Enhanced acyl radical formation in the Au nanoparticle-catalysed aldehydeoxidation†
Marco
Conte
a,
Hiroyuki
Miyamura
b,
Shū
Kobayashi
b and
Victor
Chechik
*a
aDepartment of Chemistry, University of York, Heslington, York, UK YO10 5DD. E-mail: vc4@york.ac.uk; Tel: +44 1904 434185
bGraduate School of Pharmaceutical Sciences, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo, 113-0033, Japan
First published on 19th October 2009
Abstract
EPR spectroscopy and spin-trapping experiments showed that polymer-encapsulated Au nanoparticles promote aldehydeoxidationvia a radical pathway by initiating formation of acyl radicals .
Aldehyde oxidation is an important reaction for the manufacture of organic acids, peracids and anhydrides, which find applications as precursors for resins and pharmaceutical products.1Aldehydes can autoxidise in air via a radical pathway;2 this reaction is facilitated by Mn3+, Cu2+ or Co2+ salts, which promote the initiation step.3Gold nanoparticles can also efficiently catalyse aldehydeoxidation,4 providing the advantage of a heterogeneous system. However, many mechanistic aspects of the catalytic cycle and possible pathways, particularly in basic media, are not well understood. This prompted us to explore aldehydeoxidation over polymer-incarcerated Au nanoparticles (PI-Au)5 in the absence and presence of base using EPR spin trapping methodology.6
Spin-trapping methodology relies on the fast selective addition (trapping) of short-lived radicals to a diamagnetic spin trap, usually a nitrone or a nitroso compound, such as 5,5-dimethyl-1-pyrroline-N-oxide (DMPO). The product of this addition (spin adduct ) is a persistent free radical (nitroxide) with sufficiently long lifetime to enable detection by conventional EPR spectroscopy. Magnetic properties of the spin adducts often make it possible to assign the structure of the original short-lived radicals. Structures of spin traps used in this study are shown in Scheme 1.
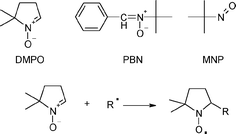 | ||
| Scheme 1 Chemical structure of DMPO, PBN and MNP spin traps (top) and trapping of a free radical (bottom). | ||
PI-Au is a catalyst containing gold nanoparticles trapped in a polymer matrix.5 The catalyst is prepared by reduction of Au(I) with sodium borohydride in the presence of polymer solution (full preparation details are in the ESI† ). This material is an efficient alcohol oxidationcatalyst.7 Reaction of benzaldehyde with air in toluene at 50 °C in the presence of a DMPO or MNP spin trap, gave rise to a persistent free radical which was identified by X-band EPR as an acyl adduct by comparison with the literature data, e.g., DMPO–COR adduct (aN = 14.03 G, aH = 15.73 G)8 or MNP–COR adduct (aN = 8.1 G).9EPR spectra recorded with an MNP spin trap additionally contain a triplet with coupling constant aN = 15.4 G, which is characteristic of the di-tert-butyl aminoxyl radical (DTBA), an impurity present in MNP samples (see ESI† ).10 However, as the amount of DTBA is constant in all samples, it can be used to estimate the relative amount of MNP–COR adduct formed under different conditions. When PBN was used as a spin trap, a peroxyl PBN–OOCR3adduct was observed (aN = 13.4, aH = 1.72 G)11 in aerobic conditions, while a PBN–COR adduct (aN = 14.3, aH = 4.57 G)11 was observed in partially deoxygenated mixtures (details are in the ESI† ).
Observation of acyl and peroxyl adducts is consistent with the free radical mechanism of aldehyde autoxidation reported in the literature.2 This mechanism includes: (i) an initiation step which leads to acyl radical formation, (ii) addition of oxygen to acyl radicals to form peroxyl radicals, (iii) abstraction of a hydrogen atom from the aldehyde by the peroxyl radical (giving a peracid) during the propagation step, and (iv) carboxylic acid formation from the aldehyde and peracidvia Baeyer–Villiger reaction.2 Consequently, using different spin traps with different affinity for different radicals, we could monitor the overall reaction.
When the catalyst PI-Au was introduced into the system, two effects were observed: a detectable increase in the acyl adduct intensity (by 32% relative to DTBA), and increased sharpening of the spectral lines, which is diagnostic of oxygen consumption (Fig. 1a). This is consistent with the catalytic action of the PI-Au nanocomposite. Control tests confirmed that the catalyst cannot oxidise solvent (toluene) and hence the observed radicals originate from the substrate.
