Electrocatalytic oxidation and nanomolar detection of hydrazine by luteolin electrodeposited at a multi-walled carbon nanotube and ionic liquid composite modified screen printed carbon electrode
Shao-Hua
Wu
,
Fa-Hui
Nie
,
Qi-Zhen
Chen
and
Jian-Jun
Sun
*
Ministry of Education Key Laboratory of Analysis and Detection for Food Safety, College of Chemistry and Chemical Engineering, Fuzhou University, Fuzhou, Fujian 350002, China. E-mail: jjsun@fzu.edu.cn; Fax: +86 591 22866136; Tel: +86 591 22866136
First published on 27th September 2010
Abstract
A new electrode was fabricated by electrodepositing luteolin at a screen printed carbon electrode (SPCE) modified with a composite of multi-walled carbon nanotube (MWNT) and ionic liquid (IL). Cyclic voltammograms of the modified electrode in phosphate buffer solution showed a pair of stable and reversible redox couple of luteolin with surface confined characteristics. This electrode possessed excellent electrocatalytic ability towards hydrazine oxidation. The overpotential was decreased significantly and the peak current was increased dramatically compared to those at bare SPCE. This enhancement of the responses was mainly contributed to the combination of the unique electrocatalytic and electronic properties of luteolin, MWNT and IL. The modified electrode was employed to the amperometric detection of hydrazine at an applied potential of 0.31 V vs. SCE with fast response, high sensitivity, good stability and reproducibility. Two linear ranges of hydrazine were from 2.0 × 10−8 to 2.0 × 10−7 M and from 2.0 × 10−7 to 1.2 × 10−4 M. Nanomolar detection limit of 6.6 × 10−9 M (S/N = 3) could be obtained, which is at least one magnitude lower than any others with electrochemical detection of hydrazine. The proposed method was also used to determine hydrazine residues in spiked drinking water and river water with average recoveries of 101.7% and 101.1%, respectively.
1. Introduction
Luteolin (see Scheme 1), a crucial member of flavonoids, widely occurs in vegetables, fruits, seeds, and medicinal herbs.1 Its biological and physiological activities have been described as vasodilation, antioxidation, inhibiting lipid peroxidations and DNA oxidative damage, anticarcinogenic and antimutagenic effects.2–4 Therefore, investigating the redox process and electrocatalytic capability of luteolin is of great importance. Its electrochemical properties had been investigated at a glassy carbon electrode.5 But there was no report about the electrocatalytic activity of luteolin. | ||
| Scheme 1 Structure of luteolin | ||
Carbon nanotubes (CNT), which were invented in 1991,6 exhibit large specific surface area, high electrical conductivity, chemical stability and significant mechanical strength.7 Because of the subtle electronic properties, they can be used as electrode materials to promote electron transfer reactions.8–11 Furthermore, their huge specific surface area and graphene-sheet structure endow them excellent adsorption abilities and can be applied as support for immobilization electron transfer mediators onto electrode surfaces12–16 such as luteolin in this study.
Ionic liquids (IL) are compounds consisting entirely of ions that exist in the liquid state around room temperature. They have attracted great attention in recent years because of their specific characteristics including high chemical and thermal stability, negligible vapor pressure, high viscosity and solubility, good ionic conductivity and wide potential windows.17,18 So far, IL have been applied in many fields including organic synthesis,19 catalysis,20 extraction,21 and material science.22 In the field of electrochemistry and electroanalysis, IL can be used as media without addition of an external supporting electrolyte due to their high ionic conductivity.23 On the basis of their high viscosity and solubility, IL have also been used as a binder to fabricate high-performance carbon paste electrodes24–26 and as a material to construct composite films to modify the traditional electrodes27–30 for the direct electrochemistry of redox proteins and the detection of various low molecular compounds. Especially, the composite of CNT and IL (CNT-IL) has received much attention and was used to modify graphite,30 glassy carton28,29 and Au28 electrodes. But there is no report, on the modification of CNT-IL composite on a disposable screen printed carbon electrode (SPCE).
Hydrazine and its derivatives have found wide spread usage in rocket fuels, missile systems, weapons of mass destruction and fuel cells.31 They are also widely used as catalyst, corrosion inhibition, emulsifier, antioxidant, oxygen scavenger in boiler, dye stuffs, insecticide and plant-growth regulators, etc.32 Despite its wide application, hydrazine has been recognized as a carcinogenic and hepatotoxic substance.33 Therefore, the determination of hydrazine is of industrial and pharmacological significance. Different methods for determination of hydrazine have been described in the literature, including spectrophotometric,34 coulometric,35 titrimetric,36 potentiometric,37 fluorimetric,38 chemiluminescence,39 and flow-injection electrogenerated chemiluminescence40 methods.
For electrochemical detection of hydrazine, because of the large overpotential of hydrazine at conventional electrodes, various substances have been adopted to fabricate chemically modified electrodes to enhance the electron transfer rate for the oxidation of hydrazine, including metal nanoparticle,41 metal oxide,42 hexacyanoferrate salt,43 phthalocyanine complex,44 overoxidized polypyrrole,45 and organic mediator.33,46–49 Especially, due to the high conductivity and adsorption capability, carbon nanotube has been used as a good material to immobilize various mediators such as curcumin,50 indenedione derivative,51 catechol derivatives,52 and hematoxylin16 for electrocatalytic oxidation of hydrazine.
