In situ vapor generation inductively coupled plasma spectrometry for determination of iodine using a triple-mode microflow ultrasonic nebulizer after alkaline solubilization
Henryk
Matusiewicz
* and
Mariusz
Ślachciński
Politechnika Poznańska, Department of Analytical Chemistry, 60-965, Poznań, Poland. E-mail: Henryk.Matusiewicz@put.poznan.pl
First published on 8th September 2010
Abstract
The analytical potential of a coupled continuous-microflow ultrasonic nebulizer triple-mode micro-capillary system (µ-USN/TCS) for the determination of iodine in biological samples by direct iodine vapor generation inductively coupled plasma optical emission spectrometry (VG-ICP-OES) has been investigated. The iodine atomic emission line at 183.038 nm was selected as the analytical line of interest. An extremely short oxidation reaction time between sample, acid and oxidant and a rapid separation of the reaction products is obtained by mixing the sample, sulfuric acid, hydrogen peroxide, and the sodium nitrite solution at the quartz oscillator, converting liquids into aerosol at the entrance to the spray chamber. A univariate approach and simplex optimization procedures were used to achieve optimized conditions and derive analytical figures of merit. Results showed that the analytical performance of the new system was superior to that of pneumatic nebulizer. Analytical performance of the ultrasonic nebulization system was characterized by determination of the limits of detection (LODs) and precision (RSDs) with the µ-USN/TCS-VG-ICP-OES observed at a 15 µL min−1 flow rate. The experimental concentration detection limits for iodine determination, calculated as the concentration giving a signal equal to three times of the standard deviation of the blank (LOD, 3σblank criterion, peak height), were 1.6 ng mL−1 for iodine. The method offers relatively good precision (RSD ranged from 2 to 4%) for liquid analysis and microsampling capability. Samples were prepared by solubilization with tetramethylammonium hydroxide (TMAH), permits complete sample solubilization and significantly reduces the risk of iodine evaporation, before iodine was quantified by USN-VG-ICP-OES. The accuracy of the method was verified using certified reference materials (NIST 1549 and NIST 1566b) and using a simple external calibration technique. The measured contents of elements in reference materials were in satisfactory agreement with the certified value (I). The method was applied to the determination of total iodine in different samples with satisfactory results.
Introduction
Iodine (I2) is an essential micronutrient, which is utilized by the thyroid gland for the biosynthesis of the thyroid hormones and triiodothyronine, which have an important influence on an extended range of biochemical reactions. Iodine deficiency may lead to various malfunctions in the living organism. Therefore, highly efficient and reliable analytical techniques are necessary for iodine determination in foodstuffs, animal feed and biological materials.Iodine is very volatile and can be easily oxidized or reduced. Typically present in very low concentrations in biological samples, it consequently presents one of the most complex analytical challenges to mineral analysis. There are many methods which are applied for the determination of iodine in biological samples including atomic absorption spectrometry (AAS),1 inductively coupled plasma optical emission spectrometry (ICP-OES)2 and mass spectrometry (ICP-MS),3 microwave induced plasma optical emission spectrometry (MIP-OES),4 neutron activation analysis (NAA),5 energy dispersive X-ray fluorescence spectrometry (EDXRF),6 cathodic and anodic stripping voltammetry,7 ion chromatography (IC)8 and selective electrodes.9
One of the methods for reducing the detection limits in ICP-OES is to increase the efficiency of the sample nebulization and analyte excitation. In ICP-OES, the efficiency excitation does not exceed 1–2%. When analytes are injected into a plasma as vapor, the efficiency excitation essentially increased. Therefore, hyphenation of ICP-OES and the vapor generation (VG) technique enables significant improvement of the detection limit and elimination of the main interferences. Oxidation vapor generation is a widely used method of iodide transfer to the gas phase. However, the data on the use of vapor generation with ICP-OES are scant.10–13 The feasibility of the use of a silicone–rubber gas–liquid separator for the determination of iodide in waters has been investigated.10 Iodide is determined after oxidation to iodine, the gaseous species are separated from solution across the silicone-rubber membrane and are swept into the injector stream of an inductively coupled plasma for analysis.10 Treatment of test solution has been used to form the volatile molecular species of iodine before nebulization into the plasma; oxidation of I− in acidified test solution to I2 by using hydrogen peroxide for Ar ICP-OES was evaluated.11 Application of gas generation for the determination of iodine has been reported, applying a commercial hydride generation device for generation of iodine using an aqueous solution of KMnO4 as oxidizing agent without any optimization of the parameters.12 Total iodine is determined directly by ICP-OES using iodine vapor generation; dissolved I− is oxidized in situ to I2 by adding NaNO2 in H2SO4 in a simplified continuous flow manifold.13 VG-ICP-OES was used for the determination of the iodine content in milk samples. The vapor of elemental iodine was generated by means of oxidation of iodine by hydrogen peroxide in sulfuric acid.14 Recently, Vtorushina et al.15,16 have developed a method to generate volatile iodine by oxidation of iodide15 and reduction of iodate16 in solutions and determination of iodine by ICP-OES using the vapor generation technique.
Ultrasonic nebulizers (USNs) have been shown to be more efficient of aerosol formation and can produce aerosols with smaller and more uniform droplet size. Generating a primary aerosol with a very small mean drop size (ideally <5 µm) is an alternative to direct injection to the plasma, as such fine aerosol drops should pass to the plasma with high efficiency and the amount of solvent entering the plasma can be drastically reduced and sensitivity improved. This results in more efficient atomization, higher sensitivity and reduced matrix effects. It has been reported that ultrasonic nebulizers are capable of operating at flow rates down to 20 µL min−1, without the need for any makeup solvent.17,18 The ability to provide very high transport efficiency at flow rates in the range of 2–20 µL min−1 suggests good potential for ultrasonic micronebulizers use with plasma spectrometry. Ultrasonic nebulizers become popular with inductively coupled plasma (ICP) optical emission (OES) and mass spectrometry (MS). However, over the past decades, only one ultrasonic nebulizer design has been developed for use with ICP-OES for the determination of iodine.10
The principal aim of this study was to evaluate, for the first time, the performance of specially designed compact continuous microflow ultrasonic nebulizer triple-mode micro-capillary system (µ-USN/TCS) to operate stably with liquid flows as low as ca. 15 µL min−1 for iodine vapor generation in ICP-OES. Iodine is generated in situ, taking advantage of very rapid iodine vapor generation and fast separation of the vapor from the liquid phase, and determined by inductively coupled plasma spectrometry. The optimization of variables affecting the process was carried out by using univariate and simplex optimization approach. Samples were prepared by solubilization with tetramethylammonium hydroxide (TMAH), permits complete sample solubilization and significantly reduces the risk of iodine evaporation, before iodine was quantified by USN-VG-ICP-OES.
The overall plan was to develop the present technique for determining iodine in biological samples, using USN-VG coupled to ICP-OES.