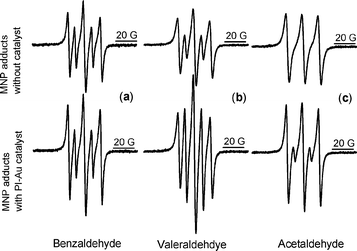 | ||
| Fig. 1 Spin adducts formed from MNP in an oxidation reaction in the absence (top) and presence (bottom) of PI-Au with: (a) benzaldehyde, (b) valeraldehyde and (c) acetaldehyde. The three lines with bigger coupling constant in all spectra are due to di-tert-butyl aminoxyl radicals (DTBA). | ||
We subsequently tested the oxidation of aliphatic aldehydes, e.g., valeraldehyde and acetaldehyde, both in the presence and absence of PI-Au. Enhanced acyl radical generation was detected for valeraldehyde within the first 5 min of the reaction (spin adduct intensity more than doubled, Fig. 1b). For acetaldehyde, acyl radicals could be detected only in the presence of PI-Au catalyst (Fig. 1c).
In order to confirm that the intensity of the radical spin adducts is related to the catalytic efficiency, bulk tests were carried out. A detectable increase in the yield of carboxylic acids was observed for PI-Au catalysed benzaldehyde and valeraldehydeoxidations, consistent with the EPR results (Table 1, entries 1–4; full experimental details are in the ESI† ).
| Entry | Substratea | Catalyst b | Reaction mediumc | Acid (%)d |
|---|---|---|---|---|
| a Reaction conditions: substrate (0.5 ml), solvent (0.5 ml), T = 50 °C, P = 1 atm, reaction time 5 h. b Catalyst (5 mg). c TEMPO (100 ppm) in toluene. d Determined by 1H-NMR spectroscopy. | ||||
| 1 | Benzaldehyde | — | Toluene | 29 |
| 2 | Benzaldehyde | PI-Au | Toluene | 50 |
| 3 | Valeraldehyde | — | Toluene | 46 |
| 4 | Valeraldehyde | PI-Au | Toluene | 54 |
| 5 | Benzaldehyde | — | Toluene/TEMPO | 1.2 |
| 6 | Benzaldehyde | PI-Au | Toluene/TEMPO | 1.5 |
| 7 | Valeraldehyde | — | Toluene/TEMPO | 3.6 |
| 8 | Valeraldehyde | PI-Au | Toluene/TEMPO | 4.4 |
These data suggest that the PI-Au catalyst can promote the initiation step of the aldehydeoxidation by increasing the rate of acyl radical formation. In order to confirm that the catalysed oxidation indeed occurs via a free radical mechanism, we carried out oxidations in the presence of 2,2′,6,6′-tetramethylpiperidine N-oxyl (TEMPO). While relatively unreactive towards oxygen centred radicals, TEMPO is a highly efficient quencher for carbon centred radicals.12TEMPO efficiently suppressed oxidation both in the presence and absence of catalyst. For instance, for both benzaldehyde and valeraldehyde, conversion values in the presence of TEMPO dropped from ca. 50% to ca. 3%. (Table 1, entries 5–8). Control tests using catalyst pre-treated with TEMPO, showed a very similar activity to the untreated catalyst (e.g., conversion of benzaldehyde in a 24 h reaction with treated and untreated catalysts was ca. 56 and 59%, respectively). Thus, the suppression of oxidation by TEMPO is not due to the catalyst deactivation by the nitroxide. We conclude therefore that the catalytic oxidation occurs via a radical mechanism.
The radical chain mechanism explains the rather moderate increase in conversion in catalytic vs. non-catalytic reactions (Table 1, entries 1–4). The turnover in the chain reactions may be determined by the rate of termination rather than that of initiation. For instance, simply replacing glass reaction vials with polyethylene vials led to significantly increased conversions (67% when PI-Au was used, see ESI† ). This suggests that termination can be controlled by the walls of the reaction vessel.13 A related phenomenon was observed when ZnO-supported gold catalyst (Au/ZnO)14 was used. The oxidation in the presence of this catalyst was partially suppressed. The conversion under conditions reported in Table 1, was ca. 15%, which is much lower than that in a catalyst-free reaction. We believe this is due to the termination of radicals by the catalyst. This effect was confirmed by the very low conversion observed when Au-free ZnO was added to the uncatalysed reaction (7%, see ESI† ). ZnO is a material rich in neutral oxygen vacancies15 that could possibly quench peroxyl radicals. In fact, spin trapping experiments in the presence of ZnO did not reveal any significant amount of peroxyl adduct .
