossRef CAS.
- F. E. Alemdaroglu, M. Safak, J. Wang, R. Berger and A. Herrmann, Chem. Commun., 2007, 1358–1359 RSC.
- F. E. Alemdaroglu and A. Herrmann, Org. Biomol. Chem., 2007, 5, 1311–1320 RSC.
- E. S. Andersen, M. Dong, M. M. Nielsen, K. Jahn, R. Subramani, W. Mamdouh, M. M. Golas, B. Sander, H. Stark, C. L. P. Oliveira, J. S. Pedersen, V. Birkedal, F. Besenbacher, K. V. Gothelf and J. Kjems, Nature, 2009, 459, 73–76 CrossRef CAS.
- H. G. Borner and H. Schlaad, Soft Matter, 2007, 3, 394–408 RSC.
- H. Kukula, H. Schlaad, M. Antonietti and S. Forster, J. Am. Chem. Soc., 2002, 124, 1658–1663 CrossRef CAS.
- J. K. Oh, H. Dong, R. Zhang, K. Matyjaszewski and H. Schlaad, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4764–4772 CrossRef CAS.
- H. Schlaad, in Peptide Hybrid Polymers, ed. H.-A. Klok and H. Schlaad, Series: Advances in Polymer Science vol. 202, Springer, 2006, pp. 53-73 Search PubMed.
- H. Schlaad, L. You, R. Sigel, B. Smarsly, M. Heydenreich, A. Mantion and A. Masic, Chem. Commun., 2009, 1478–1480 RSC.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- S. Forster and M. Konrad, J. Mater. Chem., 2003, 13, 2671–2688 RSC.
- H. I. Lee, J. A. Lee, Z. Y. Poon and P. T. Hammond, Chem. Commun., 2008, 3726–3728 RSC.
- K. V. Bernaerts, C.-A. Fustin, C. Bomal-DeHaese, J.-F. Gohy, J. C. Martins and F. E. Du Prez, Macromolecules, 2008, 41, 2593–2606 CrossRef CAS.
- C. Y. Hong, Y. Z. You, D. C. Wu, Y. Liu and C. Y. Pan, J. Am. Chem. Soc., 2007, 129, 5354–5355 CrossRef CAS.
- G. J. Chen, L. Tao, G. Mantovani, J. Geng, D. Nystrom and D. M. Haddleton, Macromolecules, 2007, 40, 7513–7520 CrossRef CAS.
- B. Li, A. L. Martin and E. R. Gillies, Chem. Commun., 2007, 5217–5219 RSC.
- A. L. Martin, B. Li and E. R. Gillies, J. Am. Chem. Soc., 2009, 131, 734–741 CrossRef CAS.
- J. A. Opsteen, R. P. Brinkhuis, R. L. M. Teeuwen, D. W. P. M. Lowik and J. C. M. v. Hest, Chem. Commun., 2007, 3136–3138 RSC.
- Z. S. An, W. Tang, M. H. Wu, Z. Jiao and G. D. Stucky, Chem. Commun., 2008, 6501–6503 RSC.
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS.
- S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966–1972 CrossRef CAS.
- H. Lomas, I. Canton, S. MacNeil, J. Du, S. P. Armes, A. J. Ryan, A. L. Lewis and G. Battaglia, Adv. Mater., 2007, 19, 4238–4243 CrossRef CAS.
- Y. Li, B. S. Lokitz and C. L. McCormick, Angew. Chem., Int. Ed., 2006, 45, 5792–5795 CrossRef CAS.
- A. I. Triftaridou, M. Vamvakaki and C. S. Patrickios, Biomacromolecules, 2007, 8, 1615–1623 CrossRef CAS.
- M. Vamvakaki and C. S. Patrickios, Soft Matter, 2008, 4, 268–276 RSC.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 2774–2776 RSC.
- M. Ehrbar, S. C. Rizzi, R. G. Schoenmakers, B. San Miguel, J. A. Hubbell, F. E. Weber and M. P. Lutolf, Biomacromolecules, 2007, 8, 3000–3007 CrossRef CAS.
- K. L. Kiick, Soft Matter, 2008, 4, 29–37 RSC.
- B. R. Saunders, N. Laajam, E. Daly, S. Teow, X. Hu and R. Stepto, Adv. Colloid Interface Sci., 2009, 147–148, 251–262 CrossRef CAS.
- A. Fernandez-Barbero, I. J. Suarez, B. Sierra-Martνn, A. Fernandez-Nieves, F. J. de las Nieves, M. Marquez, J. Rubio-Retama and E. Lopez-Cabarcos, Adv. Colloid Interface Sci., 2009, 147–148, 88–108 CrossRef CAS.
- J. D. Kretlow, L. Klouda and A. G. Mikos, Adv. Drug Delivery Rev., 2007, 59, 263–273 CrossRef CAS.
- E. S. Lee, D. Kim, Y. S. Youn, K. T. Oh and Y. H. Bae, Angew. Chem., Int. Ed., 2008, 47, 2418–2421 CrossRef CAS.