There is a catechol structure in the molecular structure of luteolin (Scheme 1), so it was anticipated to have the normal reversible quinone/hydroquinone electrochemistry and good electrocatalytic activity. Inspired by this, in this paper, combining the advantages of luteolin, multi-walled carbon nanotubes (MWNT) and ionic liquids, a new luteolin, MWNT and IL modified SPCE (Lu/MWNT-IL/SPCE) was fabricated by eletrodepositing luteolin on the surface of MWNT and IL composite (MWNT-IL) modified SPCE. The electrochemical characteristics of the modified electrode were investigated in detail, and the strong electrocatalytic ability for hydrazine oxidation was also demonstrated. Under optimum conditions the Lu/MWNT-IL/SPCE was then used to determine trace amount of hydrazine with amperometry and nanomolar detection limit as the lowest value for electrochemical determination of hydrazine was obtained. This method was also successfully employed to analyze real water samples. Finally, the usage of SPCE provides great potential for mass production, low cost, miniaturization and compatibility with on-site monitoring towards hydrazine.
2. Experimental
2.1. Chemicals
An IL of 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide (BMIM·NTf2, 99%) was purchased from Chengjie Chemical Co., Ltd. Shanghai, China. Carbon ink (No. CH-1) was obtained from Jujo Chemical Co., Ltd. Tokyo, Japan. Nafion (5% ethanol solution) was purchased from Sigma-Aldrich. Luteolin was obtained from the National Institute for Control of Pharmaceutical & Biological Products, Beijing, China. Hydrazine hydrate (85%) and the other chemicals were of analytical grade and received from Sinopharm Chemical Reagent Co., Ltd. All above mentioned chemicals were used without further purification. Stock solution (3.49 × 10−3 M) of luteolin was prepared with ethanol absolute and stored in the dark at 4 °C. Other solutions were prepared with deionized water (Millipore Milli-Q system, USA). Multi-wall carbon nanotubes were obtained from Shenzhen Nanotech Port Co., Ltd., Shenzhen, China. Prior to use, MWNT were refluxed in concentrated nitric acid, filtered and washed with deionized water, and then dried under vacuum.2.2. Apparatus
The cyclic voltammetric (CV) and amperometric measurements were performed with an electrochemical analyzer (CHI440, Chenhua, China). The electrochemical impedance spectroscopy measurements were performed with a multi-channel electrochemical workstation (PARC, USA). An electrochemical cell with conventional three-electrode system was used, an unmodified or modified SPCE acted as working electrode, a platinum foil as counter electrode and a saturated calomel electrode (SCE, Chenhua, China) as reference electrode. A 0.2 M phosphate buffer solution (PBS, pH 7.0) was used unless otherwise mentioned. Field emission scanning electron microscopy (FSEM) measurements were carried out on a Nano SEM 232 scanning electron microscope (American FEI company).2.3. Preparation of SPCEs
Preparation of SPCEs was based on a screen printing technique.53 In brief, one piece of printed circuit board with single-faced copper foil was masked and etched to leave one conducting strip. At one end of the board, carbon ink was manually printed three times with 4 h drying period at 65 °C in between onto the copper foil as the working electrode (3 mm × 3 mm). The other end was soldered with a copper wire serving as the electrical contact. The copper wire and solders were covered by epoxy resin for insulation.2.4. Fabrication of the modified SPCEs
Fig. 1 shows the scheme of different electrode configuration. Prior to modification, the SPCEs were subjected to cyclic scanning in a saturated Na2CO3 solution between 0.9 and 1.5 V for 12 cycles with scan rate 10 mV s−1 for activation. The viscous MWNT-IL composite was formed by thoroughly grinding the mixture of 8 mg MWNT and 44 μL IL in an agate mortar for 20 min. For MWNT-IL modified SPCE (MWNT-IL/SPCE), MWNT-IL composite was cast on the SPCE by mechanical rubbing with a spatula to get a very thin film. MWNT and Nafion composite (MWNT-Nafion) was formed by sonicating the mixture of MWNT and 0.5% Nafion ethanol solution. For MWNT-Nafion modified SPCE (MWNT-Nafion/SPCE), 6 μL (optimized amount) MWNT-Nafion was cast on the SPCE. For all luteolin modified electrodes (Lu/SPCE, Lu/MWNT-IL/SPCE and Lu/MWNT-Nafion/SPCE), luteolin was modified by continuous CV scanning 20 cycles between 0 and 0.6 V with 20 mV s−1 in 0.2 M PBS (pH 7.0) containing 1 × 10−5 M luteolin.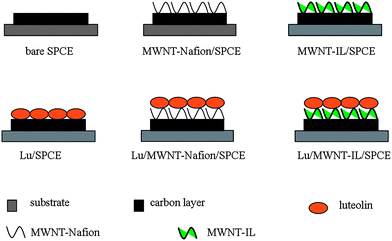 | ||
| Fig. 1 Scheme of different electrode configuration. | ||
3. Results and discussion
3.1. FSEM characterization of MWNT-IL/SPCE
The morphology of MWNT-IL/SPCE was displayed in Fig. 2 by FSEM. The crosslinked and uniformly distributed MWNTs covering the surface of SPCE was clearly observed, indicating successful modification of MWNT-IL composite onto the SPCE due to the high viscosity of IL. | ||