In order to further test the catalytic efficiency of PI-Au in aldehydeoxidation, we carried out reactions in sealed vials in the presence of TEMPO. The sensitivity of TEMPOEPR signals to the oxygen concentration16 makes it possible to assess the conversion from the EPR data as TEMPO line broadening is linearly proportional to the oxygen concentration. The presence of excess TEMPO efficiently breaks the radical chain process, so that the rate of deoxygenation is determined by the initiation reaction. The rate of deoxygenation in the presence of catalyst was ca. 2-fold higher than that in the absence of catalyst, thus providing strong evidence that the catalyst enhances the rate of initiation in aldehydeoxidations (see ESI for further details† ).
The overall reaction mechanism can thus be proposed as follows (Scheme 2). It is likely that the reaction occurs via H abstraction from the C–H bond by O2 activated over the catalyst surface, probably via AuOO˙ species. Activation of molecular oxygen by Au catalysts has been proposed in other similar reactions.6,17
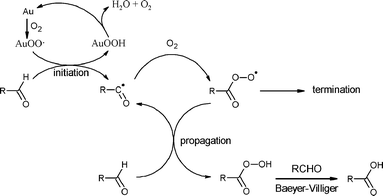
The activation of the C–H bond would also be consistent with a recent kinetic study of aldehydeoxidation over Au/TiO2,19via activated oxygen over the catalyst surface, which illustrates that activation of the C–H bond takes place in the rate-determining step.
Interestingly, different results were obtained when the oxidation was carried out in the presence of base. Base is often added to Au-catalysed oxidation reactions.20Oxidation of benzaldehyde, valeraldehyde and acetaldehyde in the presence of NaOH, PI-Au and DMPO gave a weak DMPO–H adduct (aN = 14.49, aH(1) = aH(2) = 18.69 G).21 In the absence of catalyst, only a DMPO–CHR2adduct was identified (aN = 14.55, aH = 21.07 G),8 (Fig. 2 and ESI† ). To explore the origin of the DMPO–H adduct , perdeuterated acetaldehyde was oxidised in the presence of base and PI-Au catalyst. A DMPO–D adduct (aN = 14.54, aH = 18.79, aD = 2.84 G)22 was observed, thus suggesting that the hydrogen adduct is formed as a consequence of C–H bond cleavage (see ESI† ).
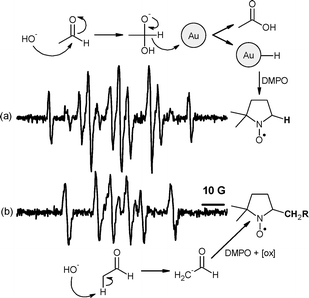 | ||
| Fig. 2 DMPO spin adducts formed in a reaction of acetaldehyde in basic media. (a) DMPO–H formation in the presence of PI-Au and (b) DMPO–CH2R adduct formation in the absence of catalyst (R = COH). | ||
Observation of a DMPO–H adduct in a related Au-catalysed alcohol oxidation reaction was recently attributed to the intermediate formation of Au–H as a consequence of hydride transfer from the C–H bond to gold.23 We propose therefore that the detection of DMPO–H during aldehydeoxidation in basic medium is due to the intermediate formation of geminal diol (Fig. 2a) which undergoes hydride transfer to the Au surface. Detection of a DMPO–CHR2adduct in the uncatalysed reaction is probably due to the nucleophilic addition of the enolate to the spin trap (Fig. 2b) followed by oxidation to a nitroxide24 (not detected for benzaldehyde which does not form the enolate).
In order to probe whether hydride transfer from the geminal diol is the predominant oxidation pathway in basic medium, we added radical scavengerTEMPO to the reaction mixture. In the presence of base, typical conversions for benzaldehydeoxidation were 29% and 21% in the presence and absence of catalyst, respectively, with ca. 90% selectivity for benzoic acid (see Table S1 in the ESI† ). When TEMPO was added, the conversion dropped in both cases to ca. 6% with selectivity >70% (the by-product was benzyl alcohol which was formed viaCannizzaro reaction). The significant suppression of reaction by TEMPO, both in the presence and absence of catalyst, strongly suggests that the radical pathway is dominant for both reactions in the presence of base.
In conclusion, gold catalyses aldehydeoxidation by accelerating the initiation step in the radical pathway leading to enhanced formation of acyl radicals . In the presence of base, hydride transfer from the geminal diol is possible, however, this is likely to be a minor secondary pathway as the radical route appears to be dominant.
