Quantification of α-polylysine: a comparison of four UV/Vis spectrophotometric methods†
Andrea
Grotzky
,
Yuichi
Manaka
,
Sara
Fornera
,
Martin
Willeke
and
Peter
Walde
*
Department of Materials, Swiss Federal Institute of Technology (ETH) Zürich, Wolfgang-Pauli-Strasse 10, CH-8093, Zürich, Switzerland. E-mail: peter.walde@mat.ethz.ch
First published on 13th August 2010
Abstract
Four UV/Vis spectrophotometric methods for the quantification of α-polylysine (α-PL) are described and discussed. The methods are based on different chemical reactivities of α-PL allowing the indirect determination of α-PL concentrations in aqueous solutions down to 1–2 µg mL−1. The four methods are the trypan blue (TB) assay, the 2,4,6-trinitrobenzene sulfonate (TNBS) assay, the ortho-phthalaldehyde (OPA) assay and the bicinchoninic acid (BCA) assay. The TB assay is based on the polycationic character of α-PL in acidic aqueous solutions, in which the non-covalent binding with the oligoanionic dye TB leads to a precipitation of the dye and a concomitant decrease in the intensity of the blue color of the solution. Both the TNBS and the OPA assay utilize the nucleophilicity of the amino groups in alkaline solutions resulting in trinitrophenylated amino groups and isoindole derivatives, respectively. Finally, in the BCA assay the reductive properties of the peptide bonds are used to reduce copper(II) to copper(I) which eventually forms a stable, purple 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complex with bicinchoninic acid. For each method, mechanistic aspects are discussed and detailed experimental protocols are provided. With respect to the level of minimum quantification the TB and the OPA assays are best.
:2 complex with bicinchoninic acid. For each method, mechanistic aspects are discussed and detailed experimental protocols are provided. With respect to the level of minimum quantification the TB and the OPA assays are best.
Introduction
The polypeptide α-polylysine, abbreviated as α-PL, is the linear homopolymer of the amino acid lysine in which the peptide bonds are formed via the α-amino groups (Scheme 1). Each lysine residue carries one ε-amino group (pKa(R–NH3+) = 9–10),1 therefore, in neutral or acidic aqueous solutions α-PL is a polycation. In literature the general term “polylysine” is often used for α-PL as well as for ε-polylysine, abbreviated as ε-PL. In contrast to α-PL, in ε-PL the lysine repeating units are linked via the ε-amino groups (Scheme 1). The focus here is on α-polylysine only. Since lysine has one chiral center, the α-carbon, two enantiomeric forms exist: the naturally occurring L-lysine and its mirror image D-lysine (Scheme 1). Depending on whether α-PL is formed from L-lysine or from D-lysine, the polypeptides are α-poly-L-lysine (α-PLL) or α-poly-D-lysine (α-PDL). In the following, four methods for the spectrophotometrical determination of α-PL are described. If the stereochemistry of α-PL is not specified it may apply to the L- and/or the D-form. Since no chiral reagents were used, α-PLL and α-PDL cannot be distinguished by the four methods and the results are, within the limits of error, the same. However, the focus will be on α-PDL because of possible proteolytic degradation of α-PLL under non-sterile working conditions. Therefore, the data shown here refer to α-PDL hydrobromide (Mw = 15000–30000 g mol−1).
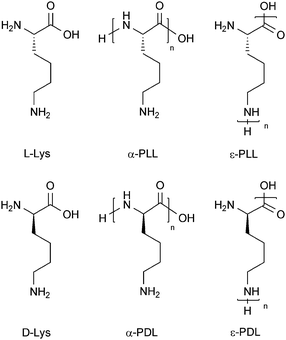 | ||
| Scheme 1 Structural formulae of L-lysine (L-Lys), D-lysine (D-Lys), α-poly(L-Lys) (α-PLL), α-poly(D-Lys) (α-PDL), ε-poly(L-Lys) (ε-PLL), and ε-poly(D-Lys) (ε-PDL). | ||
Furthermore, the conformation of α-PL in aqueous solution varies depending on the experimental conditions such as pH value (which determines the degree of ε-amino group protonation), temperature, α-PL and salt concentration.2,3 If all amino groups are in their neutral form (not protonated), α-PL may form an α-helix (e.g. at pH 11 and 25 °C) or an anti-parallel β-sheet (e.g. at pH 11, if an α-PL solution is heated for 15 min at 52 °C and then cooled down again to 22 °C).3 In the case of α-helix formation at alkaline pH, the helix is right-handed for α-PLL and left-handed for α-PDL. Upon protonation of the ε-amino groups, α-PL turns into a polycationic polymer which does not adopt a secondary structure, i.e. random coils form (e.g. at pH 5.7 and 25 °C).3