| Fig. 2 FSEM image of MWNT-IL/SPCE. Accelerating voltage: 15 kV. | ||
3.2. Electrochemical impedance spectroscopy of the modified electrodes
Electrochemical impedance spectroscopy (EIS) is an effective method to probe the interfacial electron-transfer resistance at modified electrodes. The semicircle diameter of the impedance spectra at higher frequencies corresponds to the electron-transfer resistance (Ret), and the linear portion at lower frequencies corresponds to the diffusion process.55Fig. 3 shows the EIS at bare SPCE (curve a), MWNT-Nafion/SPCE (curve b) and MWNT-IL/SPCE (curve c). The Randles circuit (Fig. 3 inset) was chosen to fit the obtained impedance data. There is a remarkable semicircle portion in curve a (Ret = 92 Ω), indicating a slow electron-transfer rate at bare SPCE. Compared to curve a, the diameter of the semicircle portion of curve b (Ret = 60 Ω) is noticeably decreased due to the excellent electronic conductivity of the modified material MWNT, which can effectively promote electron transfer reaction. While curve c exhibits a nearly straight line (Ret = 0.01 Ω) at higher frequencies, which means the electron-transfer resistance of the MWNT-IL/SPCE is relative small and even can be ignored. So the presence of ionic liquids (ionic conductivity) instead of Nafion (not conductive) can greatly accelerate the electron transfer. In addition, the double-layer capacitance (Cdl) at MWNT-IL/SPCE was 18 times as that at bare SPCE, which was perhaps mainly resulted from the large specific surface of MWNT.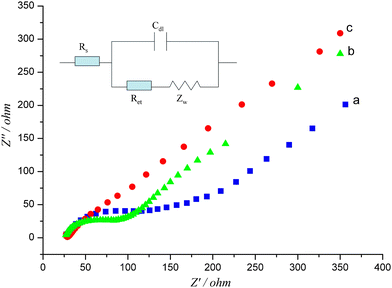 | ||
| Fig. 3 Electrochemical impedance spectra at (a) bare SPCE, (b) MWNT-Nafion/SPCE, (c) MWNT-IL/SPCE in 5 mM Fe(CN)63−/4− + 0.5 M KCl solution. Frequency range: from 100 kHz to 0.01 Hz. Perturbation amplitude: 5 mV. | ||
3.3. Electrochemical behavior of Lu/MWNT-IL/SPCE
The electrodeposition of luteolin onto MWNT-IL/SPCE was clearly observed with cyclic voltammetry. Immersing the electrode into PBS containing 1.0 × 10−5 M luteolin and recording the cyclic voltammograms continuously, a pair of reversible redox wave at potential about + 0.29 V (not shown) was observed, which corresponds to the oxidation of the 3′,4′-dihydroxy substituent on the B-ring of luteolin and the reduction of the oxidation product 3′,4′-diquinone, respectively.54 The peak current was increased and finally reached constant (ca. 20 cycles) with cycle number increasing, suggesting the continuous deposition of luteolin onto the surface of the electrode. The redox wave was still clearly observed after the electrode was taken out from the luteolin solution, rinsed with deionized water and re-cycled in a blank PBS. These facts indicated that luteolin was deposited onto the surface of the electrode and a Lu/MWNT-IL/SPCE was prepared.The electrochemical behavior of Lu/MWNT-IL/SPCE was further examined in 0.2 M PBS (pH 7.0). Cyclic voltammograms of Lu/MWNT-IL/SPCE in various scan rates (v) were shown in Fig. 4. As seen, the peak current was proportional to the scan rate from 40 to 600 mV s−1 (Fig. 4A), which demonstrated a surface confined redox process. The linear equation was ipa (μA) = 28.803ν (V s−1) + 2.9147 with r2 = 0.9916 and ipc (μA) = −31.172ν (V s−1) + 2.0828 with r2 = 0.9960. The surface coverage of luteolin was calculated according to ref. 55 to be ca. 2.96 × 10−10 mol cm−2 from the slope of the anodic peak currents versus scan rates plot (Fig. 4A (a)). This value was much higher than that of Lu/SPCE (1.84 × 10−11 mol cm−2), indicating that the large specific surface area of MWNTs can significantly increase the luteolin modification amount. As shown in Fig. 4B, with the scan rate increasing the anodic peak potential shifts positively, the cathodic peak potential shifts negatively and the peak separation increases, indicating that the redox reaction becomes more and more irreversible. At higher scan rates, the peak potential and logarithm of ν has a linear relationship. The linear equation was Epa (V) = 0.0625 log ν (V s−1) + 0.3346 with r2 = 0.9941 and Epc (V) = −0.0814 log ν (V s−1) + 0.1966 with r2 = 0.9932. Linear regress analysis yielded the charge transfer coefficient α as 0.67 and apparent charge transfer rate constant ks as 0.013 s−1 for luteolin oxidation.56
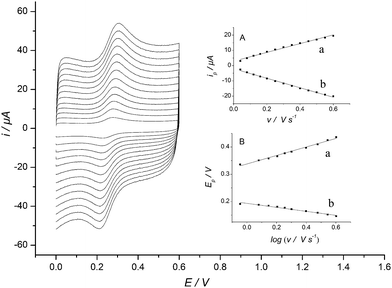 | ||
| Fig. 4 Cyclic voltammograms of Lu/MWNT-IL/SPCE in 0.2 M PBS (pH 7.0) with different scan rates (ν) from inner to outer, 0.04, 0.08, 0.12, 0.16, 0.2, 0.25, 0.3, 0.35, 0.4, 0.45, 0.5, 0.55, and 0.6 V s−1. Insets: (A) plot of anodic (a) and cathodic (b) peak current vs. scan rate; (B) plot of anodic (a) and cathodic (b) peak potential vs. log ν. | ||
