Energy transfer with gold nanoparticles for analytical applications in the fields of biochemical and pharmaceutical sciences
Jian
Ling
ab and
Cheng Zhi
Huang
*b
aKey Laboratory of Medicinal Chemistry for Natural Resource, Ministry of Education, School of Chemical Science and Engineering, Yunnan University, Kunming, 650091, P. R. China
bEducation Ministry Key Laboratory on Luminescence and Real-Time Analysis, College of Pharmaceutical Sciences, Southwest University, Chongqing, 400715, P. R. China. E-mail: chengzhi@swu.edu.cn.; Fax: +86-23-68254659; Tel: +86-23-68254659
First published on 17th September 2010
Abstract
Gold nanoparticles (AuNPs) are the most interesting nanomaterials for analytical purposes owing to their unique optical and electrochemical properties resulting from the localized surface plasmon resonance (LSPR) of the electrons, displaying strong absorption and scattering of light from visible to near-infrared region by tuning the particles sizes and shapes. In fluorescence resonance energy transfer (FRET) process, AuNPs have been identified to act as excellent acceptors to replace traditional organic quenchers. Starting from quenching advantages of AuNPs in FRET system and energy transfer mechanism of donor to AuNPs, herein we try to summarize and discuss typical AuNPs based FRET methods in terms of DNA hybridizations, immunoreactions, specific molecular bindings or adsorptions, and provide their analytical applications in biochemical and pharmaceutical analysis.
 Jian Ling | Dr Jian Ling is an Assistant Professor of Analytical Chemistry at Yunnan University, PR China. He received his PhD degree in analytical chemistry from the College of Chemistry and Chemical Engineering, Southwest University, in June 2009. His research interests are the development of analytical methods for biomolecules, metal ions based on localized surface plasmon resonance of metal nanoparticles. |
 Cheng Zhi Huang | Dr Cheng Zhi Huang is a Professor of Analytical Chemistry in Southwest University, PR China. He received his PhD from Peking University in 1996, and was a postdoctoral scholar in the Central Research Laboratory of Hitachi Limited, Tokyo, in 1998-1999, the Loeb Laboratory of Ottawa University in 2000-2001, and the Pharmaceutical Department of Tokyo University in 2001-2003. Previously his research interests involved the analytical applications of light scattering, fluorescence and absorption spectroscopy of small organic molecules, biomolecules and nanoparticles. Currently, his research focuses on localized surface plasmon resonance spectroscopy, particularly long range resonance energy transfer (LrRET) involving the preparation and assembly of metal nanoparticles. |
1. Introduction
Gold nanoparticles (AuNPs) have found wide applications in the fields of biochemical sensing, medicinal diagnosis and cellar imaging.1–6 AuNPs can be seen in atom clusters, dots and particles with the sizes ranging from several atom to hundred nanometres.1 Due to the quantum size effect, they exhibit various optical properties which strongly depend on the particle size, shape, interparticle distance and the nature of particle surroundings. For example, gold clusters of several atoms have strong fluorescence.7,8 Much like semiconductor quantum dots, gold clusters have size-tunable emission maxima, which shifts to longer wavelengths with increasing nanocluster size. However, 5∼100 nm AuNPs have unique localized surface plasmon resonance (LSPR)9–12 properties, which show strong absorption and scattering of light from visible to near-infrared region by tuning the particles sizes and shapes.Fluorescence resonant energy transfer (FRET) is a powerful and sensitive spectroscopic technique to study nanoscale interactions, conformational and distance changes between molecules and nanosystems. Traditional FRET pairs (e.g., organic dyes, fluorescent proteins and fluorescent polymers) suffer from the inherent limitations of low quantum yields, narrow excitations and broad emissions bound, chemical and photo-degradation, reducing the efficiency of FRET process. The development of modern nanotechnology produces nanomaterials like quantum dots (QDs) and AuNPs, which greatly promote the development of FRET technology theoretically and practicably.13–17
AuNPs can be used as excellent quenchers in a FRET system as substitutes for traditional organic quenchers. The quenching abilities of metal particles have been well studied theoretically,18–20 however, the practical performances of AuNPs in FRET system have only been studied for several years,21 making AuNPs as acceptors to quench the fluorescence of dyes,14 quantum dots,22 fluorescent particles23 and polymers.24 Recent investigations have demonstrated that AuNPs have following advantages in FRET systems acting as acceptors/quenchers: (1) High fluorescence quenching efficiency. AuNPs of 1.4-nm diameter can quench fluorescence as much as 100 times higher than traditional organic quenchers and have higher quenching efficiency up to 100% for dyes emitting from visible to near-infrared (VIS-NIR) region. AuNPs, with a size larger than 5 nm, for instance, have strong surface plasmon resonance optical properties with the molar extinction coefficients up to 1010 cm−1 M−1 at the plasmon resonance wavelength maximum, which assist the mechnism of nonradiative energy transfer to metal surface. (2) Tunable quenching property. Large AuNPs have unique surface plasmon resonance absorption that can be tuned precisely and easily by changing the shape, size and composition. Thus, the quenching of fluorescence by AuNPs can be optimized to a specific wavelength near the emission maximum of fluorophore. (3) Stable optical property. Compared with organic quenchers, AuNPs have chemical, thermal stability and nonsusceptibility to photobleaching. It is feasible to stand long-time or real-time researches taking account of photobleaching and time. (4) Ease of labelling. Biomolecules containing exposed thiol groups can be easily attached to the gold surface through gold–sulfur bonds. Other functional groups such as carboxyl and amine groups can also attach on gold surface via sulfur containing ligands with special terminal groups. Therefore, AuNPs are useful nanomaterials in FRET technology for its unique optical and chemical properties.
In this review, we will discuss the energy transfer mechanism of AuNPs in FRET system, and summarize typical AuNPs based FRET methods for analytical applications in biochemical and pharmaceutical analysis.
