Recycle of tin thiolate compounds relevant to ammonia–borane regeneration†
Andrew D.
Sutton
*a,
Benjamin L.
Davis
a,
Koyel X.
Bhattacharyya
a,
Bobby D.
Ellis
b,
John C.
Gordon
*a and
Philip P.
Power
*b
aLos Alamos National Laboratory, Los Alamos, NM 87545, USA. E-mail: adsutton@lanl.gov; Fax: (+1) 505 667 9905; Tel: (+1) 505 665 2931
bUniversity of California, Davis, One Shield Ave, Davis, CA 96363, USA. E-mail: pppower@ucdavis.edu; Fax: (+1) 530 752 8995; Tel: (+1) 530 752 6913
First published on 13th November 2009
Abstract
The use of benzenedithiol as a digestant for ammonia–borane spent fuel has been shown to result in tin thiolate compounds which we demonstrate can be recycled, yielding Bu3SnH and ortho-benzenedithiol for reintroduction to the ammonia–borane regeneration scheme.
A necessary target in realizing a hydrogen (H2) based economy, especially for the transportation sector, is its storage for controlled delivery, presumably to an energy producing fuel cell.1 Chemical hydrogen storage has been dominated by one appealing material, ammonia–borane (H3N–BH3, AB), due to its high gravimetric capacity of hydrogen (19.6 wt%) and low molecular weight (30.7 g mol−1). In addition, AB has both hydridic and protic moieties, yielding a material from which H2 can be readily released in contrast to the loss of H2 from C2H6, which is substantially endothermic.2 A number of publications have described H2 release from amine boranes, yielding various rates depending on the method applied.3–6 The viability of any chemical hydrogen storage system is critically dependent on efficient recyclability, but reports on the latter subject are sparse, invoke the use of high energy reducing agents, and suffer from low yields.1,7–10
We recently described an energy efficient regeneration process for polyborazylene (PB) spent fuel.11 In this scheme, PB is digested with ortho-benzenedithiol to yield [NH4][B(C6H4S2)2] (1) and (C6H4S2)BH·NH3 (2). 1 can then be converted into 2 using Bu3SnH as a reductant with C6H4SH(S–SnBu3) (3) as a by-product, thus resulting in a single boron containing compound that can subsequently be converted to AB using a slight excess of Bu2SnH2.11 Modification of this process due to the relative instability of Bu2SnH2 enables us to carry out the reduction chemistry with Bu3SnH via an amine exchange step with Me3N. In such a scheme, the use of Bu3SnH as the reductant produces C6H4(S–SnBu3)2 (4) as a by-product. In order to maximize the overall efficiency of an AB regeneration process which utilizes the Sn–H moiety in a reduction step, recycle of the tin-containing by-products is essential, and herein we report a recycle scheme for the regeneration of both Bu3SnH and the digesting reagent ortho-benzenedithiol from 3 and 4 (Fig. 1).
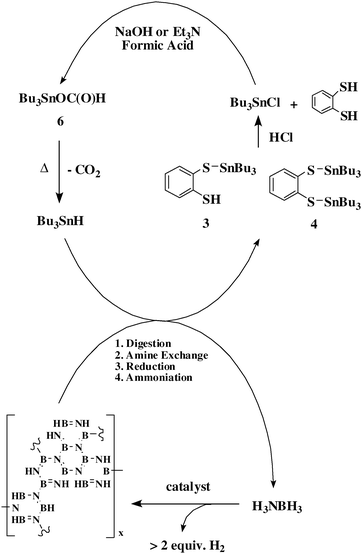 | ||
| Fig. 1 Regeneration of AB with tin recycle and ortho-benzenedithiol recovery. | ||
Our focus in this regard was centered on the conversion of 3 and 4, i.e., species containing Sn–SR bonds, to a species containing a formate moiety, (Bu3Sn–OC(O)H), as this compound has been described as a precursor to Bu3SnH via thermal decarboxylation.12 Our initial investigations focused on a relatively simple system, i.e., Bu3SnSPh (5), which can be formed by refluxing (Bu3Sn)2O and PhSH in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio in toluene with a Dean–Stark apparatus (to collect the water generated). After refluxing for 12 h the toluene was removed in vacuo and the product distilled under reduced pressure to yield 5 in near quantitative yield. Direct reaction of 5 with formic acid does not result in a clean reaction, and although some evidence does exist for the production of Bu3Sn–OC(O)H, an equilibrium appears to exist which predominantly favors the presence of 5 at ambient temperature. No enhancement of Bu3Sn–OC(O)H is observed by NMR spectroscopy at increased temperatures although a slight increase of 5 is observed with increasing concentrations of formic acid. In support of this fact is the observation that independently prepared Bu3SnOC(O)H and PhSH react instantly to form 5 at room temperature, which indicates that the equilibrium lies heavily in favor of the Sn–SPh species.
:2 molar ratio in toluene with a Dean–Stark apparatus (to collect the water generated). After refluxing for 12 h the toluene was removed in vacuo and the product distilled under reduced pressure to yield 5 in near quantitative yield. Direct reaction of 5 with formic acid does not result in a clean reaction, and although some evidence does exist for the production of Bu3Sn–OC(O)H, an equilibrium appears to exist which predominantly favors the presence of 5 at ambient temperature. No enhancement of Bu3Sn–OC(O)H is observed by NMR spectroscopy at increased temperatures although a slight increase of 5 is observed with increasing concentrations of formic acid. In support of this fact is the observation that independently prepared Bu3SnOC(O)H and PhSH react instantly to form 5 at room temperature, which indicates that the equilibrium lies heavily in favor of the Sn–SPh species.
However, when Bu3SnSPh was dissolved in an ethereal solution of HCl (1.0 M, 0.5 mL), this resulted in clean conversion to Bu3SnCl and PhSH. Upon removal of solvent and subsequent redissolution in deutero solvent, this gave an approximately 1:1 ratio of these two species according to 1H NMR spectroscopy with no evidence for any residual 5, indicating near quantitative conversion to the desired products.
After satisfactorily studying a model system, we then pursued the same conversion with the by-products of AB regeneration. Initial studies of C6H4SH(S–SnBu3)11 (3) in HCl–Et2O in the above manner indeed yielded Bu3SnCl and C6H6S2 in a 1:1 ratio, as expected. On scale-up 3 can be converted to Bu3SnCl (83% isolated yield) following washing and drying the resultant solid several times with Et2O. 3 was not observed in either the 1H or 11B NMR on redissolution of the solid. Compound 4 can be readily synthesised via a salt metathesis route in THF from [C6H4S2][Na]2 and two equivalents of Bu3SnCl in high yield (93%). Similarly to the model compound 3, 4 can also be converted to Bu3SnCl and ortho-benzenedithiol as expected with Bu3SnCl being isolated (87% yield), again with no indication of residual 4 present. This shows that both the Sn–SR by-products of digestion, 3 and 4, can be readily converted to Bu3SnCl, which allows the following stages to be applied to either digestion scheme. The ortho-benzenedithiol can also be recovered by vacuum distillation at this stage under mild conditions (b.p. 119–120 °C, 17 torr). Attempts to achieve accurate isolated yields of ortho-benzenedithiol on removal of the solvent were hampered due to its volatility under dynamic vacuum and slight loss of the isolated compound following the rigorous washing and drying of the resultant Bu3SnCl.
After the ortho-benzenedithiol has been removed, the resulting Bu3SnCl that is subsequently formed can then be converted to Bu3SnOC(O)H (6) via the reaction of Bu3SnCl with formic acid in the presence of either NaOH or Et3N which presumably proceeds via the formation of (Bu3Sn)2O. 6 is quantitatively formed as shown by 119Sn NMR spectroscopy (Fig. 2) at room temperature. 6 has also been synthesised independently by refluxing a toluene solution of (Bu3Sn)2O with formic acid, followed by removal of solvent and distillation of the product giving 78% yield of 6. The distillation of 6 through a column of 3 mm Raschig rings under reduced pressure (112 °C, 0.3 torr) afforded 60% isolated yield of Bu3SnH, which can re-enter the AB regeneration process.
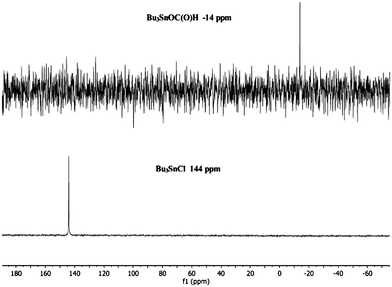 | ||
| Fig. 2 119Sn {1H} NMR spectra of Bu3SnCl (bottom) conversion to Bu3SnOC(O)H (top) following reaction with base and formic acid. | ||
In conclusion, we have demonstrated that Bu3SnH and ortho-benzenedithiol components can be recycled following their use in a scheme to recycle the tin reductant for AB regeneration which is a necessary component for a viable AB regeration scheme.
This work was funded by the US Department of Energy, Office of Energy Efficiency and Renewable Energy.
Notes and references
- F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 2613 RSC.
- M. H. Matus, K. D. Anderson, D. M. Camaioni, S. T. Autrey and D. A. Dixon, J. Phys. Chem. A, 2007, 111, 4411 CrossRef CAS.
- M. E. Bluhm, M. G. Bradley, R. Butterick III, U. Kusari and L. G. Sneddon, J. Am. Chem. Soc., 2006, 128, 7748 CrossRef CAS.
- M. C. Denney, V. Pons, T. J. Hebden, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 12048 CrossRef CAS.
- R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844 CrossRef CAS.
- F. H. Stephens, R. T. Baker, M. H. Matus, D. J. Grant and D. A. Dixon, Angew. Chem., 2007, 119, 760 CrossRef; F. H. Stephens, R. T. Baker, M. H. Matus, D. J. Grant and D. A. Dixon, Angew. Chem., Int. Ed., 2007, 46, 746 CrossRef CAS.
- L. G. Sneddon, Amineborane Hydrogen Storage in Department of Energy Hydrogen Program Review. Can be found at http://www.hydrogen.energy.gov/pdfs/review06/st_3_sneddon.pdf 2006 Search PubMed.
- L. G. Sneddon, Amineborane-Based Chemical Hydrogen Storage in Department of Energy Hydrogen Program Review. Can be found at http:/www.hydrogen.energy.gov/pdfs/review07/st_27_sneddon.pdf 2007 Search PubMed.
- S. Hausdorf, F. Baitalow, G. Wolf and F. O. R. L. Mertens, Int. J. Hydrogen Energy, 2008, 33, 608 CrossRef CAS.
- P. V. Ramachandran and P. D. Gagare, Inorg. Chem., 2007, 46, 7810 CrossRef CAS.
- B. L. Davis, D. A. Dixon, E. B. Garner, J. C. Gordon, M. H. Matus and F. H. Stephens, Angew. Chem., Int. Ed., 2009, 48, 6812 CrossRef CAS.
- R. J. Klingler, I. Bloom and J. W. Rathke, Organometallics, 1985, 4, 1893 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental data. See DOI: 10.1039/b919383a |
| This journal is © The Royal Society of Chemistry 2010 |