From the CV data (not shown) of Lu/MWNT-IL/SPCE obtained in PBS over the pH range of 3.0 to 9.0, the relationship between the anodic peak potential (Epa) of luteolin and pH value was achieved, which can be described as Epa (V) = −0.0547 pH + 0.679 with r2 = 0.991. The slope is close to the theoretical value 0.059 as predicted by the Nernst equation. It can be concluded that the proton number involved is equal to the electron-transfer number in the electrode reaction, consisting with the anticipated two-electron-two-proton oxidation of the catechol 3′,4′-dihydroxyl group in the luteolin molecule to the quinone form.57 The anodic peak currents almost keep unchanged from pH 3.0 to 7.0, however, when pH exceeds 7.0 the anodic peak currents begin to decrease. This may be mainly related to the proton taking part in the electrode reaction. The more basic the solution is, the more difficult the electrochemical reaction becomes due to the shortage of proton. In addition, with high pH luteolin turns to anions and becomes more soluble in aqueous solution, probably leading to the desorption of luteolin from the electrode surface.
The peak-to-peak separation of the anodic and cathodic waves was 46 mV. The anodic Full Width Half Maximum (ΔEpa, 1/2) was 101 mV (see Fig. 5, curve a), close to the theoretically calculated value of ΔEpa, 1/2 = 62.5/[(1 − α)n] = 95 mV (α = 0.67, n = 2).55 This was consistent with a surface confined two electron process.
 | ||
| Fig. 5 Cyclic voltammograms of Lu/MWNT-IL/SPCE (a and b), Lu/MWNT-Nafion/SPCE (c and d), MWNT-IL/SPCE (e and f), Lu/SPCE (g and h) and bare SPCE (i and j) in 0.2 M PBS (pH 7.0) in the absence (a, c, e, g, i) and presence (b, d, f, h, j) of 2.0 × 10−4 M hydrazine. Scan rate: 0.1 V s−1. | ||
3.4. Electrocatalytic properties of Lu/MWNT-IL/SPCE for hydrazine oxidation
The electrocatalytic ability of the Lu/MWNT-IL/SPCE to the oxidation of hydrazine was examined by cyclic voltammetry. Fig.5 shows the cyclic voltammograms at Lu/MWNT-IL/SPCE, Lu/MWNT-Nafion/SPCE, MWNT-IL/SPCE, Lu/SPCE, and bare SPCE in the absence (curve a, c, e, g, i) and presence (b, d, f, h, j) of 2.0 × 10−4 M hydrazine. In the absence of hydrazine, there was no electrochemical response at bare SPCE (curve i). But in the presence of hydrazine, there was a broad, weak and irreversible oxidation peak of hydrazine at 0.75 V (curve j), indicating a sluggish electrochemical reaction process. At MWNT-IL/SPCE, the hydrazine oxidation peak potential shifted negatively to 0.42 V (curve f). This was probably mainly contributed to the subtle electronic conductivity of MWNT and which was beneficial to promoting electron transfer reaction. At the same time, the capacitive current was increased significantly, indicating the electrode active area (Aea) increased. In fact, the Aea of MWNT-IL/SPCE was calculated as 0.8 cm2, about 9 times of its geometrical area.55 At Lu/SPCE, a pair of redox peak of luteolin was observed (curve g) in the absence of hydrazine. However, the anodic peak current (the peak potential as 0.29 V) increased noticeably and the cathodic peak current decreased after addition 2.0 × 10−4 M hydrazine (curve h), which was typically characteristic of an electrocatalytic oxidation process, indicating the electrocatalytic activity of luteolin for hydrazine oxidation. Under the same experimental conditions, the redox peak current of luteolin at Lu/MWNT-IL/SPCE (curve a) was greatly increased compared with that at Lu/SPCE (curve g), but the peak potential was almost unchanged. This was mainly ascribed to the large specific surface area, porosity effects and strong adsorption ability of MWNT, which were favorable to immobilizing more electron transfer mediator luteolin to the electrode surface. In addition, the high viscosity and ionic conductivity of IL enabled it to effectively bind MWNT onto the surface of the SPCE but not to sacrifice the modified electrode's conductivity. Accordingly, the catalytic oxidation peak current (curve b) was also greatly enhanced. By comparison, at Lu/MWNT-Nafion/SPCE, using Nafion (not conductive) instead of IL as a binder, the corresponding response currents were obviously decreased (curve c and d). Thus, the combined action of luteolin, MWNT and IL was responsible for the response enhancement of the Lu/MWNT-IL/SPCE. These results were consistent with that in section 3.2.Fig. 6 displays the typical cyclic voltammetric responses for the electrocatalytic oxidation of hydrazine in successive addition of hydrazine. As shown, with the continuous injection of hydrazine, correspondingly, the anodic peak current increased and the cathodic peak current decreased. The clear increase of anodic peak current and the almost disappearance of cathodic peak current indicated that the anodic catalytic current was mainly obtained from the hydrazine concentration in the solution.46 The catalytic current was linear with the concentration of hydrazine (Fig. 6, inset), the linear equation was i (μA) = 20.80c (mM) + 7.90 with r2 = 0.998, so it can be used for hydrazine measurement.