Applications of α-PL include (i) its use as a model system for protein aggregation studies: basic studies on the α-helix → β-sheet transition,2–4 and fiber-formation with amyloid-like morphology;5,6 (ii) the coating of glass surfaces for the non-covalent adhesion of cells;7 (iii) its use for the PLL-induced condensation of DNA for designing and developing DNA complexes used for gene therapy;8 (iv) its use as polycationic polypeptide in combination with polyanionic polypeptides for the preparation of alternating polymeric multilayers;9 (v) the preparation of α-PL–drug and α-PL–protein conjugates for increased cellular uptake of drugs and proteins;10,11 and (vi) the preparation of cross-linked enzyme-α-PL films to achieve superior enzyme stability.12
Due to its wide application range there is a necessity for methods for the determination of unknown α-PL concentrations in aqueous solutions. This is especially the case after manipulation, modification reactions, separation and purification steps e.g. after partial modification of the amino groups or after size exclusion chromatography or ultrafiltration. A gravimetric analysis is then highly inaccurate. Spectrophotometric methods are more suitable. However, since α-PL itself does not absorb light in the UV and visible region it cannot be quantified directly with UV/Vis spectroscopy. Nevertheless, the concentration of α-PL in aqueous solutions can be determined indirectly with different spectrophotometric methods; we compared four of them. Thereby, different reactivities of α-PL are utilized to form products which allow a spectrophotometric quantification. Under the experimental conditions used the minimal concentration in an aqueous solution – which we call “analyte solution” – is 1–2 µg mL−1 α-PDL corresponding to 4.8–9.6 µmol L−1 lysine repeating units. The lower limit of quantitation very much depends on the type of method and on the actual experimental conditions used (e.g. reagent concentration and reaction time).
The four methods considered here include the trypan blue (TB) assay, the 2,4,6-trinitrobenzene sulfonate (TNBS) assay, the ortho-phthalaldehyde (OPA) assay and the bicinchoninic acid (BCA) assay. We have optimized the methods with respect to the minimum level of quantification of α-polylysine and to minimize the extent of unwanted side reactions. For each method mechanistic aspects are discussed and detailed experimental procedures are provided.
Experimental
Materials
α-Poly-D-lysine hydrobromide (α-PDL, M(monomer, Lys·HBr, C6H13N2OBr) = 209.09 g mol−1, Mw = 15000–30000 g mol−1, calculated average polymerization degree = 108) was purchased from Sigma-Aldrich. Trypan blue was obtained from Acros Organics and 2,4,6-trinitrobenzene sulfonate as a 5 w/v% solution in methanol from Thermo Fisher Scientific, Pierce Biotechnology. All other chemicals, including o-phthalaldehyde (OPA, IUPAC name phthalaldehyde), 2-mercaptoethanol, BCA assay kit, and buffer salts were purchased from Sigma or Fluka in BioChemika or BioUltra quality (purity ≥99.0%). For all experiments ultrapure water (filtered with a Synergy water preparation apparatus from Millipore) was used; all solutions were prepared in plastic reaction vessels since α-PL adheres to glass surfaces. The solutions were stored at 4 °C after preparation. The UV/Vis measurements were carried out on a Lambda 20 double beam UV/Vis-spectrophotometer from Perkin-Elmer.
Experimental procedures
All measurements were carried out with α-poly-D-lysine hydrobromide (Mw = 15000–30000 g mol−1). If α-PL of other molar mass ranges are analyzed appropriate calibrations are needed. Because of the eventual removal of the counter ion upon manipulations of the polylysine solution, it is useful to use either molar concentration of polylysine or molar concentration of lysine repeating units instead of mass concentration of polylysine. Furthermore, we define the analyte solution as the solution which contains the polylysine in the appropriate measuring concentration but without the reagents needed for the assay. In turn, the sample solution contains polylysine and the reagents. In general, the absorbance of the reference cell (Aref) refers to a solution containing all reagents but replacing the α-PDL solution by the appropriate buffer solution. All samples were prepared at least three times. Additionally, it is important that the buffer of the polylysine containing solution to be analyzed does not contain any substances with free amines since they interfere with the detection. Therefore, quantification of α-PL in complex biological matrices with the four methods shown here is not possible.