Experimental
ICP-OES instrumentation
A Thermo Jarrel-Ash Iris HR (Franklin, USA) ICP emission spectrometer, equipped with an argon purged Echelle monochromator (175–900 nm), was used in combination with a microflow ultrasonic nebulizer as a system for direct sample injection into plasma. Instrument settings and operational parameters used for the experimental ICP-OES system are summarized in Table 1. A schematic diagram of the entire experimental set-up (i.e., sample introduction system-ICP-OES) is shown in Fig. 1.| ICP-OES parameters | |
| RF generator frequency/MHz | 27.12 |
| RF power/W | 1150 |
| Spectral range/nm | 175–900 |
| Argon flow rate/L min−1 | |
| Outer | 14.0 |
| Intermediate | 0.5 |
| Nebulizer | 0.4 |
| Replicates | 3.0 |
| Integration time/s | 20 |
| Measurement mode | Axial view, peak height |
| Analytical wavelength/nm | 183.038 |
| Three-mode capillary ultrasonic nebulization parameters | |
| Instrument | Triple-mode ultrasonic nebulizer |
| Solution flow mode | Continuous |
| Transducer frequency/MHz | 1.65 |
| Acoustic power/W | 80 |
| Transducer type/W | Piezoelectric quartz plate, water cooled |
| Spray chamber | Cyclonic |
| Nebulizer carrier gas (Ar) flow rate/L min−1 | 0.4 |
| VG sample introduction | |
| H2SO4 solution concentration/mol L−1 | 3.5 |
| H2SO4 solution flow rate/µL min−1 | 15 |
| H2O2 solution concentration/mol L−1 | 3.0 |
| H2O2 solution flow rate/µL min−1 | 15 |
| Sample solution flow rate/µL min−1 | 15 |
| NaNO2 solution concentration/mmol L−1 | 1.0 |
| Fe3+ solution concentration/mg L−1 | 1.0 |
| Rinse time between the samples/s | 360 |
 | ||
| Fig. 1 Schematic diagram of the elaborated µ-USN/TCS-VG-ICP-OES system. | ||
Vapor generation three capillary mode sample introduction system
Vapor generation was accomplished in the continuous mode micro-flow ultrasonic nebulizer. The nebulizer is the modified NOVA-2 ultrasonic nebulizer having three solution channels.19 Those channels were used for delivery of the hydrogen peroxide, sample (NaNO2 and Fe3+) solution and sulfuric acidic solution, respectively. The integrated internal water-cooled ultrasonic nebulizer was operated at 1.65 MHz ultrasonic wave with a forward power of 80 W. Fig. 2 shows the main components of the continuous-type µ-USN/TCS.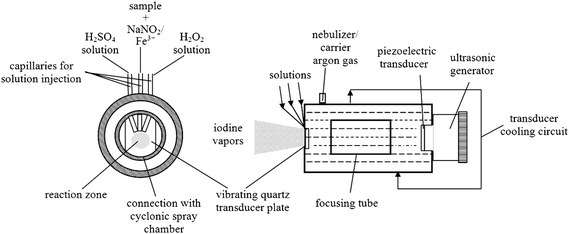 | ||
| Fig. 2 Schematic diagram of the capillary vapor generation µ-USN/TCS system. | ||
Nebulization was achieved by placing three identical solution injection capillaries (narrow PFA tubes with 0.4 mm orifices) in contact with the nebulizer surface (the vibrating plate). Two capillaries were positioned at an angle of ca. 45°, third one perpendicularly and placed in almost direct contact with the quartz surface of the ultrasonic transducer. The precise position is not important; it makes no difference whether the end of the capillaries touches the quartz plate or not. In this manner three independent solutions are pumped simultaneously onto the surface of a quartz piezoelectric transducer (1.65 MHz resonant frequency) at a flow rate in the range 5–50 µL min−1 using a peristaltic pump, where they were effectively mixed and dispersed into droplets of ca. 2.0–2.5 µm in mean droplet diameter (0.1–4.5 µm size distribution),10 (a very fine aerosol is formed as a result of the interaction between ultrasonic waves and the liquid film). The ultrasonic nebulizer is directly connected to the cyclonic glass spray chamber. The transducer quartz plate was cooled with a closed circuit water cooling system. The cyclonic spray chamber made it possible to introduce argon in a pseudoconcentric manner, thereby minimizing turbulence. The system was used without desolvation, unlike most higher flow ultrasonic nebulization systems. A 40 mL cyclonic spray chamber was held in the PTFE nut, a tight connection being provided by a rubber sealing ring.
The liquid samples were introduced through the ultrasonic nebulizer by means of a Perimax12 peristaltic pump (SPETEC, Erding, Germany). The gas flow rate was controlled by means of a mass flow controller (DHN, Warsaw, Poland) with a pressure regulator. Argon was used as the nebulizing-carrier gas, purge gas and as the plasma gas. The flow rate of nebulizing Ar was maintained with an external mass flow controller.
Reagents
Compressed, pure argon gas (N-50 purity, 99.999%) obtained from BOC GAZY (Poznań, Poland) used in this investigation served in a triple role, being a plasma-sustaining gas for the ICP discharge, carrier gas for introduction of the separated molecular iodine into the MIP source and the purge gas for the measurement in vacuum UV region.A standard stock solution of iodide (1000 mg L−1) (Sigma-Aldrich, Steinheim, Germany) prepared from potassium iodide (KI dried at 105 °C for 2 hours) before use. Working standard solutions were freshly prepared daily by diluting appropriate aliquots of the stock solution in water. Iodide solution was kept in a refrigerator and protected from light.
Tetramethylammonium hydroxide (TMAH, 25% in water) of high purity was obtained from Fluka (Buchs, Switzerland) and was used for the solubilization of samples.
Iron solution, used as a catalyst of iodine vapor generation reaction, was prepared from 1000 mg L−1 iron atomic absorption standard (Titrisol grade, Merck, Darmstadt, Germany).
10 mM solution of sodium nitrite were freshly prepared daily by diluting appropriate amount of NaNO2 (Sigma-Aldrich, Steinheim, Germany) in pure water.
Ultra-high purity commercial acids (HCl and H2SO4) (Extra pure, Merck, Darmstadt, Germany) were used to prepare all reagents and samples. Hydrogen peroxide 30% (v/v) was obtained from POCh (Gliwice, Poland). Water was initially deionized (DEMIWA 5 ROSA, Watek, Czech Republic) and then doubly distilled in a quartz apparatus (Heraeus Bi18, Hanau, Germany).
Certified reference materials and real samples
Applicability of the method described in this work was assessed using two reference materials, which were chosen to represent solid sample matrices: NIST 1566b (Oyster Tissue) and NIST 1549 (Non-Fat Milk Powder) supplied by NIST (USA). The certified reference values are available for iodine element for assessment of the method accuracy. All solid reference materials were used as bottled, without further grinding and sieving.To check the applicability of the technique, the iodine content (information concentrations of iodine) of some commercial mineral water, non-fat milk powder, iodized salt and Jodid 100 tablets (Merck KGaA, Darmstadt, Germany) was also analyzed.
Analytical procedures
Approximately 250 mg of NIST SRM 1549 (Non-Fat Milk Powder) were weighted and moistened with 1 mL of 25% TMAH and 4 mL of water. When working with NIST SRM 1566b (Oyster Tissue), approximately 250 mg of powdered sample were moistened with 2.5 mL of 25% TMAH and 2.5 mL of water. The samples were sonicated continuously for 3 min at 60 W. After 2 h solubilized samples were transferred quantitatively to volumetric flask and filled up to the final volume of 10 mL with water. No internal standard was used. This solution can be measured directly or after dilution, depending on the amount of matrix present in the sample. Calibration solutions were prepared in TMAH of adequate concentration.
Solubilization procedure of milk powder NAN 2 (Nestle, Vevey, Switzerland) was identical to that used for NIST SRM 1549.
Jodid 100 tablet (Merck KGaA, Darmstadt, Germany) was moistened with 10 mL of 4% HCl, mixed well and the upper clear solution was diluted with water to appropriate volume.
Iodized salt and mineral water were dissolved in water and made up to appropriate volume.
In all cases, a corresponding blank was also prepared according to the above solubilization and dissolution procedures.
Results and discussion
Sample preparation
The TMAH as a reagent for the alkaline solubilization of biological materials is highly recommended for iodine determination,21 however, its use requires large digestion times, commonly 4–6 h. Therefore, of speeding up the alkaline solubilization of samples, ultrasonic agitation was applied. TMAH being a strong base prevents iodine losses, as it forms tetramethylammonium iodate salt in the reaction mixture. After solubilization, samples were diluted to yield a final solution of 1.0% v/v TMAH to provide the optimum medium for vapor generation.Triple-mode ultrasonic micronebulizer system for vapor generation
The new micro-flow system was used to generate the volatile iodine. Operation of the triple-mode system consisted of pumping the H2O2 solution with a peristaltic pump through one capillary, the sample solution (containing NaNO2 and Fe3+ ions) was delivered with the same pump through another capillary and the acidic solution (H2SO4) was delivered by third capillary. Sample, H2SO4 and H2O2 solutions were introduced and a reaction occurred in situ on the very localized aerosol generation area and in the quartz piezoelectric transducer to concurrently produce the iodine volatile species.In order to test the applicability of the triple-mode system for ultrasonic nebulization vapor generation to ICP-OES, an optimization of the I emission signal intensities as a function of the different parameters was carried out.
Optimization of operating parameters for VG-ICP-OES
Based on results of a preliminary study, the atomic line 183.038 nm in the vacuum UV range was chosen for iodine showing no spectral interference of phosphorus,11,22,23 and the best raw intensity to background ratio. There was no significant spectral line overlap in the vicinity of the I(I) 183.038 nm line. Therefore, the more suitable analytical line for the determination of iodine was I(I) 183.038 nm with respect to not only detection limits24–27 but also spectral interference from some contaminants, i.e., from phosphorus.Three different types of experimental variables affect the method. These are as follows: first, variables controlling the emission response in the plasma, i.e. the power of the generator; secondly, variables, such as the argon carrier flow and sample uptake rate, that regulate transport; and thirdly, variables controlling the chemical generation such as concentration and acidity of oxidizing solution. Table 1 lists the optimum values that the univariate and simplex optimization experiments indicated for each of the three factors studied.
| Parameter (variable) | Boundary limits of parameters, range | Univariate method | Simplex method | Chosen values |
|---|---|---|---|---|
| a Response, peak height of the element emission intensity. | ||||
| RF power/W | 950–1750 | 1150 | 1160 | 1150 |
| Nebulizer gas flow rate/mL min−1 | 200–600 | 400 | 390 | 400 |
| H2SO4 solution concentration/mol L−1 | 1–5 | 3.5 | 3.4 | 3.5 |
| H2O2 solution concentration/mol L−1 | 1–5 | 3.0 | 3.2 | 3.0 |
| Sample/H2SO4/H2O2 solution flow rate/µL min−1 | 5–25 | 15 | 15 | 15 |
 | ||
| Fig. 3 Effect of the variables on the iodine normalized emission intensity for µ-USN/TCS-VG-ICP-OES system. Influence of (a) power; (b) nebulizer gas flow rate; (c) H2SO4 concentration; (d) H2O2 concentration; and (e) solutions flow rate on the emission intensity of the iodine in the vapor generation technique. The experimental conditions employed are detailed in the Experimental. | ||
The rate of generation of volatile vapors also depends on the concentration of NaNO2, as the oxidizing reagent to oxidize iodide and other iodine species to iodine vapor. The concentration of 1.0 mmol L−1 NaNO2 was adopted.
Nakahara and Wasa25 reported enhancement by factors of 5 to 16, iodine emission intensity in the presence of iron(III). Consequently, iron(III) was considered to behave as an oxidizing agent in a manner similar to the prior-oxidation procedure. Based on this, 0.1% (m/v) Fe(III) was satisfactory to achieve maximum signals of iodine.
Analytical figures of merit
The analytical performance characteristics were evaluated for the iodine element. The limits of detection (LODs) calculated using the IUPAC recommendation (based on a 3σblank criterion), determined by 10 repetitive measurements of the blank involving the entire process, and obtained under optimal operating conditions are summarized in Table 3, based on the raw, unsmoothed data and compared with those of several other analytical methods. As shown in this table the combined use of the iodine 183.038 nm line and the iodine ultrasonic vapor generation with sodium nitrite appears particularly attractive in view of its lower detection limits when compared with other vapor generation approaches.10–12,14,15,27 However, the LOD found to be 1.6 µg L−1 is slightly less than similar method reported in the literature for the determination of I by oxidation- or reduction vapor generation-ICP-OES.16| Parameter | I(I) | References | ||||||
|---|---|---|---|---|---|---|---|---|
| 10 | 11 | 12 | 14 | 15 | 16 | 27 | ||
| a mg L−1. b For sample weights of 250 mg. c Continuous solution flow rate of 15 µL. | ||||||||
| Analysis wavelength/nm | 183.038 | 178.276 | 183.04 | 183.04 | 178.218 | 183.038 | 178.276 | 183.038 |
| Detection limit (3σ)/µg L−1 | 1.6 | 4.5 | 5 | 3.4 | 20 | 31 | 0.28–1.27 | 2–7a |
| Detection limitb (3σ)/µg g−1 | 0.05 | 0.029–0.072 | ||||||
| Absolute detection limitc/pg | 0.02 | |||||||
| Precision (% RSD) | 4.2 | 3.5 | 2–3 | |||||
The precision of replicate determinations was calculated from the RSD (%) of the mean of five replicate measurements of element standards using a concentration 50-fold above the LOD. Precision was at approximately 3.9% (evaluated as the peak height). These values may be considered satisfactory, especially owing to the large number of parameters governing the performance of the coupled technique. In other words, this reflects the cumulative imprecision of all of the sample solution handling, sample ultrasonic vapor generation, transfer of vapors, excitation and detection steps.
Validation of the method by analysis of reference materials
To evaluate the accuracy and precision of the sample introduction system tested on the determination of volatile vapor species (I2), two certified reference materials were selected because they were closest in nature to real biological samples. The results obtained for the analysis of reference materials by the USN-VG-ICP-OES method, using the external calibration technique, are summarized in Table 4. Table 4 gives the I values for CRMs, along with information or certified values and acceptable ranges. The results obtained by the external calibration technique agree with certified values for the two reference materials, indicating that simple external calibration (without matrix matching) could produce accurate results. All experimental concentrations agreed fairly well with the certified interval element values, which confirm the accuracy of the developed procedure. Although no interference study was undertaken, it is obvious that there are no systematic errors due to the presence of the matrices. These results also indicate clearly that the applied sample alkaline solubilization protocol was effective in breaking down organic and inorganic biological and environmental matrices. This indicates also that the total amount of iodine present in these reference materials is available in a readily soluble form, which can be extracted by an alkaline reagent such as TMAH.Total iodine determination in selected real samples
Finally, in order to evaluate the usefulness of the proposed method in determining total iodine contents, some real samples were analyzed using the experimental conditions previously optimized. The results for the samples analyzed using the evaluated method are given in Table 5. In all cases, the calibration was achieved using the aqueous standard calibration curves. The found concentration is in good agreement with information values given in the analyzed samples.Conclusions
The applicability and reliability of the ultrasonic nebulization vapor generation technique with ICP detection for iodine determination have been demonstrated in previous works and in our recent work. These methodologies have certain advantages compared to the methods usually applied for the determination of iodine, including better limit of detection, lower time and chemical consumption and alkaline solubilization, which reduces the risk of contamination as well as iodine loss by evaporation. By monitoring the most sensitive spectral lines when possible, using ultrasonic nebulizer and plasma with axial view of observation, concentration at µg g−1 or ng mL−1 levels can be accurately determined. The measured values are in good agreement with the certified values given for the CRMs or information values and %RSD is <4%. The presented method also demonstrates the possibility of using external calibration for quantification, which is more convenient than, i.e., standard addition technique. ICP-OES technique has been successfully applied to the determination of iodine in biological samples (milk powder, jodid tablets, iodised salt, and mineral water) using oxidation ultrasonic vapor generation after TMAH solubilization of samples.Since the use of the combination of both TMAH solubilization and USN-VG-ICP-OES leads to assess total iodine contents in samples the methodologies developed throughout this work can be a good starting point to study iodine speciation in biological materials, when USN-VG-ICP-OES as the final I-detector.
Acknowledgements
Financial support by Ministry of Science and Higher Education, Poland (Grant No. N N204 130935) is gratefully acknowledged. The assistance and cooperation of A. Ramsza, OPTOlab, Warsaw, Poland in obtaining the ultrasonic nebulizer are acknowledged.References
- P. Bermejo-Barrera, M. Aboal-Somoza and A. Bermejo-Barrera, J. Anal. At. Spectrom., 1999, 14, 1009–1018 RSC.
- Z. Zhu, Q. He, Q. Shuai, H. Zheng and S. Hu, J. Anal. At. Spectrom., 2010, 25, 1390–1394 RSC.
- J. V. Dyke, P. K. Dasgupta and A. B. Kirk, Talanta, 2009, 79, 235–242 CrossRef CAS.
- F. Camuña, J. E. Sanchez Uria and A. Sanz Medel, Spectrochim. Acta, Part B, 1993, 48, 1115–1125 CrossRef.
- E. Andrási, J. Kučera, Cs. Bélavári and J. Mizera, Microchem. J., 2007, 85, 157–163 CrossRef CAS.
- J. Pavel, M. Fille and U. Frey, Fresenius' Z. Anal. Chem., 1988, 331, 51–54 CrossRef CAS.
- V. A. Demin, A. I. Kamenev, N. P. Zveryak and V. I. Zarembo, J. Anal. Chem., 2010, 65, 87–90 CrossRef CAS.
- T. K. Malongo, S. Patris, P. Macours, F. Cotton, J. Nsangu and J.-M. Kauffmann, Talanta, 2008, 76, 540–547 CrossRef CAS.
- R.-I. Stefan, S. G. Bairu and J. F. van Staden, Anal. Lett., 2003, 36, 1493–1500 CrossRef CAS.
- M. R. Cave and K. A. Green, J. Anal. At. Spectrom., 1989, 4, 223–225 RSC.
- S. P. Dolan, S. A. Sinex, S. G. Capar, A. Montaser and R. H. Clifford, Anal. Chem., 1991, 63, 2539–2542 CrossRef CAS.
- R. Wennrich, A. Mroczek, K. Dittrich and G. Werner, Fresenius' J. Anal. Chem., 1995, 352, 461–469 CrossRef.
- K. A. Anderson and P. Markowski, J. AOAC Int., 2000, 83, 225–230 CAS.
- E. Niedobová, J. Machát, V. Otruba and V. Kanický, J. Anal. At. Spectrom., 2005, 20, 945–949 RSC.
- E. A. Vtorushina, A. I. Saprykin and G. Knapp, J. Anal. Chem., 2008, 63, 643–648 CrossRef CAS.
- E. A. Vtorushina, A. I. Saprykin and G. Knapp, J. Anal. Chem., 2009, 64, 129–135 CrossRef CAS.
- J. F. Karnicky and L. T. Zitelli, Anal. Chem., 1987, 59, 327–333 CrossRef CAS.
- M. A. Tarr, G. Zhu and R. F. Browner, Anal. Chem., 1993, 65, 1689–1695 CrossRef CAS.
- A. Tyburska, K. Jankowski, A. Ramsza, E. Reszke, M. Strzelec and A. Andrzejczuk, J. Anal. At. Spectrom., 2010, 25, 210–221 RSC.
- G. Knapp, B. Maichin, P. Fecher, S. Hasse and P. Schramel, Fresenius' J. Anal. Chem., 1998, 362, 508–513 CrossRef CAS.
- K. Tagami, S. Uchida, I. Hirai, H. Tsukada and H. Takeda, Anal. Chim. Acta, 2006, 570, 88–92 CrossRef CAS.
- E. Niedobová, J. Machát, V. Kanický and V. Otruba, Microchim. Acta, 2005, 150, 103–107 CrossRef CAS.
- I. Varga, Microchem. J., 2007, 85, 127–131 CrossRef CAS.
- O. Buresch, W. Hönle and H. G.v. Schnering, Fresenius' Z. Anal. Chem., 1986, 325, 607–610 CAS.
- T. Nakahara and T. Wasa, Anal. Sci., 1988, 4, 223–225 CAS.
- K. Krengel-Rothensee, U. Richter and P. Heitland, J. Anal. At. Spectrom., 1999, 14, 699–702 RSC.
- J. Naozuka, M. A. Mesquita Silva da Veiga, P. V. Oliveira and E. de Oliveira, J. Anal. At. Spectrom., 2003, 18, 917–921 RSC.
| This journal is © The Royal Society of Chemistry 2010 |