2. Energy transfer mechanism of AuNPs
Förster resonant energy transfer (FRET) theory describes the excitation energy from a donor fluorophore to an acceptor through nonradiative dipole–dipole interaction. The efficiency of dipole–dipole energy transfer (EFRET) depends on Förster distance(R0): | (1) |
 | (2) |
The use of AuNPs in FRET systems, however, does not seem to accord with the Förster mechanism. A clear evidence is that 1.4-nm AuNPs, which have no sign of absorption in visual and near-infrared region, however, have excellent ability to quench many fluorescence dyes, e.g. fluorescein, rhodamine 6G, Texas red and Cy5.26 To reveal the nature of AuNPs in FRET system, researchers want to find a theory revealing the energy transfer mechanism between AuNPs and fluorescent dyes.27–29
Surface energy transfer (SET), which describes the energy transfer from fluorescent dyes to a metallic surface, has also been theoretically studied.30 Different from FRET, SET is not a dipole–dipole energy transfer, but dipole to a metallic surface. The efficiency of dipole-surface energy transfer, ESET, expresses as:
 | (3) |
 | (4) |
Clear differences can be seen in FRET and SET mechanisms, where, in the case of dipole–dipole energy transfer, EFRET depends on (r/R0)6, R0 mainly depends on the absorption of acceptor, while, for dipole–surface energy transfer, ESET depends on (r/d0)4, d0 mainly depends on angular frequency and Fermi wavevector of metal. What the dominant quenching mechanism will be when the donor fluorophore is near a nanosized gold, especially 1.4-nm gold nanoparticle, which has no sign of absorption in VIS-NIR region and is similar in size with traditional dyes?
Strouse's group appended a 1.4-nm gold nanoparticle to the 5′ end of one DNA strand as the energy acceptor and a fluorescein (FAM) to the 3′ end of the complementary strand as the energy donor.27 They studied the distance dependence of energy transfer between gold and FAM by tuning the lengths of DNA strand from 62 Å (15 base pairs) to 232 Å (60 base pairs), and found that energy transfer efficiency is in precise agreement with SET mechanism which is 1/R4 distance dependence. Efficient energy transfer distance range was estimated to be from 50 to 250 Å, which doubled the distances (10 to 100 Å) achievable in FRET system using traditional organic pairs (as showed in Fig. 1).
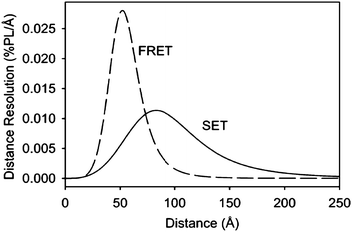 | ||
| Fig. 1 Separation distance-dependent length resolution of the FRET and SET mechanisms. (Reprinted with permission from ref. 27. Copyright 2005 American Chemical Society). | ||
In their further studies, distance dependent energy transfer from two dyes of different energies to 1.5-nm nanoparticles were also studied by tuning the length of linking double stranded DNA between dyes and AuNPs. The absorption, continuous-wave photoluminescence and picosecond fluorescence lifetime spectroscopy data in the fluorescence quenching process, validated the SET mechanism of gold based energy transfer.29 Other researchers have also proved the validity of the SET mechanism in their studies using different donors and linkers.31–37 Thus, the SET mechanism is accepted to describe AuNPs based energy transfer system by many researchers.38,39
Besides small 1.4-nm nanogold, energy transfer with larger AuNPs with different sizes40,41 and shapes36 have also been studied theoretically and practicably.42–45 Larger AuNPs have unique optical property originating from localized surface plasmon resonance (LSPR),9–12 which can be tuned precisely by their shape, size, and composition. Different from small nanogold, larger AuNPs have strong absorption near their surface plasmon resonance band. Thus, if AuNPs with strong light absorption property were used in a FRET system, what is the influence of AuNPs' absorption on energy transfer process, when donor's emission is near the AuNPs' absorption band, although it is independent on spectral overlap of the donor emission and AuNPs' absorption according to SET mechanism.
Mariana et al. studied fluorescence quenching of CdSe–QDs by gold nanoclusters with core diameters from 1.1 to 4.9 nm. Experimental evidence suggested that the energy transfer quenching efficiencies shows an enormous increase with the increasing of particles sizes.46 The increasing of energy transfer efficiency on larger AuNPs may be attributed to the appearance of AuNPs' surface plasmon. The surface plasmon dipole fields assist to overcome the weak electronic coupling between the emitting donor and absorbing acceptor transition exciton dipoles in the homogeneous medium.47 By using gold nanorods (AuNRs) with different LSPR absorption peaks as quenchers, Li et al. demonstrated that the quenching efficiency of QDs by AuNRs increases as AuNPs's LSPR absorption peak gets closer to the emission peak of the QDs.48 Therefore, the overlap of the donor emission and AuNPs' LSPR absorption is important to enhance the energy transfer efficiency. The quenching of fluorescence can be optimized by tuning the intensity, position, and the number of the AuNPs' LSPR absorption.