TB assay
First, a α-PDL stock solution at a concentration of 2 mg α-poly-D-lysine hydrobromide in 1 mL MES buffer (0.1 M MES, 0.15 M NaCl) pH 4.7 was prepared. The analyte solutions which contained a final MES concentration of 0.01 M and a final NaCl concentration of 0.015 M were prepared by diluting the stock solution. The final polylysine concentrations were between 1 and 9 µg mL−1 α-poly-D-lysine hydrobromide or 4.8 to 43.2 µmol L−1 lysine. The working reagent was a 1 mg mL−1 trypan blue solution in MES buffer (0.01 M MES, 0.015 M NaCl) pH 4.7 which was stable for at least 6 months. For the Standard TB assay, to 1250 µL of the analyte solution 50 µL working reagent solution were added to get a final sample solution volume of 1.3 mL. The mixture was incubated at 37 °C for 1 h, cooled to room temperature and centrifuged at 8000 rpm for 20 min to sediment the precipitate. The supernatant was pipetted off carefully. The absorption spectrum of the supernatant was measured against MES buffer (0.01 M MES, 0.015 M NaCl) pH 4.7 between 200 nm and 800 nm using a 1.5 mL quartz cell with a path length of 1 cm. The absorbance at λmax = 580 nm was plotted as a function of the α-PDL concentration. For the Micro TB assay, 125 µL of analyte solution and 5 µL working reagent solution were mixed. The final volume of the sample solution was 130 µL. After following the procedure for the standard TB assay, the absorption spectrum of the supernatant was measured in a 100 µL, 1 cm cell (see ESI†).TNBS assay
A 2 mg mL−1 α-PDL stock solution was first prepared in MES buffer (0.1 M MES, 0.15 M NaCl) pH 4.7. The analyte solutions were prepared by diluting the stock solution with MES buffer (0.1 M MES, 0.15 M NaCl) pH 4.7 and contained between 10 and 60 µg mL−1 α-PDL hydrobromide corresponding to 48–288 µmol L−1 lysine. The working reagent, a 0.025 w/v% 2,4,6-trinitrobenzene sulfonic acid (TNBS) solution, was prepared by diluting a 5 w/v% TNBS solution in methanol with borate buffer (0.1 M borate) pH 8.5. This solution was freshly prepared every day. To 500 µL analyte solution 250 µL working reagent were added. Then, the pH was adjusted to pH 9.5 by adding 50 µL 1 M NaOH. For that the amount NaOH necessary was determined by mixing the working reagent with buffer not containing polylysine with 1 M NaOH solution to get a pH value of 9.5. Since polylysine adheres to the pH glass electrode the pH of the analyte solution containing polylysine could not be measured directly. The reaction mixture was incubated at 37 °C for 30 min and cooled to room temperature. Then, 250 µL 10 w/v% sodium dodecyl sulfate (SDS) in deionized water and 125 µL 1 M HCl were added to get the sample solution. The mixture was shaken carefully to avoid foaming. The absorption spectrum was measured against borate buffer (0.1 M borate) pH 8.5 between 250 nm and 600 nm using a 1.5 mL, 1 cm quartz cell. The absorbance at λmax = 344 nm was plotted as a function of the α-PDL concentration (see ESI†).OPA assay
A 2 mg mL−1 α-PDL stock solution in phosphate buffer saline (0.1 M sodium phosphate, 0.15 M NaCl) pH 7.2 was prepared. The analyte solution contained between 20 and 670 µg mL−1 α-PDL hydrobromide corresponding to 96–3216 µmol L−1 lysine, and was prepared by diluting the stock solution with phosphate buffer saline (0.1 M sodium phosphate, 0.15 M NaCl) pH 7.2. The working reagent was freshly prepared every day by mixing 1 mL 40 mg mL−1 OPA (o-phthalaldehyde) in ethanol, 25 mL borate buffer (0.4 M borate) pH 9.5, 12.5 mL 20 w/w% sodium dodecyl sulfate (SDS) in deionized water, and 0.1 mL 2-mercaptoethanol in a volumetric flask and filling it up to a volume of 50 mL. The sample solution was directly prepared in a 1.5 mL, 1 cm quartz cell by gentle mixing 25 µL analyte solution with 1 mL working reagent. Immediately after mixing the absorbance at 337 nm was recorded for 300 s against the working reagent. The absorbance measured at λmax = 337 nm after 300 s was plotted as a function of the α-PDL concentration (see ESI†).BCA assay
First, an α-PDL stock solution at a concentration of 2 mg α-poly-D-lysine hydrobromide in 1 mL MES buffer (0.1 M MES, 0.15 M NaCl) pH 4.7 was prepared. The analyte solution contained between 200 and 1000 µg mL−1 α-PDL hydrobromide corresponding to 0.96–4.8 mmol L−1 peptide bonds and was prepared by diluting the stock solution with MES buffer (0.1 M MES, 0.15 M NaCl) pH 4.7. The working reagent for the BCA assay was freshly prepared every day by mixing the bicinchoninic acid solution (composition listed in ESI†) from the BCA assay kit with the 4 w/v% copper(II) sulfate solution. The ratio of BCA solution to copper(II) solution was 50 to 1. The mixture was shaken until the solution was green and transparent. Then, to 60 µL of analyte solution were added 1.2 mL working reagent. The sample solution was incubated at room temperature overnight (∼12 h) and the absorption spectrum was measured between 450 nm and 750 nm using a 1.5 mL, 1 cm quartz cell against working reagent. The absorbance at λmax = 562 nm was plotted against the α-PDL concentration (see ESI†).Results and discussion
TB assay
Trypan blue (TB) is a large oligoanionic dye containing two azo groups (R–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–R′) and four sulfonate groups (Scheme 2). The dye belongs to a group of trypan dyes known since Paul Ehrlich (1854–1915) which kill trypanosomes, the parasites that cause sleeping sickness.13
N–R′) and four sulfonate groups (Scheme 2). The dye belongs to a group of trypan dyes known since Paul Ehrlich (1854–1915) which kill trypanosomes, the parasites that cause sleeping sickness.13
 | ||
| Scheme 2 Structural formula of trypan blue (TB). | ||
Dilute aqueous solutions of TB are blue with an absorption maximum at λmax = 580 nm with a molar absorption coefficient of ε580 = 2.0 × 104 M−1 cm−1, as estimated from the spectrum given by Shen et al.14 This value is in agreement with our own determination within the limits of error. The use of TB to determine the polylysine concentration in aqueous solutions was first proposed in 1984.14 The negatively charged dye interacts with the polycationic form of α-PL leading to a quantitative precipitation and a coinstantaneous decrease in intensity of the blue color of the supernatant solution14 proportional to the amount of added α-PDL (see Fig. 1(a)). Under the experimental conditions used (see Protocol 1 in ESI†), there is a linear relationship between the absorbance at λmax = 580 nm and the α-PDL concentration in the analyte solution between 1 and 9 µg mL−1 (Fig. 1(b)). The assay was carried out in MES buffer (0.01 M MES, 0.015 M NaCl) pH 4.7. It is also possible to use higher concentrated buffers up to 0.1 M buffer salt and 0.15 M NaCl. The pH range may be from about 5 to 7.
 | ||
| Fig. 1 Quantification of α-PDL with TB. (a) Absorption spectra of supernatants of aqueous TB solutions (40 µM in the cuvette) with different concentrations of α-PDL hydrobromide: 0, 1, 2, 3, 4, 5, 6, 7, 8, and 9 µg mL−1 in the analyte solution at pH 4.7 measured against 0.01 M MES, 0.015 M NaCl buffer pH 4.7. The absorbance A decreases with increasing α-PDL concentration (for reaction conditions see Protocol 1 in ESI†). (b) Dependence of the absorbance A of the supernatant at λmax = 580 nm on the concentration of (i) α-PDL in the analyte solution: linear regression between 1 and 9 µg·mL−1: r = −0.99892, (ii) lysine residues in α-PDL in the analyte solution: linear regression between 4.8 and 43.2 µmol L−1 and (iii) lysine residues in α-PDL in the sample solution: linear regression between 4.6 and 41.6 µmol L−1. | ||
However, due to the increased proportion of charged lysine residues at lower pH values the sensitivity of the assay is increased at lower pH values. On the other hand the pH should not be too low; TB must be negatively charged. Usually, the volume of the solution to be measured was 1.3 mL. Alternatively, the volume may be scaled down to 130 µL. We define the latter procedure to be the Micro TB assay. This method is especially useful if the concentration of α-PDL in the sample is too low for further dilution e.g. after a preparative size exclusion chromatography. A further scaling down was not possible, since the amount of precipitate was too low to sediment with a table centrifuge. The minimum level of quantitation for both volumes was 4.8 µmol L−1 lysine, although the actual amount can be lowered from 6.2 nmol lysine to 0.62 nmol lysine per sample with the Micro TB assay.