 | ||
| Fig. 6 Cyclic voltammograms of Lu/MWNT-IL/SPCE in 0.2 M PBS (pH 7.0) with different concentrations of hydrazine from (a) to (f), 0, 0.1, 0.2, 0.4, 0.6, 0.8 mM. Scan rate: 0.1 V s−1. Inset: plot of anodic peak current vs. concentrations of hydrazine. | ||
By recording cyclic voltammograms of 5.0 × 10−5 M hydrazine solution in various scan rates from 0.02 to 0.3 V s−1 at Lu/MWNT-IL/SPCE (data not shown), the electrocatalytic oxidation current of hydrazine was found to be linear to the square root of scan rate. This result indicated that at sufficient overpotential, the electrode reaction was diffusion controlled and related to hydrazine concentration, which was ideal case for quantitative applications.46 According to the data of the rising part of the current–voltage curve at a scan rate of 20 mV s−1, a Tafel plot was obtained (not shown) and the linear equation was log i (μA) = 6.6096E (V) − 0.9876 with R2 = 0.9992. The slope of 6.6096 V−1 indicated that one electron was involved in the rate-determining step assuming a charge transfer coefficient of α = 0.61.16
In addition, the electrocatalytic oxidation of hydrazine at Lu/MWNT-IL/SPCE was investigated in PBS containing 5.0 × 10−5 M hydrazine with different pH values by cyclic voltammetry (Fig. 7). As can be seen from Fig. 7A, the catalytic oxidation peak potential was proportional to the pH value. The linear equation was Epa (V) = 0.058 pH + 0.701 with r2 = 0.997. The slope of 58 mV was very close to Nernstian slope 59 mV, indicating an equal number of electrons and protons involving in the electrode process. While the catalytic current increased with the pH value increasing from 3.0 to 7.0, and reached the maximum value in pH 7.0, then decreased for higher pH value (Fig. 7B). At lower pH values, the hydrazine would be protonated, which might be disadvantage to the electrocatalytic oxidation of hydrazine. Therefore, pH dependence of luteolin stability at the surface of the modified electrode and the pH dependence of electrocatalytic oxidation efficiency of hydrazine were both responsible for the above results. Thus, pH 7.0 was selected as the optimum pH value for the following amperometric determination of hydrazine.
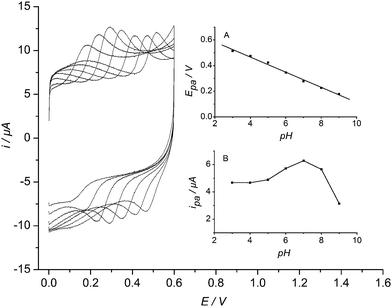 | ||
| Fig. 7 Cyclic voltammograms of Lu/MWNT-IL/SPCE in 0.2 M PBS containing 5.0 × 10−5 M hydrazine with different pH values from left to right, 3, 4, 5, 6, 7, 8, 9, 10. Scan rate: 0.1 V s−1. Insets: (A) plot of catalytic oxidation peak potential vs. pH; (B) plot of catalytic oxidation peak current vs. pH. | ||
3.5. Amperometric detection of hydrazine at Lu/MWNT-IL/SPCE
The Lu/MWNT-IL/SPCE was further evaluated for amperometric detection of hydrazine. Fig. 8 displays the typical steady-state electrocatalytic oxidation current time response of the modified electrode with successive addition of hydrazine into a magnetically stirred 0.2 M PBS (pH 7.0) at an applied potential 0.31 V (optimized). As shown, upon successive injection of hydrazine, a well-defined response was observed. The catalytic current was promptly increased for each addition of hydrazine within a response time less than 2 s. These demonstrated the efficient and stable catalytic ability of the modified electrode. The catalytic current was found to be proportional to hydrazine concentration in the range of 2.0 × 10−8 to 2.0 × 10−7 M, the regression equation was i (μA) = 0.190c (μM) − 0.225 with r2 = 0.995. There was another linear relationship when the hydrazine concentration was changed from 2.0 × 10−7 to 1.2 × 10−4 M, the regression equation was i (μA) = 0.0625c (μM) + 0.258 with r2 = 0.997. Obviously, the slope of the latter was smaller than that of the former, which was mainly ascribed to a change of the electrode surface condition due to the generation of more molecular nitrogen bubbles with higher hydrazine concentration during its catalytic oxidation,47 which affected the diffusion of new hydrazine molecules to the electrode surface. The detection limit was estimated to be nanomolar level of 6.6 × 10−9 M (S/N = 3). In Table 1, some of the response characteristics obtained in this research are compared to those presently reported in ref. 16,33,41–52. Indeed, the proposed modified electrode is dramatically superior with the detection limit and quantitation limit being at least one magnitude lower than any others with electrochemical determination of hydrazine, further verifying the powerful capability of the modified electrode.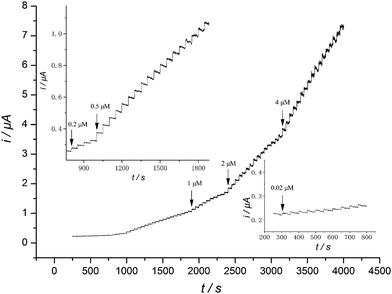 | ||