3. DNA hybridizations
The detection of DNA is of great importance in clinical diagnostics, gene therapy and forensic analysis. DNA hybridization between a DNA probe and its complementary target can be used to establish a FRET system.3.1. Labeling with small AuNPs
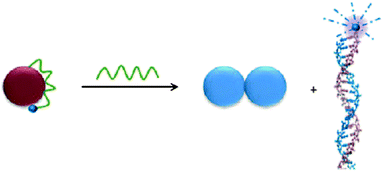
Dubertret et al. firstly appended a 1.4-nm AuNP and a fluorophore to the 5′ and 3′end of single-stranded DNA probe with hairpin shaped structure.26Fig. 2 shows the schematic drawing of this FRET system. Self-hybridization of the DNA probe, brings the fluorophore and AuNP into close proximity, and efficient fluorescence quenching of AuNP can be observed. Dubertret found that 1.4-nm AuNPs have excellent quenching ability which can advantageously replace DABCYL as quenchers of fluorescence. The fluorescence restoration of this AuNPs based molecule beacon increases by a factor of as much as several thousand as it binds to a complementary target DNA. Detection sensitivity and single mismatch detection ability was enhanced up to 100-fold and 8-fold respectively, compared with traditional molecular beacons. | ||
| Fig. 2 Schematic drawings of the two conformations of the dye–oligonucleotide–gold conjugate. On the left, the hairpin structure brings the fluorophore and the gold particle in close proximity (within a few angstroms). In this conformation, the gold cluster quenches the fluorescence of the dye. Through sequence-specific hybridization with a single-stranded target DNA, the hairpin structure changes to a rod-like structure (on the right), which maintains the fluorophore and the quencher far apart and thus restores the fluorescence. (Reprinted with permission from ref. 26. Copyright 2001 Nature Publishing Group). | ||
Using a similar principle, a oligonucleotide molecule labeled with a thiol group at one end and a fluorophore at the other was immobilized on a 2.5-nm gold nanoparticle through Au-thiol bond. Unlike molecular beacons, this oligonucleotide was found to spontaneously assemble into a constrained arch-like conformation on the particle surface, and bring the fluorescence quenching of fluorophore labeled at the other end. Binding with target molecules results in a conformational change, which restores the fluorescence of the quenched fluorophore.49
The detection of DNA using fluorescence quenching of 1.4-nm AuNPs is further improved by employing QDs, which have highly efficient fluorescence and size dependent band-gap emission, as a fluorescence donor.50,51 The hybridization of two complementary single-stranded DNA (ssDNA) linked to QDs and AuNPs respectively, brings the QDs and AuNPs into close proximity. The emission quenching of QDs by AuNPs can be tuned by the length of double-stranded DNA (dsDNA) spacer.50 Using this scheme, Dyadyusha et al. established a similar DNA sensing approach using oligonucleotide tagged CdSe QDs and gold.51 Their results showed that QD–AuNPs DNA conjugates have significant potential for the detection of single molecule of DNA in the solution phase or in cellular systems.
3.2 Quenching on the surface of larger AuNPs
AuNPs with various shapes and particle sizes range from 5 to 100 nm have advantages such as handy preparation, facile modification, unique optical property, and more importantly, large surface area for ssDNA adsorption. Based on the finding that ssDNA has propensity to adsorb on citrate-coated AuNPs while dsDNA does not,52 Li et al. designed a novel fluorescent quenching assay for DNA hybridization by the adsorption of dye-labeled ssDNA on AuNPs.53 Fluorescent dye-tagged probe ssDNA sequences have their fluorescence efficiently quenched when they adsorb on the surface of AuNPs, however, dsDNA formed by hybridization with complementary target sequences does not adsorb on gold surface and the fluorescence persists. The results showed that target sequences in complex mixtures of DNA and single-base mismatches in DNA sequences could be sensitively and easily detected using this method. By the same principle, Ray et al. demonstrated a hybridization detection method using multicolor dyes-tagged ssDNA as probes.54 Multiplexed detections of two different target sequences could be achieved using two different dyes-tagged ssDNA probes complementary to the two targets respectively.This fluorescence assay based on adsorption of dye-ssDNA on AuNPs was also reported to screen the RNA sequence relative to hepatitis C virus (HCV).38 Based on fluorescnce recovery of dye-RNA, departs from AuNP surface after hybridization (scheme showed in Fig. 3), a linear correlation between the fluorescence intensity and concentration of the target RNA was found over the range of 15∼550 pM. The effect of fluorescence quenching efficiency of this system was further studied by turning the size of AuNPs and the length between the fluorescent label and gold surface. The results showed that the quenching efficiency increases by three orders of magnitude, as the gold nanoparticle size increases from 5 to 70 nm, and the efficient fluorescence quenching distance ranges from 8 to 40 nm according to the length of RNA immobilized on gold surface.
 | ||
| Fig. 3 Schematic representation of the RNA hybridization process, when dye–RNA is adsorbed on AuNP. Dye–RNA departed from the AuNP surface after hybridization, thus restores the fluorescence. (Reprinted with permission from ref. 38. Copyright 2009 Wiley-VCH Verlag GmbH & Co. KGaA). | ||
Different from the above schemes when the fluorescence is directly quenched by the adsorption of dyes–ssDNA on AuNPs surface, Wu et al. developed a homogeneous fluorescence quenching assay for the detection of DNA hybridization by immobilizing thiol–ssDNA on AuNPs surface.55 The hybridization of target dye–ssDNA on AuNPs with the immobilized detection ssDNA will brings dyes into close proximity with AuNPs. Fluorescence quenching increases with the target DNA concentration and provides a quantitative detection of target DNA with the range from 1.4 to 92 nM. This fluorescence quenching method based on hybridization of dye–ssDNA on AuNPs surface, has a lower background signal compared with the method using the adsorption of dye–ssDNA on AuNPs, furthermore, this homogeneous assay has potential use for DNA monitoring in real time. Other scheme based on the fluorescence quenching and restoring of dyes-tagged DNA on AuNPs can also be established by hybridization of oligonucleotides labeled with gold and fluorophore.56
4. Aptamer binding
Aptamers are novel oligonucleotides which can bind with small molecules, metal ions and proteins specially. Detection of small molecules, metal ions and proteins using aptamer–AuNPs based FRET systems have been frequently reported.55–59 For example, Huang et al. established a aptamer–AuNPs based fluorescence switching system for detection of cancer marker protein (platelet-derived growth factor, PDGF).57 They found that PDGF binding aptamer has a unique structure that also allows the binding of a fluorescence dye (N,N′-dimethyl-2,7-diazapyrenium dication, DMDAP). The fluorescence of DMDAP is almost completely quenched by AuNPs when it intercalates with the aptamers on aptamer–AuNPs surface, however the competitive binding of PDGF with aptamer will release DMDAP from aptamer–AuNPs surface, thus restore the florescence. Their results showed that this aptamer based fluorescence switching approach could detect various PDGFs specifically and sensitively.Wang et al. established an aptamer biosensor for thrombin by AuNPs with three different strategies.58 As showed in Fig. 4, in strategies A and B, thiol–DNA/thiol–aptamer were immobilized on AuNPs and hybridized with dye–aptamer/dye–DNA for fluorescence quenching. The detections are based on the competition between duplex formation and thrombin–aptamer recognition. In strategy C, dye–aptamer was adsorbed on AuNPs directly. After binding with thrombin, dye–aptamer did not tend to adsorb on AuNPs, and fluorescence recovered. Comparison with these three strategies, they found that strategy A was the best way for thrombin detection with highest affinity and sensitivity, although it was complicated in probe preparation.