- A. K. Udit, C. Everett, A. J. Gale, J. R. Kyle, M. Ozkan and M. G. Finn, ChemBioChem, 2009, 10, 503–510 CrossRef CAS.
- S. Sen Gupta, K. S. Raja, E. Kaltgrad, E. Strable and M. G. Finn, Chem. Commun., 2005, 4315–4317 RSC.
- H. Shen and A. Eisenberg, Angew. Chem., Int. Ed., 2000, 39, 3310–3312 CrossRef CAS.
- D. E. Discher, V. Ortiz, G. Srinivas, M. L. Klein, Y. Kim, D. Christian, S. Cai, P. Photos and F. Ahmed, Prog. Polym. Sci., 2007, 32, 838 CrossRef CAS.
- M. M. Santore, D. E. Discher, Y. Y. Won, F. S. Bates and D. A. Hammer, Langmuir, 2002, 18, 7299–7308 CrossRef CAS.
- G. Battaglia and A. J. Ryan, Nat. Mater., 2005, 4, 869–876 CrossRef.
- D. A. Hammer, G. P. Robbins, J. B. Haun, J. J. Lin, W. Qi, L. A. Smith, P. P. Ghoroghchian, M. J. Therien and F. S. Bates, Faraday Discuss., 2008, 139, 129–141 RSC.
- G. Pasparakis and C. Alexander, Angew. Chem., Int. Ed., 2008, 47, 4847–4850 CrossRef CAS.
- H.-C. Chiu, Y.-W. Lin, Y.-F. Huang, C.-K. Chuang and C.-S. Chern, Angew. Chem., Int. Ed., 2008, 47, 1875–1878 CrossRef CAS.
- A. Kishimura, S. Liamsuwan, H. Matsuda, W. F. Dong, K. Osada, Y. Yamasaki and K. Kataoka, Soft Matter, 2009, 5, 529–532 RSC.
- D. M. Vriezema, P. M. L. Garcia, N. S. Oltra, N. S. Hatzakis, S. M. Kuiper, R. J. M. Nolte, A. E. Rowan and J. C. M. van Hest, Angew. Chem., Int. Ed., 2007, 46, 7378–7382 CrossRef CAS.
- M. Nallani, H.-P. M. de Hoog, J. J. L. M. Cornelissen, A. R. A. Palmans, J. C. M. van Hest and R. J. M. Nolte, Biomacromolecules, 2007, 8, 3723–3728 CrossRef CAS.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Adv. Mater., 2009, 21, 2787–2791 CrossRef CAS.
- M. A. Bedau, Trends Cognit. Sci., 2003, 7, 505–512 Search PubMed.
- A. P. de Silva, I. M. Dixon, H. Q. N. Gunaratne, T. Gunnlaugsson, P. R. S. Maxwell and T. E. Rice, J. Am. Chem. Soc., 1999, 121, 1393–1394 CrossRef CAS.
- T. Gunnlaugsson, D. A. Mac Donaill and D. Parker, J. Am. Chem. Soc., 2001, 123, 12866–12876 CrossRef CAS.
- D. Margulies, G. Melman, C. E. Felder, R. Arad-Yellin and A. Shanzer, J. Am. Chem. Soc., 2004, 126, 15400–15401 CrossRef CAS.
- S. Uchiyama, N. Kawai, A. P. de Silva and K. Iwai, J. Am. Chem. Soc., 2004, 126, 3032–3033 CrossRef CAS.
- S. D. Straight, J. Andreasson, G. Kodis, S. Bandyopadhyay, R. H. Mitchell, T. A. Moore, A. L. Moore and D. Gust, J. Am. Chem. Soc., 2005, 127, 9403–9409 CrossRef CAS.
- J. Andreasson, S. D. Straight, G. Kodis, C. D. Park, M. Hambourger, M. Gervaldo, B. Albinsson, T. A. Moore, A. L. Moore and D. Gust, J. Am. Chem. Soc., 2006, 128, 16259–16265 CrossRef CAS.
- D. Margulies, G. Melman and A. Shanzer, J. Am. Chem. Soc., 2006, 128, 4865–4871 CrossRef CAS.
- L. M. Adleman, Science, 1994, 266, 1021–1024 CrossRef CAS.
- A. Saghatelian, N. H. Volcker, K. M. Guckian, V. S. Y. Lin and M. R. Ghadiri, J. Am. Chem. Soc., 2003, 125, 346–347 CrossRef CAS.
- G. Paun, Theor. Comput. Sci., 2000, 231, 275–296 CrossRef.
- A. V. Garibotti, S. P. Liao and N. C. Seeman, Nano Lett., 2007, 7, 480–483 CrossRef CAS.
- R. Nguyen, L. Allouche, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2009, 48, 1093–1096 CrossRef CAS.
- C. Ritchie, G. J. T. Cooper, Y. F. Song, C. Streb, H. Yin, A. D. C. Parenty, D. A. MacLaren and L. Cronin, Nat. Chem., 2009, 1, 47–52 Search PubMed.
- C. A. Hutchison, H. O. Smith and J. C. Venter, Scientist, 2006, 20, 38 Search PubMed.
- M. Bedau, Leonardo, 2002, 35, 395–400 CrossRef.
- Artificial Life II, ed. C. G. Langton, C. Taylor, J. D. Farmer and S. Rasmussen, Addison-Wesley, 1991 Search PubMed.
- J. Haseloff, IET Synth. Biol., 2007, 1, 1–2 Search PubMed.
- A. M. Turing, Philos. Trans. R. Soc. London, Ser. B, 1952, 237, 37–72 CrossRef.
- J. Von Neumann, Theory of Self-Reproducing Automata, University of Illinois Press, Champaign, IL, USA, 1966 Search PubMed.
- A. Lindenmayer, J. Theor. Biol., 1968, 18, 300 CrossRef CAS.
- E. Pennisi, Science, 2005, 310, 769 CrossRef CAS.
- J. I. Glass, N. Assad-Garcia, N. Alperovich, S. Yooseph, M. R. Lewis, M. Maruf, C. A. Hutchison, H. O. Smith and J. C. Venter, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 425–430 CrossRef CAS.
- R. Blanco, I. Inza and P. Larranga, Int. J. Intell. Syst., 2003, 18, 205–220 CrossRef.
- T. Gabaldon, J. Pereto, F. Montero, R. Gil, A. Latorre and A. Moya, Philos. Trans. R. Soc. London, Ser. B, 2007, 362, 1751–1762 CrossRef CAS.
- F. M. Menger and M. I. Angelova, Acc. Chem. Res., 1998, 31, 789–797 CrossRef CAS.
- G. von Kiedrowski, L. H. Eckardt, K. Naumann, W. M. Pankau, M. Reimold and M. Rein, Pure Appl. Chem., 2003, 75, 609–619 CrossRef CAS.
- R. V. Sole, A. Munteanu, C. Rodriguez-Caso and J. Macia, Philos. Trans. R. Soc. London, Ser. B, 2007, 362, 1727–1739 CrossRef CAS.
- T. Rocheleau, S. Rasmussen, P. E. Nielsen, M. N. Jacobi and H. Ziock, Philos. Trans. R. Soc. London, Ser. B, 2007, 362, 1841–1845 CrossRef CAS.
- P. M. Gardner, K. Winzer and B. G. Davis, Nat. Chem., 2009, 1, 377–383 Search PubMed.
- M. B. Miller and B. L. Bassler, Annu. Rev. Microbiol., 2001, 55, 165 CrossRef CAS.
- L. Cronin, N. Krasnogor, B. G. Davis, C. Alexander, N. Robertson, J. H. G. Steinke, S. L. M. Schroeder, A. N. Khlobystov, G. Cooper, P. M. Gardner, P. Siepmann, B. J. Whitaker and D. Marsh, Nat. Biotechnol., 2006, 24, 1203–1206 CrossRef CAS.
- P. J. Hoogerbrugge and J. M. V. A. Koelman, Europhys. Lett., 1992, 19, 155–160 CrossRef.
- G. Paun and G. Rozenberg, Theor. Comput. Sci., 2002, 287, 73–100 CrossRef.
- R. D. Groot and K. L. Rabone, Biophys. J., 2001, 81, 725–736 CrossRef CAS.
- R. D. Groot and P. B. Warren, J. Chem. Phys., 1997, 107, 4423–4435 CrossRef CAS.
- P. Espanol and P. Warren, Europhys. Lett., 1995, 30, 191–196 CrossRef CAS.
- A. Grafmuller, J. Shillcock and R. Lipowsky, Phys. Rev. Lett., 2007, 98, 218101 CrossRef.
- V. Ortiz, S. O. Nielsen, D. E. Discher, M. L. Klein, R. Lipowsky and J. Shillcock, J. Phys. Chem. B, 2005, 109, 17708–17714 CrossRef CAS.
- F. J. Romero-Campero, J. Twycross, M. Camara, M. Bennet, M. Gheorghe and N. Krasnogor, Int. J. Found. Comput. Sci., 2009, 20, 427–442 CrossRef.
- J. Fisher and T. A. Henzinger, Nat. Biotechnol., 2007, 25, 1239–1249 CrossRef CAS.
- D. T. Gillespie, Annu. Rev. Phys. Chem., 2007, 58, 35–55 CrossRef CAS.
- J. Smaldon, J. Blake, N. Krasnogor and D. Lancet, Proceedings of the Genetic and Evolutionary Computing Conference (GECCO 2008), (Atlanta, GA, USA, July 12–16, 2008), ed. M. Keijzer, GECCO ’08, ACM, New York, NY, pp. 249–256, DOI: http://doi.acm.org/10.1145/1389095.1389134 Search PubMed.
- J. Smaldon, N. Krasnogor, C. Alexander and M. Gheorghe, Proceedings of the 2009 Genetic and Evolutionary Computation Conference (GECCO 2009), (Montreal, Quebec, Canada, July 08-12, 2009), GECCO ’09, ACM, New York, NY, pp. 161–168, DOI: http://doi.acm.org/10.1145/1569901.1569924 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |