Switch of channel geometry by 1D component slide responding to slight structural stimuli of adsorbed guest†
Satoshi
Takamizawa
*ab,
Takamasa
Akatsuka
a and
Ryosuke
Miyake
a
aDepartment of Nanosystem Science, Graduate School of Nanobioscience, Yokohama City University, Kanazawa-ku, Yokohama, Kanagawa 236-0027, Japan. E-mail: staka@yokohama-cu.ac.jp; Fax: +81 45-787-2187; Tel: +81 45-787-2187
bPRESTO, Japan Science and Technology Agency (JST), Honcho, Kawaguchi, Saitama, 332-0012, Japan
First published on 8th September 2009
Abstract
The adsorption behavior of a flexible single-crystal host [Cu2(bza)4(pyz)]n was studied for a planar triangular molecule and investigated for large host structural changes including a switch of channel geometry responding to included guest molecules.
Porous materials are one of the most attractive functional materials for gas separation, molecular recognition and catalysis.1,2 In particular, flexible porosities can produce advanced properties, such as switching and/or controlling the properties of porosity by external stimuli, due to their dynamic nature.3,4,5 To develop flexible porosities as sophisticated functional materials, precise control of structural changes is important. Guest inclusions are useful stimuli because of their variety. If a host structure can dramatically change responses based on the kind of guest molecules, a flexible host can control the properties of porosities by selecting guest molecules with diverse structures. Although there are many reports of dynamic adsorption behavior, the study of structural changes by the function of the guest variety was limited to single structural transformation because of the observational limitation of the method of host transformation and includable guest.4 In a molecular crystal of a flexible cyclic complex, a pioneering work of multi-conformational changes responding to guest inclusion was reported by Barbour et al.5
We previously reported a flexible single-crystal host [Cu2(bza)4(pyz)]n (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :bza = benzoate, pyz = pyrazine) in which 1D metal complexes assembled by π–π interaction form a molecular crystal with a channel diameter of 2–4 Å (Chart 1).6 The single-crystal host reversibly adsorbed various gases and organic vapors by weak interaction accompanying crystal phase transition.7,8 However, the difference of the transformation induced by adsorption was unclear. Since 1 can transform to adjust to the guest structure and change selectivity,9,10 there is a possibility of controlling the host structure sensitively triggered by the inclusion of guest molecules.
:bza = benzoate, pyz = pyrazine) in which 1D metal complexes assembled by π–π interaction form a molecular crystal with a channel diameter of 2–4 Å (Chart 1).6 The single-crystal host reversibly adsorbed various gases and organic vapors by weak interaction accompanying crystal phase transition.7,8 However, the difference of the transformation induced by adsorption was unclear. Since 1 can transform to adjust to the guest structure and change selectivity,9,10 there is a possibility of controlling the host structure sensitively triggered by the inclusion of guest molecules.
![One-dimensional assembled structure of [Cu2(bza)4(pyz)]n1.](/image/article/2010/CE/b917263g/b917263g-c1.gif) | ||
| Chart 1 One-dimensional assembled structure of [Cu2(bza)4(pyz)]n1. | ||
In this report, we studied the reversible adsorption behavior and inclusion structure of “a planar triangular molecule” for the single-crystal host 1, which is a simple and symmetric molecule with a projection, and used single-crystal X-ray analysis to investigate large anisotropic changes in the host structure with dramatic switching of channel geometry depending on the guest structure. To control host structural changes by guest inclusion, sufficient vapor pressure and a rigid structure was favored. Thus, acetone was used for this study to influence host structural changes by guest inclusion.
First, the adsorption behavior of the flexible host 1 was measured for acetone vapor at 293 K (Fig. 1). With increasing vapor pressure, the adsorption amount increased to ca. 2.5 molecules per Cu2 unit through an abrupt increase in the adsorption amount at ca. 20 Torr (relative pressure 0.1), indicating host crystal transition11 as was observed in the adsorption of other guests.7,8 The crystal transition was induced at a low adsorption amount (ca. 0.3 mol per Cu2 unit), indicating that the mechanism inducing a cooperative structural change would enhance the transformation of the neighboring structure.9
 | ||
| Fig. 1 Adsorption isotherm curve of acetone at 293 K: adsorption process (closed square) and desorption process (open square). A: amount of adsorption in crystal 1. | ||
For clarification of the acetone-including structure, we first synthesized 1 by using acetone as the solvent instead of methanol to obtain an acetone-including crystal suitable for X-ray analysis6,12 since, after adsorbing acetone vapor, the single crystal 1 was not suitable for X-ray single-crystal analysis.131·2(acetone) was obtained as a blue plate crystal which crystallizes in the monoclinic space group C2/m (Table S1†).14 With the same 1D chain structure as previously reported for the structure of 1, a slipped-stack arrangement included acetone molecules as a crystal solvent (Fig. 2b,d). The calculated pattern of a powder X-ray diffraction measurement from 1·2(acetone) fitted the measured pattern of 1 after the adsorption of acetone.15,16 The XRPD pattern changed reversibly to that of an empty crystal of 117 following the removal of acetone (Fig. S2).† Thus, the host structure of 1 reversibly switched to the structure of an as-synthesized crystal of 1 by acetone inclusion. After adsorption or desorption, a single crystal of 1 changed to differently oriented microcrystals, indicating the large structural changes induced by acetone adsorption.
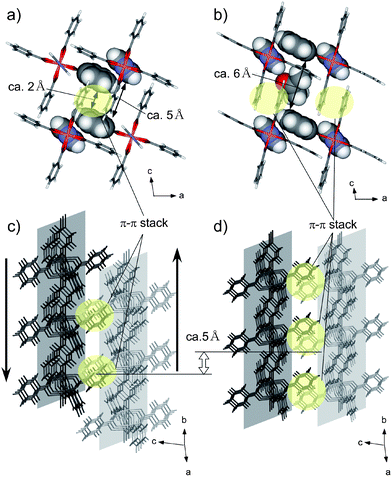 | ||
| Fig. 2 Comparison of the crystal structure of empty crystal 1 (left) and the acetone-including crystal 1·2(acetone) (right) at 90 K: top view (a), (b), side view showing the relative position of neighbor 1D chain in the crystal (c), (d). One possible crystal structure was shown as an acetone-including crystal. Disordered parts of the structure (also see Fig. 3c) and some included acetone molecules are omitted for clarity. The parts of the host structure that form the channel wall are shown as space fill models in contrast to the others, which are shown as stick models (a), (b). The distances shown in (a), (b) were calculated by subtracting the van der Waals radius from the distance between the aromatic rings which were measured by using MPLA restraints in the SHELXL program. | ||
Actually, a large change of crystal volume including switching of channel geometry was revealed by comparison with those of an empty crystal structure of 1 at 90 K.18 The acetone-including crystal increased its crystal volume by 14%, which is a much larger value than that of the other inclusion crystals (4–8%).8,19,20 The expansion of the crystal was observed for the direction for which the 1D chain was assembled by π–π interaction (Fig. 2a,b, also see Fig. S3†). Interestingly, the expansion of the crystal volume was observed for only one axis: the distance between the 1D chains for the c-axis increased by 13% (from 9.5 to 10.8 Å) while that for the a-axis increased very little (2% (from 8.7 to 8.9 Å)). This difference between the two directions was apparently bigger than that observed in other simple guest inclusion structures (Table S3†). As shown in Fig. 2c,d, the neighboring 1D chain slides alternatively ca. 5 Å in the opposite direction during the transformation. This sliding motion, which should be supported by cooperative structural changes with the neighboring structure, helped expand the distance between the neighbor chains along the c-axis, by switching the combination of benzoate, which interacts with neighbor 1D chains by π–π interaction. In contrast to the expansion in the c-axis direction, new channel-like spaces were generated along the b-axis by this slide motion (Fig. 3a,b).
 | ||
| Fig. 3 Comparison of empty crystal 1 (a) and acetone-including crystal 1·2(acetone) (b) at 90 K showing the switch of channel direction (red arrow) with slide motion of the 1D chain and possible crystal structures of the acetone-including crystal of 1 by excluding irrelevant contacts with neighbor host or guest molecules (c). Inset in (c): possible configurations of disordered parts when benzoate is at the center of the figure (set as blue configurations). Disordered parts of the structure in possible positions were drawn in stick style. | ||
In this channel generated along the b-axis, included acetone molecules were arrayed in a linear way (Fig. 3b, also see Fig. S4†). Acetone molecules placed the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups alternatively to the direction perpendicular to the channel and, consequently, their widest part (C2S–C1S–C3S: ca. 6 Å21) was placed in the direction of the expanded c-axis (Fig. 2a,b). The acetone molecules and benzoate ligand are disordered in two groups with identical occupancy (blue and red in Fig. 3c). The possible crystal structures were translated to three structures, shown in Fig. 3c, by excluding irrelevant distances between benzoate and acetone.22 The host structure was disordered to avoid host–guest contact in the acetone-including crystal.
O groups alternatively to the direction perpendicular to the channel and, consequently, their widest part (C2S–C1S–C3S: ca. 6 Å21) was placed in the direction of the expanded c-axis (Fig. 2a,b). The acetone molecules and benzoate ligand are disordered in two groups with identical occupancy (blue and red in Fig. 3c). The possible crystal structures were translated to three structures, shown in Fig. 3c, by excluding irrelevant distances between benzoate and acetone.22 The host structure was disordered to avoid host–guest contact in the acetone-including crystal.
In conclusion, flexible single-crystal host 1 adsorbed planar triangular molecules (acetone vapor) smoothly accompanying the crystal transition similar to the adsorption behavior of inorganic gases and alcohol vapor. Single-crystal X-ray analysis of acetone-including structures revealed that the flexible host 1 transforms by adsorption in a different way compared with other included crystals: 1 expands its structure anisotropically and changes channel geometry dramatically to adjust included acetone molecules. Thus, the observed structural switching demonstrated the responses to the slight differences in the structural information of the included guest.23 Since only a small adsorption amount was needed for phase transition, the structural transformation was supported by cooperative structural changes to the whole crystal through the motion of the 1D chain slide, which would be a useful property for molecular sensing. This clearly shows that large drastic changes can be induced by small stimuli such as slight changes in the guest structure and properties in flexible porosities. In addition, especially for molecular crystals which have a potentially high degree of freedom, flexible hosts have the potential to transform their structures in various ways in response to the selected guest. Since the channel topology and structure should be closely related to the guest diffusion, the adsorption-induced structural switching in this report indicates the possibility for 1 to dynamically control guest selectivity by the guest itself. Thus, guest geometry is one of the elements used to design the properties of the flexible host in addition to modification of the host itself. The properties of controllable structural changes by guest inclusion should contribute to developing a “re-programmable” motional device by dynamic responses to included guest molecules available for active control of selectivity and method of diffusion, which contribute to the development of new methods of gas separation and catalysis.
Notes and references
- S. J. Dalgarno, P. K. Thallapally, L. J. Barbour and J. L. Atwood, Chem. Soc. Rev., 2007, 36, 236 RSC.
- (a) J. Pérez-Ramírez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530 RSC; (b) T. Bein and S. Mintova, Stud. Surf. Sci. Catal., 2005, 157, 263 CAS; (c) S. Mohanty and A. V. McCormick, Chem. Eng. J., 1999, 74, 1 CrossRef CAS.
- For examples, see (a) S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109 RSC; (b) C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828 CrossRef CAS; (c) S. Ma, D. Sun, X. S. Wang and H. C. Zhou, Angew. Chem., Int. Ed., 2007, 46, 2458 CrossRef; (d) A. J. Fletcher, E. J. Cussen, D. Bradshaw, M. J. Rosseinsky and K. M. Thomas, J. Am. Chem. Soc., 2004, 126, 9750 CrossRef CAS; (e) H. Kim, D. G. Samsonenko, M. Yoon, J. W. Yoon, Y. K. Hwang, J. S. Chang and K. Kim, Chem. Commun., 2008, 4697 RSC; (f) L. G. Beauvais, M. P. Shores and J. R. Long, J. Am. Chem. Soc., 2000, 122, 2763 CrossRef CAS.
- (a) J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottel, Science, 2002, 298, 1000 CrossRef CAS; (b) D. Braga, M. Curzi, A. Johansson, M. Polito, K. Rubini and F. Grepioni, Angew. Chem., Int. Ed., 2006, 45, 142 CrossRef CAS; (c) S. Galli, N. Masciocchi, G. Tagliabue, A. Sironi, J. A. R. Navarro, J. M. Salas, L. Mendez-Liñan, M. Domingo, M. Perez-Mendoza and E. Barea, Chem.–Eur. J., 2008, 14, 9890 CrossRef CAS; (d) B. D. Chandler, G. D. Enright, K. A. Udachin, S. Pawsey, J. A. Ripmeester, D. T. Cramb and G. K. H. Shimizu, Nat. Mater., 2008, 7, 229 CrossRef CAS; (e) K. Takaoka, M. Kawano, M. Tominaga and M. Fujita, Angew. Chem., Int. Ed., 2005, 44, 2151 CrossRef CAS.
- L. Dobrzańska, G. O. Lloyd, C. Esterhuysen and L. J. Barbour, Angew. Chem., Int. Ed., 2006, 45, 5856 CrossRef CAS.
- S. Takamizawa, E. Nakata and H. Yokoyama, Inorg. Chem. Commun., 2003, 6, 763 CrossRef CAS.
- (a) S. Takamizawa, E. Nakata and T. Akatsuka, Angew. Chem., Int. Ed., 2006, 45, 2216 CrossRef CAS; (b) S. Takamizawa, E. Nakata, T. Akatsuka, C. Kachi-Terajima and R. Miyake, J. Am. Chem. Soc., 2008, 130, 17882 CrossRef CAS; (c) S. Takamizawa, E. Nakata and R. Miyake, Dalton Trans., 2009, 1752 RSC.
- (a) S. Takamizawa, T. Saito, T. Akatsuka and E. Nakata, Inorg. Chem., 2005, 44, 1421 CrossRef CAS; (b) S. Takamizawa, C. Kachi-Terajima, M. Kohbara, T. Akatsuka and T. Jin, Chem.–Asian J., 2007, 2, 837 CrossRef CAS.
- S. Takamizawa, M. Kohbara and R. Miyake, Chem.–Asian J., 2009, 4, 530 CrossRef CAS.
- S. Takamizawa and R. Miyake, Chem. Commun., 2009, 4076 RSC.
- S. Takamizawa, E. Nakata, H. Yokoyama, K. Mochizuki and W. Mori, Angew. Chem., Int. Ed., 2003, 42, 4331 CrossRef CAS.
- Synthesis of [Cu2(bza)4(pyz)]n·2 acetone (1·2(acetone)): To the suspension of acetate copper(II) monohydrate 100 mg (0.50 mmol) in acetone 100 ml, benzoic acid 2.44 g (20 mmol, 20 eq) was added. After the solution was stirred at room temperature until all acetate copper dissolved in the acetone, pyrazine was added by vapor diffusion. The resulting blue plate crystal was filtered and dried in vacuo to obtain 160 mg (yield: 96%). The sample without the drying process was used for single crystal X-ray analysis.
- Single-crystal X-ray diffraction data was measured at 90 K on a Bruker SMART APEX CCD area detector (graphite-monochromated Mo Kα radiation (λ = 0.71073 Å)) with a nitrogen flow temperature controller. A well-formed single crystal 1 was sealed inside a glass capillary with its saturated solution. Empirical absorption corrections were applied using SADABS. The structure was solved by direct methods (SHELXS-97) and refined by full-matrix least-squares calculations on F2 (SHELXL-97). Restraints were used for all atoms (SIMU and ISOR for all atoms and SADI and FLAT for benzene ring). The non-hydrogen atoms were refined anisotropically; the hydrogen atoms were fixed at calculated positions.
- Crystal data for 1·2(acetone): C38H36Cu2N2O10, MW = 807.79 g mol−1, monoclinic, space group C2/m, T = 90 K, a = 17.757(3), b = 9.6260(16), c = 10.8050(18) Å, β = 95.603(4)°, V = 1838.1(5) Å3, Z = 2, ρcalcd = 1.460 Mg m−3, R1 = 0.0564 (0.0834), wR2 = 0.1282 (0.1402) for 1738 reflections with I > 2σ(I) (for 2430 reflections (Rint = 0.0421, 6902 total measured)), goodness-of-fit on |F|2 1.0894, largest diff. peak/hole 0.800/−0.477 e Å3, CCDC 736068.
- Powder X-ray diffraction was measured on a diffractometer (RINT-2000, Rigaku) at 298 K. The crystalline powder was mounted in sealed poly(ethylene) film to maintain the corresponding vapor atmosphere during measurement, following the adsorption step (3 h – 1 day).
- The calculated powder X-ray diffraction pattern of the empty crystal and acetone-including crystal 1 was calculated from the data of single-crystal X-ray structural analyses measured at 298 K. In particular, for the acetone-including crystal 1 data was used at 298 K, which was determined only the cell parameter. The crystal structure cannot be determined because of the high measurement temperature.
- Measured XRPD pattern of empty crystal of 1 shows a similar sharp pattern of the calculated pattern from the crystal structure of the empty crystal 1 (also see Fig. S2†).
- Crystal data for 1: C32H24Cu2N2O8, MW = 691.61 g mol−1, monoclinic, space group C2/c, T = 90 K, a = 17.485(3), b = 9.6876(19), c = 19.213(4) Å, β = 98.432(4)°, V = 3219.2(11) Å3, Z = 4, ρcalcd = 1.427 Mg m−3, R1 = 0.0589 (0.0827), wR2 = 0.1342 (0.1462) for 2987 reflections with I > 2σ(I) (for 3999 reflections (Rint = 0.0560, 11425 total measured)), goodness-of-fit on | F|2 1.034, largest diff. peak/hole 2.035/−1.702 e Å3, CCDC745355.
- S. Takamizawa, E. Nakata and T. Saito, Inorg. Chem. Commun., 2004, 7, 1 CrossRef CAS.
- S. Takamizawa, E. Nakata and T. Saito, CrystEngComm, 2004, 6, 197 RSC.
- The widest part of acetone molecules was calculated by adding the van der Waals radius of H atom (1.2 Å) to the distance between H(3S1) and H(2S1) (ca.4 Å).
- As shown in the inset of Fig. 3c, the distances between benzoate and acetone in different disorder groups were irrelevant (O1S-C11*: 2.50Å). In addition, considering the irrelevant combination of the neighboring benzoate (C11-C11*: 2.80 Å, *symmetry operation: 1 − x, y, 1 − z), the possible combination of the disorder groups should all be in the same group (all blue or all red) in a 1D chain.
- S. Takamizawa, K. Kojima and T. Akatsuka, Inorg. Chem., 2006, 45, 4580 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional structural information (data of X-ray powder diffraction and detailed data of single crystal structural analyses). CCDC reference numbers 736067, 736068 and 745355. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b917263g |
| This journal is © The Royal Society of Chemistry 2010 |