Funding for this work was provided by the EPSRC (grant EP/E001629/1). The authors thank Dr Moray Stark (University of York) for helpful discussions.
Notes and references
- R. A. Sheldon and H. van Bekkum, Fine Chemicals through Heterogeneous Catalysts, John Wiley & Sons Inc., New York, 2001 Search PubMed.
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed.
- C. H. Bamford, R. G. Compton and C. F. H. Tipper, Liquid Phase Oxidation, Elsevier, Oxford, 1980 Search PubMed.
- (a) C. Marsden, E. Taarning, D. Hansen, L. Johansen, S. K. Klitgaard, K. Egeblad and C. H. Christensen, Green Chem., 2008, 10, 168 RSC; (b) S. Biella, L. Prati and M. Rossi, J. Mol. Catal. A: Chem., 2003, 197, 207 CrossRef CAS.
- (a) H. Miyamura, R. Matsubara, Y. Miyazaki and S. Kobayashi, Angew. Chem., Int. Ed., 2007, 46, 4151 CrossRef CAS; (b) H. Miyamura, R. Matsubara and S. Kobayashi, Chem. Commun., 2008, 2031 RSC.
- (a) P. Ionita, B. C. Gilbert and V. Chechik, Angew. Chem., 2005, 117, 3786 CrossRef; (b) P. Ionita, M. Conte, B. C. Gilbert and V. Chechik, Org. Biomol. Chem., 2007, 5, 3504 RSC; (c) M. Conte, K. Wilson and V. Chechik, Org. Biomol. Chem., 2009, 7, 1361 RSC.
- C. Lucchesi, T. Inasaki, H. Miyamura, R. Matsubara and S. Kobayashi, Adv. Synth. Catal., 2008, 350, 1996 CrossRef CAS.
- E. G. Janzen and J.-P. Liu, J. Magn. Reson., 1973, 9, 510 CAS.
- M. Novak and B. A. Brodeur, J. Org. Chem., 1984, 49, 1142 CrossRef CAS.
- L.-B. Luo, D.-Y. Han, Y. Wu, X.-Y. Song and H.-L. Chen, J. Chem. Soc., Perkin Trans. 2, 1998, 1709 RSC.
- E. G. Janzen, E. R. Davis and C. M. DuBoset, Magn. Reson. Chem., 1995, 33, S166 CAS.
- R. A. Moss, M. S. Platz and M. Jones, Reactive Intermediate Chemistry, John Wiley & Sons Inc., New York, 2004 Search PubMed.
- B. J. Hwang, Ind. Eng. Chem. Res., 1994, 33, 1897 CrossRef CAS.
- The Au/ZnO catalyst was prepared as in: M. Conte, T. Davies, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2007, 252, 23 Search PubMed and references therein.
- F. Leiter, H. Alves, D. Pfisterer, N. G. Romanov, D. M. Hofmann and B. K. Meyer, Phys. B, 2003, 340–342, 201 CrossRef CAS.
- (a) M. Conte, Y. Ma, C. Loyns, P. Price, D. Rippon and V. Chechik, Org. Biomol. Chem., 2009, 7, 2685 RSC; (b) A. I. Smirnov, R. B. Clarkson and R. L. Belford, J. Magn. Reson., Ser. B, 1996, 111, 149 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS.
- Decomposition of AuOOH species, leading to water, can possibly occur viahydrogen peroxide through oxygen reduction reaction, see: (a) B. Besson and P. Gallezot, Catal. Today, 2000, 57, 127 CrossRef; (b) J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 315, 220 CrossRef CAS.
- P. Fristrup, L. B. Johansen and C. H. Christensen, Chem. Commun., 2008, 2750 RSC.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS.
- P. Maillard, J. C. Massot and C. Giannotti, J. Organomet. Chem., 1978, 159, 219 CrossRef CAS.
- P. Maillard and C. Giannotti, Can. J. Chem., 1982, 60, 1402 CAS.
- M. Conte, H. Miyamura, S. Kobayashi and V. Chechik, J. Am. Chem. Soc., 2009, 131, 7189 CrossRef CAS.
- P. Ionita, B. C. Gilbert and A. C. Whitwood, J. Chem. Soc., Perkin Trans. 2, 2000, 2436 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, details of polymer incarcerated nanoparticle synthesis and characterization, 1H-NMR data, oxygen consumption trend and EPR spectra. See DOI: 10.1039/b918200d |
| This journal is © The Royal Society of Chemistry 2010 |