 | ||
| Fig. 4 Principles of detection of thrombin with three different strategies. (Reprinted with permission from ref.58. Copyright 2007 Elsevier Inc.) | ||
G-rich aptamers after binding with their ligands will form a G-quadruplexes structure. Similar to ssDNA and dsDNA, the formation of quadruplexes structure from a single stranded aptamer has a different propensity to adsorb on citrate-coated AuNPs. Jin et al. designed a simple fluorescence quenching system for selecting quadruplex binding ligands using AuNPs as a fluorescence quencher.59 Dye-tagged probe aptamer adsorbs on AuNPs, so that fluorescence from the dye-tagged single-stranded aptamer is quenched. When quadruplex-binding ligands are added, the formation of G-quadruplexes leads to release of the probe DNA from AuNPs, and fluorescence enhancement is observed. Studies of two series of Chinese medicine monomers showed that flavonoids were potential quadruplex-binding ligands. Thus, a useful method for rapid screening antitumor drugs could be provided using this FRET system.
Liu et al. first established simultaneous multiplex-detection of two analytes by aptamer-linked QDs and AuNPs nanostructures based on the fluorescence quenching of QDs by AuNPs.60 By hybridization of aptamer-linked QDs and oligonucleotides-linked AuNPs, the two particles would cross-link to aggregates, and result in fluorescence quenching of QDs by AuNPs. The addition of analytes disassembled the aggregates, and released the QDs emission. Fluorescence assay for different molecules in one pot could be established by simultaneously using different aptamer-linked QDs. Zhang et al. also established a aptamer-based multicolor fluorescent assay for multiplex detection of three analtyes (adenosine, potassium ion, and cocaine) in homogeneous solution by gold nanoprobes61 (Fig. 5). Different from the method proposed by Liu et al., the fluorescence quenching was not based on the aggregation of AuNPs and QDs, but the close proximity of dyes on the surface of AuNPs by DNA hybridization. Firstly, three 3′-thiolated DNA strands complementary to adenosine, potassium and cocaine aptamers were immobilized at the surface of AuNPs at equal molar ratio. Then, the three aptamers labeled with different dyes (Rox, FAM and Cy5) at the 5′ end were hybridized with their complementary sequences at the surface of AuNPs, forming the multicolor fluorescence quenched gold nanoprobe. The specific binding of individual aptamers with their specific targets will separate the aptamer from the AuNPs surface, thus leading to fluorescence recovery that provides quantitative measurement of the analyte concentration.
 | ||
| Fig. 5 Schematic drawings of multicolor aptamer-based gold nanoprobe for the multiplex detection of adenosine (A), potassium, and cocaine. (Reprinted with permission from ref. 61. Copyright 2009 Wiley-VCH Verlag GmbH & Co. KGaA). | ||
5. Immunoreactions
Immunoassays, which are based on the specific recognition of antigen by corresponding antibody have been applied to medical and clinical diagnostics. AuNPs are excellent immunoassay tags which can easily label a broad range of antibodies without reducing their immuno-activities. Also as excellent quenchers, AuNPs have been widely used for FRET based immunoassay.62–64Fluorescence resonance energy transfer from donor to acceptor can take place when the sandwich complex is assembled by labeled antibodies and antigen. However, the efficient energy transfer between traditional organic dyes is typically limited to donor–acceptor distances smaller than 5∼7 nm.25 Since the size of antibody–antigen complexes may reach 10 nm or more, the efficacy of these sandwich type FRET based immunoassay is limited. The use of AuNPs as fluorescence quenchers, however, has higher energy transfer efficiency in longer donor–acceptor distance up to 25 nm according to the SET mechanism.27 Homogeneous sandwich immunoassay based on FRET of gold AuNPs, therefore, has being frequently reported in recent years.
Mayilo et al. designed a homogeneous sandwich fluorescence quenching immunoassay for the detection of cardiac troponin T (cTnT) using AuNPs as fluorescence quenchers.65 As shown in Fig. 6, two monoclonal antibodies that bind to two different locations on the cTnT protein were labeled with AuNPs and fluorescent dyes respectively. The sandwich interactions between cTnT and two antibodies would bring the AuNPs and fluorescent dyes into close proximity. Using time-resolved fluorescence spectroscopy, the fluorescence quenching efficiency of AuNPs was determined as high as 95%. A limit of detection (LOD) of 0.02 nM (0.7 ng mL−1) was obtained for cTnT, which is the lowest value reported for a homogeneous sandwich assay for cTnT.
 | ||
| Fig. 6 Principle of operation of the sandwich test for cTnT. AuNPs are functionalized with anti-cTnT M11.7 antibody fragments, and anti-cTnT M7 antibody fragments are labeled with fluorescent dye molecules. Upon addition of cTnT, the M11.7–AuNP and the M7–dye bind to different positions of the cTnT molecules forming sandwich assemblies. As a result, the fluorescence of the dye is quenched by the nearby AuNP. (Reprinted with permission from ref. 65. Copyright 2009 American Chemical Society). | ||
Liang et al. reported a fluorescence quenching immunosystem for the detection of human IgG (H-IgG) by goat anti-human IgG functionalized NIR-emitting QDs (GAH–IgG–QDs) and rabbit anti-human IgG functionalized Au nanorods (RAH–IgG–AuNPs).66 The fluorescence of GAH–IgG–QDs could be efficiently quenched by immunoattached on the RAH–IgG–AuNRs in presence of H–IgG. H–IgG over the range of 0.05∼2.5μM can be quantitatively detected under optimal conditions. Compared with AuNPs and other organic quenchers, Au nanorods (AuNRs) have two absorption peaks assignable to transverse and longitudinal surface plasmon resonance, which can be tuned from VIS to NIR region by changing their aspect ratio. Thus, AuNRs can be used as a suitable acceptor for energy transfer from VIS to NIR emission, due to their strong plasmon resonance absorption.
Meng et al. established sandwich-type immunoassay for the detection of goat anti-human immunoglobulin G (GAH–IgG) using NaYF4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Yb,Er NIR-to-VIS upconversion nanoparticles (UCNPs) as energy donors, and AuNPs as energy acceptors.67 The quenching of UCNPs' luminescence intensity was correlated to the concentration of the goat antihuman IgG in the range of 3·67 μg mL−1 with detection limit of 0.88 μg mL−1. This result did not show better sensitivity, however, the use of UCNPs has prominent advantages of narrow emission peak, large Stokes shift, low toxicity, and good chemical stability and photostability. More importantly, the excitation of UCNPs often requires NIR radiation which is less harmful to biological samples.
:Yb,Er NIR-to-VIS upconversion nanoparticles (UCNPs) as energy donors, and AuNPs as energy acceptors.67 The quenching of UCNPs' luminescence intensity was correlated to the concentration of the goat antihuman IgG in the range of 3·67 μg mL−1 with detection limit of 0.88 μg mL−1. This result did not show better sensitivity, however, the use of UCNPs has prominent advantages of narrow emission peak, large Stokes shift, low toxicity, and good chemical stability and photostability. More importantly, the excitation of UCNPs often requires NIR radiation which is less harmful to biological samples.
Besides binding donor and AuNPs together through sandwich immunoassay, fluoroimmunoassay based on fluorescence quenching of fluorophore using AuNPs labeled antibodies after immunoreactions can also be established. The method of quenching the fluorescence by direct immuno-gold, needs no label to the other antibody with fluorophore, however, it needs additional steps such as separation and washing of the immunocomplex.
For example, Ao et al. established a sensitive immunoassay system for α-fetoprotein (AFP) detection based on the fluorescence quenching of fluorescein isothiocyanate (FITC) by AuNPs.68 By using anti-AFP polyclonal antibody coated magnetic nanoparticles to capture fetoprotein, AuNPs coated with anti-AFP monoclonal antibodies could bind on the AFP captured by the magnetic nanoparticle. After magnetic separation of the sandwich-type immunocomplex from solution, the unbound gold nanoparticle probes in supernatant fluid, were used to quench the fluorescence of FICT. The results showed that the method has a higher sensitivity with a limit of detection of AFP up to 0.17 nM. Peng et al. also established a sensitive immunoassay system for human immunoglobulin (IgG) detection based on the fluorescence quenching of fluorescein by AuNPs.69 Different from magnetic separation using antibody coated magnetic particles mentioned above, goat anti-human IgG was directly adsorbed on polystyrene microwells to capture the IgG analytes. After being sandwiched by antibody labeled AuNPs, the sandwich-type immunocomplex was subsequently dissociated by the mixed solution of sodium hydroxide and trisodium citrate. The AuNPs dissociated from the immunocomplex were used to quench fluorescence. This method could detect the concentration of human IgG in the dynamic range of 10∼5000 ng ml−1, with a detection limit of 4.7 ng ml−1.
6. Competitive adsorption/binding
Adsorption and specific binding of fluorescent substances to the surface of bare or functionalized AuNPs would induce the quenching of fluorescence. Disassembly of this energy transfer system can be realized by competitive binding of other molecules on AuNPs surface. Thus, fluorescence “turn on” methods based on the competitive binding of fluorescent substances and analytes on the AuNP surface can be established to detect various analytes.6.1 Metal ions
Hg2+ which has higher affinity to bind on the AuNPs surface, could release dyes from the AuNPs surface and restore florescence. Using this approach, Huang et al. established a fluorescence sensor for detecting Hg2+ ions in aqueous solution based on the competitive binding of Hg2+ ions on Rhodamine B adsorbed AuNPs.70 This approach has the advantages of rapidity, simplicity, low cost, and high sensitivity with a LOD of 2.0 ppb. The selectivity could also be improved by modifying the AuNPs surfaces with thiol ligands and adding a chelating ligand. Other similar reports showed that this method is reliable and satisfactory for Hg2+ detection.71,72The equipment for an AuNPs based fluorescence method for Hg2+ was miniaturized in a small box using a battery-operated laser pointer for inducing fluorescence.33 The miniaturized facility was successfully used in sensing Hg2+ in practical environmental samples, such as soil, water and fish, with excellent sensitivity and selectivity.
Besides the adsorption of fluorescence dyes on AuNPs, the electrostatic interaction between fictionalized QDs and AuNPs can also be used to establish a FRET assay for metal ions detection. Wang et al. proposed the positively charged cysteamine capped QDs with negatively charged 11-mercaptoundecanoic acid capped AuNPs as fluorescence donors and quenchers.73 The mixture of these pairs would induce fluorescence quenching of QDs by electrostatic attraction, however, the presence of Pb2+ would efficiently inhibit the interaction of the QD–GNP assembly. AuNPs tend to aggregate and QDs “turns on” fluorescence proportionally to the concentration of Pb2+. Under the optimum conditions, Pb2+ in the range 0.22∼4.51 ppm can be quantitatively detected with a LOD of 30 ppb.
6.2. Small molecules
Small molecules having affinity to bind on the AuNPs surface, could compete with dyes to adsorb onto gold surface. For example, in the presence of thiols, fluorescent dyes departed from the gold surface by the competitive adsorption of thiols, resulting in fluorescence recovery of adsorbed dyes. Using this principle, Shang et al. established a cysteine detection method over the range 2.5 × 10−8 to 4.0 × 10−6 M, with a LOD as low as 10 nM.74 Another example is I− which also has a higher affinity to bind on AuNPs surface. Chen et al. thus established fluorescence detection of I− using fluorescein isothiocyanate attached AuNPs. The lowest detectable concentration of I− was up to 10.0 nM.75Small molecules without affinity to bind on the AuNP surface, can also be detected using functionalized AuNPs and fluorescent substances. Tang et al. developed a AuNPs based FRET biosensor for the direct determination of glucose in serum.76 As shown in Fig. 7, the specific combination of concanavalin A (ConA) conjugated QDs and thiolated β-cyclodextrins (β-SH-CDs) modified AuNPs quenched the emission of QDs. In the presence of glucose, ConA–QDs departed from the β-SH-CDs–AuNPs surface by competition of glucose with β-SH-CDs on the binding sites of ConA, resulting in the fluorescence recovery of the quenched QDs. Concentration of glucose within the range of 0.10∼50 mM could be detected with a LOD as low as 50 nM. A similar assay for the detection of cholesterol was also established using CdTe QDs and AuNPs–β-SH-CDs.77
 | ||
| Fig. 7 Schematic illustration of QDs–ConA–β-SH-CDs–AuNPs FRET nanobiosensor. (Reprinted with permission from ref. 76. Copyright 2008 Wiley-VCH Verlag GmbH & Co. KGaA). | ||
7. Summary and outlook
Due to unique optical and chemical properties, AuNPs have found wide applications in FRET analysis. In this minireview, we discuss the energy transfer mechanism of AuNPs as acceptor in a FRET system, and introduce typical approaches which can establish AuNPs based energy transfer systems. Different from traditional FRET systems, energy transfer with AuNPs has many advantages such as excellent quenching ability, unique absorption assistance, longer energy transfer distance and specific binding affinity, and can be used for detection of DNAs, proteins, metal ions, drugs, and so on.Applications of AuNPs-FRET method are not only limited to biochemical and pharmaceutical analysis, but extend to fluorescence imaging in live cells. Traditional FRET probes are often susceptible to enzymatic degradation in live cells, however, AuNPs act as both fluorescence quencher and carrier in AuNPs–FRET probes, which assist the internalization of fluorescence probes in live cells and improve their intracellular stability. AuNPs–FRET probes have been studied for transfection visualization, mRNA detection, drug screening, protease activity determination, hydroxyl radicals detection in living cells and to monitor in vivo cancer response.78–82
Furthermore, the use of AuNPs in energy transfer systems is not only useful for their quenching ability. Fluorescent gold nanoclusters,83,84 which have size-tunable emission maxima and higher fluorescent quantum yield, can also be used as fluorescence donors. Unlike quantum dots and organic dyes, fluorescent gold nanoclusters have advantages of photostabilization, biocompatibility, ease of bioconjugation, and minimal toxicity. Thus, a “gold-only” energy transfer system using gold nanomaterials as both donor and acceptor has distinct advantages in biochemical assays.85
Additionally, AuNPs were also found to enhance the fluorescence of fluorophore near the AuNPs surface.86–89 The quenching of fluorescence needs close proximity of fluorophore to AuNPs, however, if the distance between the fluorophore and AuNPs is carefully optimized, enhancement of fluorescence can be observed.90 Therefore, both quenching and enhancement of donor's fluorescence can be realized by tuning the dyes–AuNP distance or other approaches.91,92 We expect that quenching and enhancement of donor's fluorescence by AuNPs in an energy transfer system may have higher detection sensitivity than traditional methods.
Thus, we believe that the use of AuNPs for energy transfer measurements, although still in its infancy, and novel energy transfer systems with new gold nanomaterials will find wide applications in the near future.
Acknowledgements
The authors greatly appreciate the financial support provided by the National Natural Science Foundation of China (NSFC, No. 21035005), and the Science and Engineering Foundation of Yunnan University (No. kl090007).Notes and references
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CAS.
- W. R. Algar, M. Massey and U. J. Krull, TrAC, Trends Anal. Chem., 2009, 28, 292–306 CrossRef CAS.
- Y.-W. Lin, C.-W. Liu and H.-T. Chang, Anal. Methods, 2009, 1, 14–24 RSC.
- Uwe H. F. Bunz and Vincent M. Rotello, Angew. Chem., Int. Ed., 2010, 49, 3268–3279 CAS.
- K. G. Thomas and P. V. Kamat, Acc. Chem. Res., 2003, 36, 888–898 CrossRef CAS.
- J. Zheng, J. T. Petty and R. M. Dickson, J. Am. Chem. Soc., 2003, 125, 7780–7781 CrossRef CAS.
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402 CrossRef.
- U. Kreibig and M. Vollmer, Optical Properties of Metal Clusters, Springer Verlag, Heidelberg, 1995 Search PubMed.
- P. Mulvaney, Langmuir, 1996, 12, 788–800 CrossRef CAS.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS.
- W. R. Algar and U. J. Krull, Anal. Bioanal. Chem., 2008, 391, 1609–1618 CrossRef CAS.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4588 CrossRef CAS.
- I. L. Medintz and H. Mattoussi, Phys. Chem. Chem. Phys., 2009, 11, 17–45 RSC.
- A. L. Rogach, T. A. Klar, J. M. Lupton, A. Meijerink and J. Feldmann, J. Mater. Chem., 2009, 19, 1208–1221 RSC.
- S. Saini, G. Srinivas and B. Bagchi, J. Phys. Chem. B, 2009, 113, 1817–1832 CrossRef CAS.
- K. H. Drexhage, J. Fluoresc., 1970, 1–2, 693–701.
- R. R. Chance, A. Prock and R. Silbey, in Advances in Chemical Physics, John Wiley & Sons, New York, 1978, vol. 37, pp. 1–65 Search PubMed.
- A. P. Alivisatos, D. H. Waldeck and C. B. Harris, J. Chem. Phys., 1985, 82, 541–547 CrossRef CAS.
- P. C. Ray, G. K. Darbha, A. Ray, J. Walker and W. Hardy, Plasmonics, 2007, 2, 173–183 CrossRef CAS.
- R. Wargnier, A. V. Baranov, V. G. Maslov, V. Stsiapura, M. Artemyev, M. Pluot, A. Sukhanova and I. Nabiev, Nano Lett., 2004, 4, 451–457 CrossRef CAS.
- J. Q. Gu, L. D. Sun, Z. G. Yan and C. H. Yan, Chem. Asia J., 2008, 3, 1857–1864 Search PubMed.
- C. Fan, S. Wang, J. W. Hong, G. C. Bazan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6297–6301 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 3rd edn, Springer, New York, 2006 Search PubMed.
- B. Dubertret, M. Calame and A. J. Libchaber, Nat. Biotechnol., 2001, 19, 365–370 CrossRef CAS.
- C. S. Yun, A. Javier, T. Jennings, M. Fisher, S. Hira, S. Peterson, B. Hopkins, N. O. Reich and G. F. Strouse, J. Am. Chem. Soc., 2005, 127, 3115–3119 CrossRef CAS.
- T. L. Jennings, J. C. Schlatterer, M. P. Singh, N. L. Greenbaum and G. F. Strouse, Nano Lett., 2006, 6, 1318–1324 CrossRef CAS.
- T. L. Jennings, M. P. Singh and G. F. Strouse, J. Am. Chem. Soc., 2006, 128, 5462–5467 CrossRef CAS.
- B. N. J. Persson and N. D. Lang, Phys. Rev. B, 1982, 26, 5409 CrossRef CAS.
- S. Bhowmick, S. Saini, V. B. Shenoy and B. Bagchi, J. Chem. Phys., 2006, 125.
- T. Pons, I. L. Medintz, K. E. Sapsford, S. Higashiya, A. F. Grimes, D. S. English and H. Mattoussi, Nano Lett., 2007, 7, 3157–3164 CrossRef CAS.
- G. K. Darbha, A. Ray and P. C. Ray, ACS Nano, 2007, 1, 208–214 CrossRef CAS.
- K. K. Haldar and A. Patra, Chem. Phys. Lett., 2008, 462, 88–91 CrossRef CAS.
- K. K. Haldar, T. Sen and A. Patra, J. Phys. Chem. C, 2008, 112, 11650–11656 CrossRef CAS.
- T. Sen and A. Patra, J. Phys. Chem. C, 2008, 112, 3216–3222 CrossRef CAS.
- R. Chhabra, J. Sharma, H. Wang, S. Zou, S. Lin, H. Yan, S. Lindsay and Y. Liu, Nanotechnology, 2009, 20, 485201 CrossRef.
- J. Griffin, A. K. Singh, D. Senapati, P. Rhodes, K. Mitchell, B. Robinson, E. Yu and P. C. Ray, Chem. Eur. J., 2009, 15, 342–351 CrossRef CAS.
- M. P. Singh, T. L. Jennings and G. F. Strouse, J. Phys. Chem. B, 2009, 113, 552–558 CrossRef CAS.
- S. K. Ghosh, A. Pal, S. Kundu, S. Nath and T. Pal, Chem. Phys. Lett., 2004, 395, 366–372 CrossRef CAS.
- C. K. Kim, R. R. Kalluru, J. P. Singh, A. Fortner, J. Griffin, G. K. Darbha and P. C. Ray, Nanotechnology, 2006, 17, 3085–3093 CrossRef CAS.
- J. Seelig, K. Leslie, A. Renn, S. Kuhn, V. Jacobsen, M. van de Corput, C. Wyman and V. Sandoghdar, Nano Lett., 2007, 7, 685–689 CrossRef CAS.
- E. Dulkeith, A. C. Morteani, T. Niedereichholz, T. A. Klar, J. Feldmann, S. A. Levi, F. C. J. M. van Veggel, D. N. Reinhoudt, M. Moller and D. I. Gittins, Phys. Rev. Lett., 2002, 89, 203002 CrossRef CAS.
- E. Dulkeith, M. Ringler, T. A. Klar, J. Feldmann, A. Munoz Javier and W. J. Parak, Nano Lett., 2005, 5, 585–589 CrossRef CAS.
- J. Zhang, R. Badugu and J. R. Lakowicz, Plasmonics, 2008, 3, 3–11 CrossRef CAS.
- M. Kondon, J. Kim, N. Udawatte and D. Lee, J. Phys. Chem. C, 2008, 112, 6695–6699 CrossRef CAS.
- V. K. Komarala, A. L. Bradley, Y. P. Rakovich, S. J. Byrne, Y. K. Gun'ko and A. L. Rogach, Appl. Phys. Lett., 2008, 93, 123102 CrossRef.
- X. Li, J. Qian, L. Jiang and S. L. He, Appl. Phys. Lett., 2009, 94, 063111 CrossRef.
- D. J. Maxwell, J. R. Taylor and S. Nie, J. Am. Chem. Soc., 2002, 124, 9606–9612 CrossRef CAS.
- Z. Gueroui and A. Libchaber, Phys. Rev. Lett., 2004, 93, 166108 CrossRef.
- L. Dyadyusha, S. J. H. Yin, T. Brown, J. J. Baumberg, F. P. Booy and T. Melvin, Chem. Commun., 2005, 3201–3203 RSC.
- H. Li and L. Rothberg, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14036–14039 CrossRef CAS.
- H. Li and L. J. Rothberg, Anal. Chem., 2004, 76, 5414–5417 CrossRef CAS.
- P. C. Ray, G. K. Darbha, A. Ray, W. Hardy and J. Walker, Nanotechnology, 2007, 18, 375504 CrossRef.
- Z.-S. Wu, J.-H. Jiang, L. Fu, G.-L. Shen and R.-Q. Yu, Anal. Biochem., 2006, 353, 22–29 CrossRef CAS.
- Z. H. Mo, X. C. Yang, K. P. Guo and Z. Y. Wen, Anal. Bioanal. Chem., 2007, 389, 493–497 CrossRef CAS.
- C.-C. Huang, S.-H. Chiu, Y.-F. Huang and H.-T. Chang, Anal. Chem., 2007, 79, 4798–4804 CrossRef CAS.
- W. Wang, C. Chen, M. Qian and X. S. Zhao, Anal. Biochem., 2008, 373, 213–219 CrossRef CAS.
- Y. Jin, H. Y. Li and J. Y. Bai, Anal. Chem., 2009, 81, 5709–5715 CrossRef CAS.
- J. Liu, J. H. Lee and Y. Lu, Anal. Chem., 2007, 79, 4120–4125 CrossRef CAS.
- J. Zhang, L. Wang, H. Zhang, F. Boey, S. Song and C. Fan, Small, 2010, 6, 201–204 CrossRef CAS.
- N. Kato and F. Caruso, J. Phys. Chem. B, 2005, 109, 19604–19612 CrossRef CAS.
- J.-Q. Gu, J. Shen, L.-D. Sun and C.-H. Yan, J. Phys. Chem. C, 2008, 112, 6589–6593 CrossRef CAS.
- S. Mayilo, B. Ehlers, M. Wunderlich, T. A. Klar, H.-P. Josel, D. Heindl, A. Nichtl, K. Kürzinger and J. Feldmann, Anal. Chim. Acta, 2009, 646, 119–122 CrossRef CAS.
- S. Mayilo, M. A. Kloster, M. Wunderlich, A. Lutich, T. A. Klar, A. Nichtl, K. Kürzinger, F. D. Stefani and J. Feldmann, Nano Lett., 2009, 9, 4558–4563 CrossRef CAS.
- G. X. Liang, H. C. Pan, Y. Li, L. P. Jiang, J. R. Zhang and J. J. Zhu, Biosens. Bioelectron., 2009, 24, 3693–3697 CrossRef CAS.
- M. Wang, W. Hou, C.-C. Mi, W.-X. Wang, Z.-R. Xu, H.-H. Teng, C.-B. Mao and S.-K. Xu, Anal. Chem., 2009, 81, 8783–8789 CrossRef CAS.
- L. Ao, F. Gao, B. Pan, R. He and D. Cui, Anal. Chem., 2006, 78, 1104–1106 CrossRef CAS.
- Z. Peng, Z. Chen, J. Jiang, X. Zhang, G. Shen and R. Yu, Anal. Chim. Acta, 2007, 583, 40–44 CrossRef CAS.
- C. C. Huang and H. T. Chang, Anal. Chem., 2006, 78, 8332–8338 CrossRef CAS.
- J. L. Chen, A. F. Zheng, A. H. Chen, Y. C. Gao, C. Y. He, X. M. Kai, G. H. Wu and Y. C. Chen, Anal. Chim. Acta, 2007, 599, 134–142 CrossRef CAS.
- A. F. Zheng, J. L. Chen, G. N. Wu, H. P. Wei, C. Y. He, X. M. Kai, G. H. Wu and Y. C. Chen, Microchim. Acta, 2009, 164, 17–27 CrossRef CAS.
- X. Wang and X. Q. Guo, Analyst, 2009, 134, 1348–1354 RSC.
- L. Shang, J. Yin, J. Li, L. Jin and S. Dong, Biosens. Bioelectron., 2009, 25, 269–274 CrossRef CAS.
- Y.-M. Chen, T.-L. Cheng and W.-L. Tseng, Analyst, 2009, 134, 2106–2112 RSC.
- B. Tang, L. H. Cao, K. H. Xu, L. H. Zhuo, J. H. Ge, Q. F. Li and L. J. Yu, Chem. Eur. J., 2008, 14, 3637–3644 CrossRef CAS.
- N. Zhang, Y. Y. Liu, L. L. Tong, K. H. Xu, L. H. Zhuo and B. Tang, Analyst, 2008, 133, 1176–1181 RSC.
- B. Tang, N. Zhang, Z. Z. Chen, K. H. Xu, L. G. Zhuo, L. G. An and G. W. Yang, Chem. Eur. J., 2008, 14, 522–528 CrossRef.
- S. Lee, E.-J. Cha, K. Park, S.-Y. Lee, J.-K. Hong, I.-C. Sun, Sang Y. Kim, K. Choi, I. C. Kwon, K. Kim and C.-H. Ahn, Angew. Chem., Int. Ed., 2008, 47, 2804–2807 CrossRef CAS.
- M. Oishi, A. Tamura, T. Nakamura and Y. Nagasaki, Adv. Funct. Mater., 2009, 19, 827–834 CrossRef CAS.
- D. S. Seferos, D. A. Giljohann, H. D. Hill, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 15477–15479 CrossRef CAS.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027–1030 CrossRef CAS.
- W. Chen, X. Tu and X. Guo, Chem. Commun., 2009, 1736–1738 RSC.
- G.-Y. Lan, C.-C. Huang and H.-T. Chang, Chem. Commun., 2010, 46, 1257–1259 RSC.
- C. C. Huang, C. K. Chiang, Z. H. Lin, K. H. Lee and H. T. Chang, Anal. Chem., 2008, 80, 1497–1504 CrossRef CAS.
- J. Zhang and J. R. Lakowicz, Opt. Express, 2007, 15, 2598–2606 CrossRef CAS.
- Y. Fu, J. Zhang and J. R. Lakowicz, J. Am. Chem. Soc., 2010, 132, 5540–5541 CrossRef CAS.
- Y. Shan, J.-J. Xu and H.-Y. Chen, Chem. Commun., 2009, 905–907 RSC.
- S. Kuhn, U. Hakanson, L. Rogobete and a. Sandoghdar, Phys. Rev. Lett., 2006, 97, 017402 CrossRef.
- H. Mertens, A. F. Koenderink and A. Polman, Phys. Rev. B, 2007, 76, 115123 CrossRef.
- P. Anger, P. Bharadwaj and L. Novotny, Phys. Rev. Lett., 2006, 96, 113002 CrossRef.
- K. K. Haldar and A. Patra, Appl. Phys. Lett., 2009, 95, 063103–063103 CrossRef.
| This journal is © The Royal Society of Chemistry 2010 |