As shown previously, the TB assay is limited to α-PL with a molar mass of about 3000 g mol−1 or higher as short chains will not precipitate with TB.14
TNBS assay
TNBS (2,4,6-trinitrobenzene sulfonate, 1 in Scheme 3) is a tetrasubstituted, water-soluble benzene derivative. The free acid has a pKa below 0. TNBS reacts with primary amines in a nucleophilic aromatic substitution reaction (SNAr) yielding trinitrophenyl (TNP)-labeled amino groups (3, Scheme 3). First described by Okuyama and Satake,15 this reaction is generally used for the quantification of lysine-containing proteins.16,17 Therefore it is also applicable to α-PL.17 Depending on the reaction conditions, the reaction mixture does not only contain the TNP-labeled substitution product 3 (Scheme 3, with an absorption maximum around λmax = 344 nm),17 but also to a substantial amount of the intermediate adduct 2 (Scheme 3, Fig. 2(a)). Since Jakob Meisenheimer (1876–1934) was the first to propose structural formulae for these types of molecules,18 the intermediate 2 is a so-called Meisenheimer complex with an absorption maximum around λmax = 420 nm.17 Both maxima at 344 nm and 420 nm can be considered for quantification, but the first is more suitable because of its higher molar absorption coefficient, and therefore, for its higher sensitivity (Fig. 2(a)).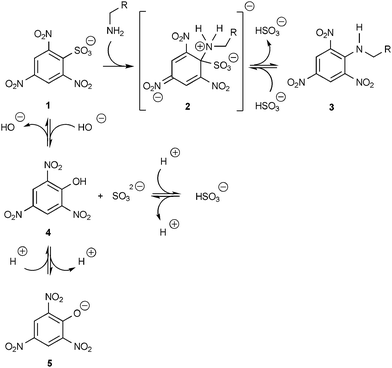 | ||
| Scheme 3 Scheme for the reaction of α-PDL with TNBS 1 involving the intermediate Meisenheimer complex 2 (λmax = 420 nm), the TNP-labeled substitution product 3 (λmax = 344 nm), and the unwanted side product picrate 5 (λmax = 355 nm). | ||
 | ||
| Fig. 2 Quantification of α-PDL with TNBS. (a) Absorption spectra of α-PDL reacted with TNBS at different concentrations of α-PDL hydrobromide in the analyte solution: 0, 10, 20, 30, 40, 50, and 60 µg mL−1 measured against buffer mixture 0.1 M MES saline buffer pH 4.7 and 0.1 M borate buffer pH 8.5 of 2 to 1 (v/v). The absorbance A increases with increasing α-PDL concentration (for reaction conditions see Protocol 2 in ESI†). (b) Dependence of the difference in absorbance A–Aref at λmax = 344 nm on the concentration of (i) α-PDL in the analyte solution: linear regression between 10 and 60 µg mL−1: r = 0.99988, (ii) lysine residues in α-PDL in the analyte solution: linear regression between 48 and 288 µmol L−1 and (iii) lysine residues in α-PDL in the sample solution: linear regression between 20.4 and 122.6 µmol L−1. | ||
Additionally, picric acid (2,4,6-trinitrophenol, 4 in Scheme 3) can be formed in an unwanted side reaction in which TNBS is attacked by hydroxide ions.17 Most of the picric acid will be in its deprotonated form 5 under the experimental conditions, since the pKa of picric acid is below 0. Aqueous solutions of the picrate ion 5 have an absorption maximum at 355 nm (ε355 = 1.44 × 104 M−1 cm−1) with a shoulder at ∼430 nm (ε430 = 5.24 × 103 M−1 cm−1), as estimated from the spectrum given by Ives and Moseley.19
Since the TNBS attacking amino groups have to be nucleophilic the reaction has to be carried out at a pH value at which at least some of the amino groups are present in their neutral form. Thus, the optimal pH value is such that the amino groups are nucleophilic and the picrate formation is reduced to a minimum. By systematic studies we found out that pH ≈ 9.5 is optimal under these contradicting boundary conditions.
The spectrum obtained in the absence of α-PDL (Fig. 2(a), bottom line) allows a rough estimation of the upper concentration limit of picrate formation. Since TNBS itself does not absorb at 430 nm,17 the concentration of picrate can be estimated directly from the bottom curve in Fig. 2(a). Hence, 5.3 mol% of the initially added TNBS underwent the unwanted side reaction.
Under the assumption that the picrate formation is unaffected by the presence of α-PDL, the amount of picric acid is the same in both the absence and the presence of α-PDL. Therefore, one can subtract the bottom line in Fig. 2(a) (reference measurement) from the spectra obtained in the presence of α-PDL. This assumption is feasible for the calibration as seen in Fig. 2(b) since the intercept of the ordinate of the linear regression is practically zero.
Moreover, as one can see in Fig. 2(a), the maximum of the TNP-labeled product shifts with increasing α-PDL concentration to higher wavelengths (red shift). As mentioned, the reaction of TNBS with amino groups yields two species: (i) the TNP-lated amino group (observed λmax = 344 nm) and (ii) the Meisenheimer-complex (observed λmax = 420 nm). With increasing α-PDL concentration the ratio of TNP-lated product to Meisenheimer-complex decreases, more Meisenheimer-complex is formed. Consequently because of overlapping bands, the contribution of the absorption of the Meisenheimer-complex to the contribution of the absorption of the TNP-lated product at 344 nm increases as well, which in turn results in the red shift.
Furthermore, the temperature is also an important factor. Since the reaction rate of picrate formation increases significantly at elevated temperatures,17 the reaction was carried out at room temperature.
Depending on the reaction conditions, the extent of amino group modification and side reaction may vary.17 In the case of α-PL we found indications that not all ε-amino groups can be modified which is most likely due to increasing steric hindrance after partial modification of the amino groups.
After the reaction it is necessary to add firstly hydrochloric acid to stop the reaction and secondly sodium dodecyl sulfate (SDS) to increase the solubility of the TNP-lated product. A systematic investigation yields a linear relationship between the measured absorbance at λmax = 344 nm and α-PDL concentration in the analyte solution between 10 and 60 µg mL−1 α-PDL hydrobromide or 48 to 288 µmol L−1 lysine, respectively (see Fig. 2(b)). The lower value of quantification was 48 µmol L−1 lysine corresponding to 24 nmol lysine in the analyte solution.
The TNBS assay can also be used for α-PL with other molar mass ranges than used here. Separate calibrations, however, are needed.
OPA assay
The quantification of amino groups with the OPA (o-phthalaldehyde, IUPAC name phthalaldehyde) assay was first described by Roth.20 It is normally used for the determination of amino acids and for the pre- or post-column derivatization in chromatographic separations of amino acids.21 The assay, which is also applicable to α-PL, is based on the following observations. The reaction of OPA 6 with 2-mercaptoethanol 7 leads to an intermediate which reacts with a primary amino group to yield the highly fluorescent isoindole derivative 8 (Scheme 4) with an absorption maximum at λmax = 337 nm (see Fig. 3(a)) and a molar absorption coefficient of ε337 = 6.0 × 103 M−1 cm−1.22–24 | ||
| Scheme 4 Scheme for the reaction of OPA 6 with 2-mercaptoethanol 7 and α-PDL to form a 1,2-disubstituted isoindol derivative 8. | ||
![Quantification of α-PDL with o-phthalaldehyde/2-mercaptoethanol. (a) Difference in absorbance A–Aref after 40 s reaction time of OPA/2-mercaptoethanol with α-PDL with at pH 9.5 and ∼22 °C, the concentrations in the sample solution were: [OPA] = 5.8 mM, [2-mercaptoethanol] = 27.8 mM, [SDS] = 173 mM, and [α-PDL] = 16 µg mL−1. (b) Increase in absorbance A at λmax = 337 nm as a function of reaction time for seven different α-PDL hydrobromide concentrations in the analyte solution: 0, 20, 85, 150, 280, 410, 540, and 670 µg mL−1. The absorbance A increases with increasing α-PDL concentration (for reaction conditions see Protocol 3 in ESI). (c) Dependence of the absorbance A at λmax = 337 nm on the concentration of (i) α-PDL in the analyte solution: linear regression between 20 and 670 µg mL−1, r = 0.99946, (ii) lysine residues in α-PDL in the analyte solution: linear regression between 96 and 3216 µmol L−1 and (iii) lysine residues in α-PDL in the sample solution: linear regression between 2.3 and 78 µmol L−1.](/image/article/2010/AY/c0ay00116c/c0ay00116c-f3.gif) | ||
| Fig. 3 Quantification of α-PDL with o-phthalaldehyde/2-mercaptoethanol. (a) Difference in absorbance A–Aref after 40 s reaction time of OPA/2-mercaptoethanol with α-PDL with at pH 9.5 and ∼22 °C, the concentrations in the sample solution were: [OPA] = 5.8 mM, [2-mercaptoethanol] = 27.8 mM, [SDS] = 173 mM, and [α-PDL] = 16 µg mL−1. (b) Increase in absorbance A at λmax = 337 nm as a function of reaction time for seven different α-PDL hydrobromide concentrations in the analyte solution: 0, 20, 85, 150, 280, 410, 540, and 670 µg mL−1. The absorbance A increases with increasing α-PDL concentration (for reaction conditions see Protocol 3 in ESI†). (c) Dependence of the absorbance A at λmax = 337 nm on the concentration of (i) α-PDL in the analyte solution: linear regression between 20 and 670 µg mL−1, r = 0.99946, (ii) lysine residues in α-PDL in the analyte solution: linear regression between 96 and 3216 µmol L−1 and (iii) lysine residues in α-PDL in the sample solution: linear regression between 2.3 and 78 µmol L−1. | ||
However, 8 has a limited stability and undergoes further conversion.24 Therefore, the optimal reaction conditions were elaborated empirically where 8 (i) is obtained within minutes and (ii) is stable for at least 20–30 minutes,24i.e. further conversion of 8 should not occur within the measuring time. Although the reaction is so widely applied,23–25 the entire reaction mechanism for the formation of 8 from 6, 7 and a primary amino group (see Scheme 4) remains unclear.23,24
The used procedure was adapted from Dinella et al.26 Since neither α-PDL nor OPA absorb at 337 nm, the formation of 8 can be directly followed by monitoring the increase in absorbance at 337 nm as a function of time. Under the conditions used a constant value is obtained after about 300 s (Fig. 3(b)). The absorbance at λmax = 337 nm measured after 300 s is linearly dependent on the amount of polylysine present in the analyte solution between 20 and 670 µg mL−1 α-PDL hydrobromide or 96 to 3216 µmol L−1 lysine (Fig. 3(c)). As in the case of the TNBS assay not all amino groups may be modified in the OPA assay because of increasing steric hindrance after partial modification. The lower level of quantitation here was 96 µmol L−1 lysine corresponding to 2.4 nmol lysine in the analyte solution.
Lower or higher molar mass ranges of α-PL may be used as well. However, appropriate calibrations are necessary.
BCA assay
Since α-PL is a polypeptide it can be quantified by a method based on the reactivity of the peptide bonds, such as the assay with bicinchoninic acid (BCA) and Cu2+ developed by Pierce Chemical Company for determining the concentration of proteins in solution.27 The assay is based on two facts. Firstly, peptide bonds as well as the side chains of cysteine, tryptophan and tyrosine, and the disulfide bonds of cystine in proteins are capable of reducing Cu2+ to Cu+.28 Secondly, bicinchoninic acid, the trivial name of 2,2′-biquinoline-4,4′-dicarboxylic acid (Scheme 5) is a highly sensitive and selective detection reagent for Cu+-ions in alkaline solutions. The basic form of BCA reacts with Cu+ to form a water soluble [CuI(BCA)2]3− complex 9 (see Scheme 5) which has an intense purple color (absorption maximum at λmax = 562 nm, ε562 = 7.7 × 103 M−1 cm−1).27,28![Reactions occurring in the BCA assay: peptide bonds of α-PDL reduce Cu2+ to Cu+ which may complex with the oxidized peptide bonds. However, in the presence of BCA, the [CuI(BCA)2]3− complex 9 is formed.](/image/article/2010/AY/c0ay00116c/c0ay00116c-s5.gif) | ||
| Scheme 5 Reactions occurring in the BCA assay: peptide bonds of α-PDL reduce Cu2+ to Cu+ which may complex with the oxidized peptide bonds. However, in the presence of BCA, the [CuI(BCA)2]3− complex 9 is formed. | ||
Since α-PL does not contain amino acids with potentially reducing side chains but only lysines, the quantification of α-PL is based entirely on the reduction of Cu2+ to Cu+ by the peptide bonds present in the α-PL-backbone (Scheme 5). During the redox reaction some of the amide bonds are oxidized to amide radicals which then directly interact with Cu+ to form a violet-blue complex. This reaction is known as biuret reaction named after a substance called biuret (“bi-urea”, H2NCONHNHCONH2),29 the simplest molecule undergoing this reaction. However, since BCA is present, Cu+ does not complex with oxidized peptide bonds but rather with BCA leading to the [CuI(BCA)2]3− complex 9. The intensity of the color of 9 is dependent on the amount of Cu+ formed which in turn is related to the amount of α-PDL present (Fig. 4(a)).
 | ||
| Fig. 4 Quantification of α-PDL with BCA/Cu2+. (a) Absorption spectra of α-PDL reacted with BCA/Cu2+ at different concentrations of α-PDL in the analyte solution: 0, 200, 400, 600, 800, and 1000 µg mL−1. The absorbance A increases with increasing α-PDL concentration (for reaction conditions see Protocol 4 in ESI†). (b) Dependence of the difference in absorbance A–Aref at λmax = 562 nm on the concentration of (i) α-PDL in the analyte solution: linear regression between 200 and 1000 µg mL−1, r = 0.99655, (ii) lysine residues in the analyte solution: linear regression between 960 and 4800 µmol L−1, and (iii) lysine residues in the sample solution: linear regression between 45 and 227 µmol L−1. | ||
Depending on the experimental conditions not all peptide bonds in α-PL may be oxidized by Cu2+. A calibration with known amounts of α-PDL is therefore needed. The resulting absorbance curve in Fig. 4(b) shows that the absorbance at 562 nm is not linearly dependent on the amount of α-PDL over the tested concentration range (0.2–2.0 mg mL−1). This is in agreement with observations in the literature,27,30 and seems to be the consequence of the complex formation equilibria between BCA with both Cu+ and Cu2+.31 Nevertheless, a linear regression between 0.2 and 1.0 mg mL−1 α-PDL hydrobromide or 0.96 to 4.8 mmol L−1 peptide bonds can be carried out. To decrease the error caused by further color development at shorter reaction times, we measured the samples after incubating them overnight. The minimal level of quantitation was 0.96 mmol L−1 or 58 nmol peptide bonds in the analyte solution.
Conclusions
We compared four methods for the UV/Vis spectrophotometric quantification of α-PDL in dilute aqueous solutions. The general principles of the methods are summarized and detailed protocols for a successful quantification of α-PDL are given in the Experimental section and in the ESI†. Each method is based on a particular chemical reactivity of one of the functional groups in this polypeptide; each method has advantages and disadvantages with respect to efforts on time for analysis and material costs.The TB, the TNBS and the OPA assay are fast and easy to perform, although in the latter case a molecular understanding of the details of the occurring reactions is still missing.23 Due to the complexity of the reactions involved, the BCA assay is the most delicate among the four methods. The dependence of the absorption strength on the concentration of α-PDL in the analyte solution clearly deviates from linearity (Fig. 4(b)) which seems to be a direct consequence of the complexity of the reactions taking place in the assay mixture, involving complex formation equilibria between BCA and both Cu2+ and Cu+,31 and a redox reaction with the peptide bonds which may be partially accessible for the reaction only.
With respect to the minimum level of quantitation the TB and the OPA assay are the best methods. The only difference is that for the OPA assay a small volume of analyte solution with a high concentration is needed. For the TB assay, however, more volume of a lower concentrated analyte solution is necessary.
Acknowledgements
The financial support by the Swiss National Science Foundation (200021-116205) is highly appreciated.References
- B. de Kruijff, A. Rietveld, N. Telders and B. Vaandrager, Biochim. Biophys. Acta, 1985, 820, 295–304 CAS.
- B. Davidson and G. D. Fasman, Biochemistry, 1967, 6, 1616–1629 CrossRef CAS.
- N. Greenfield and G. D. Fasman, Biochemistry, 1969, 8, 4108–4116 CrossRef CAS.
- M. Jackson, P. I. Haris and D. Chapman, Biochim. Biophys. Acta, 1989, 998, 75–79 CAS.
- M. Fändrich and C. M. Dobson, EMBO J., 2002, 21, 5682–5690 CrossRef.
- W. Dzwolak, R. Ravindra, C. Nicolini, R. Jansen and R. Winter, J. Am. Chem. Soc., 2004, 126, 3762–3768 CrossRef CAS.
- D. Mazia, G. Schatten and W. Sale, J. Cell Biol., 1975, 66, 198–200 CAS.
- A. Mann, R. Richa and M. Ganguli, J. Controlled Release, 2008, 125, 252–262 CrossRef CAS.
- K. Itoh, S. Tokumi, T. Kimura and A. Nagase, Langmuir, 2008, 24, 13426–13433 CrossRef CAS.
- H. J.-R. Ryser and W.-C. Shen, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 3867–3870 CAS.
- W.-C. Shen and H. J.-P. Ryser, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 1872–1876.
- P. M. Guto, C. V. Kumar and J. F. Rusling, J. Phys. Chem. B, 2007, 111, 9125–9131 CrossRef CAS.
- C.-R. Prüll, Med. Hist., 2003, 47, 332–356 Search PubMed.
- W.-C. Shen, D. Yang and H. J.-P. Ryser, Anal. Biochem., 1984, 142, 521–524 CAS.
- T. Okuyama and K. Satake, J. Biochem., 1960, 47, 454–466 CAS.
- A. F. S. A. Habeeb, Anal. Biochem., 1966, 14, 328–336.
- P. Cayot and G. Tainturier, Anal. Biochem., 1997, 249, 184–200 CrossRef CAS.
- Meisenheimer, Justus Liebigs Ann. Chem., 1902, 323, 205–247 CrossRef CAS.
- D. J. G. Ives and P. G. N. Moseley, J. Chem. Soc. B, 1966, 757–761 RSC.
- M. Roth, Anal. Chem., 1971, 43, 880–882 CrossRef CAS.
- J. R. Cronin, S. Pizzarello and W. E. Gandy, Anal. Biochem., 1979, 93, 174–179 CrossRef CAS.
- S. S. Simons, Jr and D. F. Johnson, J. Am. Chem. Soc., 1976, 98, 7098–7099 CrossRef CAS.
- P. Zuman, Chem. Rev., 2004, 104, 3217–3238 CrossRef CAS.
- P. Zuman, Anal. Lett., 2005, 38, 1213–1220 CrossRef CAS.
- K. S. Lee and D. G. Drescher, Int. J. Biochem., 1978, 9, 457–467 CrossRef CAS.
- C. Dinella, M. T. Gargaro, R. Rossano and E. Monteleone, Food Chem., 2002, 78, 363–368 CrossRef CAS.
- P. K. Smith, R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson and D. C. Klenk, Anal. Biochem., 1985, 150, 76–85 CrossRef CAS.
- A. J. Brenner and E. D. Harris, Anal. Biochem., 1995, 226, 80–84 CrossRef CAS.
- H. C. Freeman, J. E. W. L. Smith and J. C. Taylor, Nature, 1959, 184, 707–710 CrossRef CAS.
- K. J. Wiechelman, R. D. Braun and J. D. Fitzpatrick, Anal. Biochem., 1988, 175, 231–237 CrossRef CAS.
- R. D. Braun, K. J. Wiechelman and A. A. Gallo, Anal. Chim. Acta, 1989, 221, 223–238 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Composition of the BCA assay kit and detailed protocols for all assays. See DOI: 10.1039/c0ay00116c |
| This journal is © The Royal Society of Chemistry 2010 |