| Fig. 8 Amperometric response obtained at Lu/MWNT-IL/SPCE in 0.2 M PBS (pH 7.0) during successive addition of hydrazine. Insets: magnification of the former portions of the same plot. Applied potential: 0.31 V. Magnetic stirring speed: 1000 rpm. | ||
| Electrode | Modifier | Electrochemical method | Linear range/μM | Detection limit/μM | Ref. |
|---|---|---|---|---|---|
| titanium | gold nanoparticles | cyclic voltammetry | 5000–40000 | 42 | 41 |
| copper | copper (hydr)oxide | chronoamperometry | 100–1800 | — | 42 |
| carbon ceramic | nickel hexacyanoferrate nanoparticles | cyclic voltammetry | 20–2000 | 8 | 43 |
| gold | iron phthalocyanine complex | square wave voltammetry | 13–92 | 5 | 44 |
| glassy carbon | overoxidized polypyrrole | amperometry | 13–2000 | 3.6 | 45 |
| glassy carbon | quinizarine | differential pulse voltammetry | 0.2–1.0 | 0.14 | 46 |
| 2.0–10.0 | |||||
| glassy carbon | o-aminophenol | amperometry | 2.0–20.0 | 0.5 | 47 |
| glassy carbon | pyrogallol red | linear sweep voltammetry | 5–600 | 2 | 48 |
| glassy carbon | hydroquinone salophen derivatives | cyclic voltammetry | 10–400 | 1.6 | 49 |
| glassy carbon | chlorogenic acid | chronoamperometry | 50–3000 | — | 33 |
| glassy carbon | hematoxylin on MWNT | amperometry | 2–122.8 | 0.68 | 16 |
| glassy carbon | curcumin on MWNT | amperometry | 2–44 | 1.4 | 50 |
| carbon ceramic | indenedione derivative on MWNT | differential pulse voltammetry | 0.6–8.0 | 0.29 | 51 |
| 8.0–100.0 | |||||
| glassy carbon | carbon nanotubes and catechol derivatives | amperometry | 0.50–6.5 | 0.05 | 52 |
| SPCE | luteolin on MWNT-IL composite | amperometry | 0.02–0.2 | 0.0066 | this work |
| 0.2–120 | (6.6 nM) |
3.6. Stability and reproducibility of the Lu/MWNT-IL/SPCE
The stability of the Lu/MWNT-IL/SPCE was first studied by CV in pH 7.0 PBS. The results indicated that the peak current of the modified electrode decreased 0.4% after 50 continuous cycles with scan rate 0.02 V s−1 for the potential from 0 to 0.6 V. Stored in 4 °C refrigerator for one month, the peak current was decreased by only 1%. The modified electrode for amperometric detection of hydrazine was also highly stable during long periods of time towards hydrazine oxidation. By recording the amperometric response of 1.0 × 10−5 M hydrazine over 1800 s period (as shown in Fig. 9), no obvious decrease of electrocatalytic oxidation current was observed, demonstrating no inhibition effect of hydrazine and its oxidation product to the surface of the Lu/MWNT-IL/SPCE. In addition, 13 replicated amperometric measurements for 6.0 × 10−8 M hydrazine yielded a relative standard deviation (R.S.D.) of 1.73%. Thus, the stability and reproducibility of the Lu/MWNT-IL/SPCE were excellent. | ||
| Fig. 9 Amperometric response obtained at Lu/MWNT-IL/SPCE during 1800 s in (a) 0.2 M PBS, (b) 0.2 M PBS containing 1.0 × 10−5 M hydrazine. Other conditions as in Fig. 8. | ||
3.7. Interference study and real water sample analysis
An interference study was performed at Lu/MWNT-IL/SPCE in 1.0 × 10−7 M hydrazine solution in the absence and presence of a specific concentration of interfering substance by amperometry under the optimum conditions. Based on this, the amperometric response current change can be obtained. It is considered that this substance causes obvious interference when the response current change exceeds 5%. The results indicated that 1000-fold of Na+, K+, NH4+, Cl−, Br−, F−, I−, C2O4−, SO4−, CH3COO−, NO3−, glucose, fructose, sucrose and lactose had no interference on hydrazine determination. Adding excess amount of EDTA, 500 fold of Ca2+, Mg2+, Zn2+, Cu2+, Mn2+, Ba2+, Cd2+ and Pb2+ also had no interference. But electroactive substance NH2OH interfered seriously.In order to test the reliability of the modified electrode, it was applied to the determination of hydrazine in drinking water and river water samples. The results are shown in Table 2. As can be seen, the two samples were both free of hydrazine or were contaminated with concentrations below the detection limit. Therefore, a recovery study was undertaken by standard addition method. As shown, the average recoveries are 101.7% for drinking water and 101.1% for river water, respectively. This reveals that the proposed method in this work is accurate and precise and can be used to analyze real water samples.
4. Conclusion
The novel Lu/MWNT-IL/SPCE was manufactured by electrodepositing luteolin at the surface of multi-walled carbon nanotube and ionic liquid composite modified screen printed carbon electrode. The modified electrode exhibited good electrocatalytic activity towards hydrazine oxidation with a remarkable decrease of the overpotential and (or) an increase of the peak current compared with those at bare SPCE, Lu/SPCE, MWNT-IL/SPCE, and Lu/MWNT-Nafion/SPCE. The combination of the electrocatalytic and electronic properties of Lu, MWNT and IL was mainly responsible for this augmentation. This electrode was used for fast and sensitive detection of hydrazine with amperometry. The linear response ranges of hydrazine were from 2.0 × 10−8 to 2.0 × 10−7 M and from 2.0 × 10−7 to 1.2 × 10−4 M. Nanomolar detection limit of 6.6 × 10−9 M (S/N = 3) as the lowest value for electrochemical determination of hydrazine as we know was achieved. Such method was also used to determine hydrazine in drinking water and river water and satisfactory recoveries were obtained. Thus, such modified electrode would be a promising sensor for hydrazine measurement.Acknowledgements
The authors are thankful for the financial support from the National Science Foundation of China (Nos. 20975022, 20775015 and 20735002), National Basic Research Program of China (No. 2010CB732403), Specialized Research Fund for the Doctoral Program of Higher Education (20070386005) from MOE, and NCETFJ (XSJRC2007-02).Notes and references
- K. Shimoi, H. Okada, M. Furugori, T. Goda, S. Takase, M. Suzuki, Y. Hara, H. Yamamoto and N. Kinae, FEBS Lett., 1998, 438, 220 CrossRef CAS.
- J. S. Chang, Y. L. Hsu, P. L. Kuo, Y. C. Kuo, L. C. Chiang and C. C. Lin, Life Sci., 2005, 76, 1883 CrossRef CAS.
- H. Jiang, Q. Xia, X. Wang and J. F. Song, Pharmazie, 2005, 60, 444 CAS.
- R. P. Samy, P. Gopalakrishnakone and S. Ignacimuthu, Chem.-Biol. Interact., 2006, 164, 1 CrossRef CAS.
- H. P. Hendrickson, A. D. Kaufman and C. E. Lunte, J. Pharm. Biomed. Anal., 1994, 12, 325 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56 CrossRef CAS.
- B. I. Yakabson and R. E. Smally, Am. Sci., 1997, 85, 324.
- J. Wang, M. Mosameh and Y. Lin, J. Am. Chem. Soc., 2003, 125, 2408 CrossRef CAS.
- J. Wang, M. Li, Z. Shi, N. Li and Z. Gu, Anal. Chem., 2002, 74, 1993 CrossRef CAS.
- A. Salimi, M. Izadi, R. Hallaj and M. Rashidi, Electroanalysis, 2007, 19, 1668 CrossRef CAS.
- A. Salimi, C. E. Banks and R. G. Compton, Analyst, 2004, 129, 225 RSC.
- K. Zhao, H. Song, S. Zhung, L. Dai, P. He and Y. Fang, Electrochem. Commun., 2007, 9, 65 CrossRef CAS.
- Z. Li, J. Chen, D. Pan, W. Tao, L. Nie and S. Yao, Electrochim. Acta, 2006, 51, 4255 CrossRef CAS.
- F. Frehill, J. G. Vos, S. Benrezzak, A. A. Koos, Z. Konya, M. G. Ruther, W. J. Blau, A. Fonseca, J. B. Nagy, L. P. Biro, A. I. Minett and M. Panhuis, J. Am. Chem. Soc., 2002, 124, 13694 CrossRef CAS.
- R. J. Chen, Y. Zhang, D. Wang and H. Dai, J. Am. Chem. Soc., 2001, 123, 3838 CrossRef CAS.
- H. R. Zare and N. Nasirizadeh, Electrochim. Acta, 2007, 52, 4153 CrossRef CAS.
- Y. Liu, M. J. Wang, Z. Y. Li, H. T. Liu, P. He and J. H. Li, Langmuir, 2005, 21, 1618 CrossRef CAS.
- J. L. Anderson, D. W. Armstrong and G. T. Wei, Anal. Chem., 2006, 78, 2892 CrossRef.
- P. Wasserscheild and W. Keim, Angew. Chem., Int. Ed. Engl., 2000, 39, 3772 CrossRef.
- J. Dupont, G. S. Fonseca, A. P. Umpierre, P. F. P. Fichtner and S. R. Teixeira, J. Am. Chem. Soc., 2002, 124, 4228 CrossRef CAS.
- H. Luo, S. Dai, P. V. Bonnesen, A. C. Brchanan, J. D. Holbrey, N. J. Bridges and R. D. Rogers, Anal. Chem., 2004, 76, 3078 CrossRef CAS.
- Y. Zhou and M. Antonietti, J. Am. Chem. Soc., 2003, 125, 14960 CrossRef CAS.
- S. Zein El Abedin, N. Borissenko and F. Endres, Electrochem. Commun., 2004, 6, 422 CrossRef CAS.
- Norouz Maleki, Afsaneh Safavi and Fariba Tajabadi, Anal. Chem., 2006, 78, 3820 CrossRef CAS.
- Wei Sun, Ruifang Gao and Kui Jiao, J. Phys. Chem. B, 2007, 111, 4560 CrossRef CAS.
- A. Safavi, N. Maleki, O. Moradlou and M. Sorouri, Electrochem. Commun., 2008, 10, 420 CrossRef CAS.
- X. Lu, J. Hu, X. Yao, Z. Wang and J. Li, Biomacromolecules, 2006, 7, 975 CrossRef CAS.
- F. Zhao, X. Wu, M. Wang, Y. Liu, L. Gao and S. Dong, Anal. Chem., 2004, 76, 4960 CrossRef CAS.
- C. Xiang, Y. Zou, L. X. Sun and F. Xu, Electrochem. Commun., 2008, 10, 38 CrossRef CAS.
- Y. Liu, L. Huang and S. Dong, Biosens. Bioelectron., 2007, 23, 35 CrossRef CAS.
- S. D. Zelnick, D. R. Mattie and P. C. Stepaniak, Aviat., Space Environ. Med., 2003, 74, 1285 Search PubMed.
- H. F. Mark, D. F. Othmer, C. G. Overberger and G. T. Seaborg, (Ed.), Kirk-Othmer Encyclopedia of Chemical Technology, vol. 12, Wiley, New York, 2005, p. 734 Search PubMed.
- S. M. Golabi and H. R. Zare, J. Electroanal. Chem., 1999, 465, 168 CrossRef CAS.
- A. Safavi and A. A. Ensafi, Anal. Chim. Acta, 1995, 300, 307 CrossRef CAS.
- T. J. Pastor, V. J. Vajgand and N. V. Antonijeic, Mikrochim. Acta, 1983, 3, 203 CrossRef CAS.
- J. S. Budkuley, Mikrochim. Acta, 1992, 108, 103 CAS.
- J. W. Mo, B. Ogorevc, X. J. Zhang and B. Pihlar, Electroanalysis, 2000, 12, 48 CrossRef CAS.
- Greg E. Collins and Susan L. Rose-Pehrsson, Analyst, 1994, 119, 1907 RSC.
- J. C. Huang, C. X. Zhang and Z. J. Zhang, Fresenius J. Anal. Chem., 1999, 363, 126 CrossRef CAS.
- X. Zheng, Z. Zhang, Z. Guo and Q. Wang, Analyst, 2002, 127, 1375 RSC.
- Q. Yi and W.g. Yu, J. Electroanal. Chem., 2009, 633, 159 CrossRef CAS.
- G. Karim-Nezhad, R. Jafarloo and P. S. Dorraji, Electrochim. Acta, 2009, 54, 5721 CrossRef CAS.
- A. Abbaspour, A. Khajehzadeh and A. Ghaffarinejad, J. Electroanal. Chem., 2009, 631, 52 CrossRef CAS.
- K. I. Ozoemena and T. Nyokong, Talanta, 2005, 67, 162 CrossRef CAS.
- M. R. Majidi, A. Jouyban and K. Asadpour-Zeyna, Electrochim. Acta, 2007, 52, 6248 CrossRef CAS.
- M. M. Ardakani, P. E. karami, P. Rahimi, H. R. Zare and H. Naeimi, Electrochim. Acta, 2007, 52, 6118 CrossRef.
- H. M. Nassef, A.-E. Radi and C. K. O'Sullivan, J. Electroanal. Chem., 2006, 592, 139 CrossRef CAS.
- A. A. Ensafi and E. Mirmomtaz, J. Electroanal. Chem., 2005, 583, 176 CrossRef CAS.
- M. Revenga-Parra, E. Lorenzo and F. Pariente, Sens. Actuators, B, 2005, 107, 678 CrossRef.
- L. Zheng and J. F. Song, Sens. Actuators, B, 2009, 135, 650 CrossRef.
- H. R. Zare, N. Nasirizadeh, F. Chatraei and S. Makarem, Electrochim. Acta, 2009, 54, 2828 CrossRef CAS.
- A. Salimi, L. Miranzadeh and R. Hallaj, Talanta, 2008, 75, 147 CAS.
- S. A. Wring and J. P. Hart, Analyst, 1992, 117, 1281 RSC.
- M.-E. Ghica and A. M. Oliveira Brett, Electroanalysis, 2005, 17, 313 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods Fundamentals and Applications, 2nd Edition, John Willey & Sons, New York, 2001, p. 591 Search PubMed.
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19 CrossRef CAS.
- P. Janeiro and A. M. O. Brett, Anal. Chim. Acta, 2004, 518, 109 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |