Development and validation of a LC-ESI-MS/MS method in human plasma for quantification of fenofibric acid, involving chromatographic resolution of fenofibric acid acyl-β-D-glucuronide
Ajay
Kumar
,
Tausif
Monif
*,
Arshad H.
Khuroo
,
Sunil S.
Iyer
,
Abhishek Kumar
Singh
and
Debashis
Kar
Department of Clinical Pharmacology & Pharmacokinetics, Ranbaxy Laboratories Ltd., GP-5, HSIDC, Sector-18, Gurgaon, 122 015, India. E-mail: tausif.monif@ranbaxy.com; Fax: +91 124 4231002; Tel: +91 124 4768225
First published on 9th September 2010
Abstract
This is the first publication on a validated bioanalytical method for estimation of fenofibric acid in human EDTA plasma that chromatographically resolves its acyl glucuronide. An API 3000 mass spectrometer was employed in this method, and fenofibric acid-d6 served as the internal standard. Sample preparation involved solid phase extraction (SPE) using a polymer based, hydrophilic-lipophilic balanced cartridges and samples were chromatographed on a Discovery C18, (4.6 × 50 mm, 5 μm) column using a mobile phase of 0.2% formic acid solution/acetonitrile (35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :65, v/v). Negative mass transitions (m/z) of fenofibric acid and fenofibric acid-d6 were detected in multiple reactions monitoring (MRM) mode at 317.1 → 230.9 and 322.9 → 230.8, respectively. The method was validated over a concentration range of 0.150 μg mL−1 to 20.383 μg mL−1. Intra- and inter-run imprecision of fenofibric acid assay at four concentration levels was below 2.5%, and inaccuracy was within ±2.8%. Analytical recoveries for fenofibric acid and internal standard were 73.8–75.4%, and 85.9%, respectively. All other validation parameters were within acceptable limits. The back conversion of fenofibric acid from its acyl glucuronide was minimized by processing samples in an ice cold water bath under low light.
:65, v/v). Negative mass transitions (m/z) of fenofibric acid and fenofibric acid-d6 were detected in multiple reactions monitoring (MRM) mode at 317.1 → 230.9 and 322.9 → 230.8, respectively. The method was validated over a concentration range of 0.150 μg mL−1 to 20.383 μg mL−1. Intra- and inter-run imprecision of fenofibric acid assay at four concentration levels was below 2.5%, and inaccuracy was within ±2.8%. Analytical recoveries for fenofibric acid and internal standard were 73.8–75.4%, and 85.9%, respectively. All other validation parameters were within acceptable limits. The back conversion of fenofibric acid from its acyl glucuronide was minimized by processing samples in an ice cold water bath under low light.
1. Introduction
Fenofibrate is a lipid regulating agent. Fenofibric acid, the active metabolite of fenofibrate, produces a reduction in total cholesterol, low density lipoprotein (LDL) cholesterol, apolipoprotein B, total triglycerides and triglyceride rich lipoprotein (VLDL) in treated patients. In addition, treatment with fenofibrate results in an increase in high density lipoprotein (HDL) and apoproteins apoAI and apoAII. The effects of fenofibric acid, as studied in transgenic mice and in vitro human hepatocyte culture, have been reported through the activation of peroxisome proliferator activated receptor α (PPAR α). Through this mechanism, fenofibrate increases lipolysis and elimination of triglyceride-rich particles from plasma by activating lipoprotein lipase and reducing production of apoproteins C-III (an inhibitor of lipoprotein lipase activity).1–4 Several mechanisms contribute to fenofibrate's hypolipidemic action, including inhibition of fatty acid synthesis, stimulation of fatty acid β-oxidation, inhibition of triglyceride synthesis, and enhancement of lipoprotein lipase activity. Fenofibrate is metabolized in several stages. First, the carboxyl ester moiety is cleaved by hydrolysis, resulting in fenofibric acid, the main pharmacologically active compound. Fenofibric acid, in turn, undergoes carbonyl reduction, resulting in a pharmacologically active metabolite referred to as reduced fenofibric acid. Both fenofibric acid and reduced fenofibric acid may be conjugated to form glucuronides.5 There are important differences between species in the metabolism and elimination patterns of fenofibrate. In rats and dogs, fenofibric acid and reduced fenofibric acid are the principal metabolites and 70–80% of fenofibrate and its metabolites are recovered in the feces. In humans, however the glucuronide of fenofibric acid is predominant and approximately 65% of the dose is excreted in the urine, and 25% in human feces.6In previous publications7,8 on LC-MS/MS methodology, fenofibric acid has been determined in human plasma simultaneously with other statins and with a short calibration range.9 We published a method earlier for bioanalysis of fenofibric acid,10 using ultra performance liquid chromatography coupled to mass spectrometry (UPLC-MS). However, the glucuronide of fenofibric acid was not chromatographically separated in that method. The LC-MS/MS method described in this publication addresses the challenge of separation of fenofibric acid acyl-β-D-glucuronide from fenofibric acid. The current LC-MS/MS method also allows for a lower limit of quantitation (LLOQ) of fenofibric acid in plasma. The primary aim of the present study was to develop and validate a rapid, selective, and sensitive LC-MS/MS method in human plasma that would chromatographically resolve the metabolite, fenofibric acid acyl-β-D-glucuronide from fenofibric acid.
2. Experimental
2.1. Chemicals and reagents
Working standards of fenofibric acid, fenofibric acid acyl-β-D-glucuronide and fenofibric acid-d6 as internal standard were procured from Varda Biotech (P) Ltd., Mumbai, TRC, Canada, and CDN Isotopes Inc., Canada respectively. Acetonitrile and methanol was purchased from Qualigens Fine Chemicals (GSK Ltd., Mumbai, India). Human plasma containing ethylenediaminetetraacetic acid (K3EDTA) was collected from Yes Path Lab, Mumbai, India. Formic acid and ortho phosphoric acid of analytical grade were purchased from S. D. Fine Chem. Ltd., Mumbai, India. Solid phase extraction cartridges (Oasis HLB cartridge; 30 mg/1 cc) were purchased from Waters India Ltd., Bangalore, India.2.2. Instrumentation
An HPLC (Shimadzu Corporation, Kyoto, Japan) coupled to an API 3000 mass spectrometer (MDS Sciex, Canada) was employed for analysis. Chromatographic separation was achieved on a Discovery C18 column (4.6 × 50 mm, 5 μm), using isocratic mobile phase, 0.2% formic acid buffer/acetonitrile (35:65, v/v), at a flow rate of 0.5 mL min-1. Approximately 0.2 mL min-1 was allowed to go in to the ion source using a splitter. The autosampler was kept at 4 °C.
A turbo electrospray ionization (ESI) source was used as the interface. The negative ions were monitored in the multiple reaction monitoring (MRM) modes with zero air as the nebulizing gas. The chromatographic data were acquired and processed using computer based analyst software version 1.4.1.
2.3. Preparation of calibration standards and quality control samples
Stock solutions (approximately 2 mg mL−1) of fenofibric acid and fenofibric acid-d6 (approximately 1 mg mL−1) were prepared by dissolving in methanol. The stock solutions were stored in a refrigerator, protected from light. The stock dilutions of fenofibric acid were prepared in methanol/HPLC grade water, 50:50 v/v in an ice bath under low light condition. The calibration curve (CC) standards and quality control (QC) samples were prepared by spiking in blank K3EDTA human plasma with an appropriate analyte stock or stock dilutions. The final concentrations for the calibration curve were 0.150, 0.350, 0.700, 2.592, 3.200, 8.102, 16.204 and 20.383 μg mL−1.
The stock solution for spiking QC samples were prepared separately. Dilutions were used to prepare QC samples in human plasma at 0.151 μg mL−1 (limit of quantification – LOQ), 0.403 μg mL−1 (lower QC – LQC), 9.381 μg mL−1 (middle QC – MQC), and 15.635 μg mL−1 (higher QC – HQC) for fenofibric acid.
These spiked standards and QC samples were stored at −50 °C prior to analysis.
The stock solution (approximately 1 mg mL−1) of fenofibric acid acyl-β-D-glucuronide was also prepared by dissolving in methanol.
2.4. Plasma sample preparation
The requisite plasma samples were retrieved from freezer room and thawed in an ice cold water bath. A low light condition was ensured during sample processing. The thawed samples were vortexed. To 100μL of plasma samples in stoppered tubes, 50μL of IS stock dilution (5 μg mL−1 in Methanol/HPLC grade water, 50:50 v/v) was added and mixed uniformly. 100μL 10% ortho phosphoric acid was then added and vortexed. The vortexed sample was then loaded on a solid phase extraction cartridge (Oasis HLB 30 mg/1 cc) pre-conditioned with 1 mL methanol followed by 1 mL of HPLC-grade water. The cartridge was washed with 1 mL HPLC grade water. The sample was eluted with 1 mL of methanol twice. The extraction was carried out on centrifuge by running for 1 min at 1500 rpm. The eluted samples were dried under nitrogen gas at 50 °C and at about 15 psi. The dried residue was reconstituted with 500 μL of mobile phase and injected 10 μL samples.
2.5. Method validation
Method validation was conducted according to the USFDA guidance document on validation of bioanalytical methods.11
b) The quantitative evaluation of matrix effects involved measurement of the mean peak area ratio of matrix samples reconstituted with reference dilution of four replicates of quality control samples (at low, middle and high concentration) and comparison of the mean peak area ratio of reference samples at concentrations representing 100% extraction (without matrix in mobile phase) of four replicates of quality control samples at low, middle and high concentration.
The following formula was used for calculation of matrix factor:-

For autosampler stability, QC samples were processed and kept in the autosampler for over 48 h. The long term stability below −50 °C was evaluated by assaying QC samples stored at 9 weeks later. For all stability exercises, freshly processed calibration standards samples (spiked in interference free pooled plasma of the anticoagulant) were used to quantitate QC samples (LQC and HQC in replicate). The back-conversion of fenofibrate from its acyl glucuronide was minimized by analyzing samples in an ice cold water bath under low light condition. To evaluate this, fenofibric acid acyl-β-D-glucuronide was spiked in plasma at 8 μg mL−1, considering the Cmax concentration of fenofibric acid in plasma available in literature.13 This sample was processed in duplicate under two conditions, viz. room temperature and ice cold water bath.
3. Results and discussion
3.1. Optimization of LC-MS/MS condition
Fig. 1 shows the chemical structure of fenofibric acid, fenofibric acid-d6 and fenofibric acid acyl-β-D-glucuronide. The MS/MS transitions were measured in ESI negative ion mode with a dwell time of 200 ms for each fragmentation. The optimized m/z ion transitions were monitored 317.1 → 230.9 for fenofibric acid [Fig. 2(a)] and 322.9 → 230.8 for fenofibric acid-d6 [Fig. 2(b)]. The MRM state file parameters were optimized to get stable responses for the analytes. The parameters for fenofibric acid and fenofibric acid-d6 (IS) were optimized as CAD (collision gas) 4, CUR (curtain gas) 15, NEB (nebulizer gas) 10, TEM (temperature) (450 °C), ISV (ion spray voltage) (−4500 V), resolution-unit/unit for source/gas parameters and DP (declustering potential) (−45 V), FP (focusing potential) (−125 V), EP (entrance potential) (−10 V), CE (collision energy) (−40 V), CXP (collision cell exit potential) (−13 V) for compound parameters.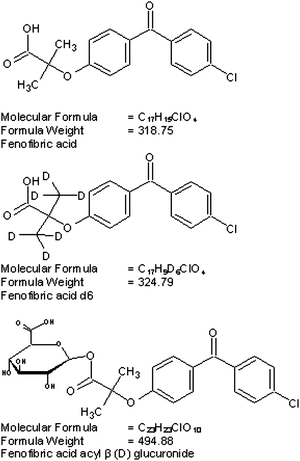 | ||
| Fig. 1 Structures of fenofibric acid, fenofibric acid-d6 and fenofibric acid acyl-β-D-glucuronide. | ||
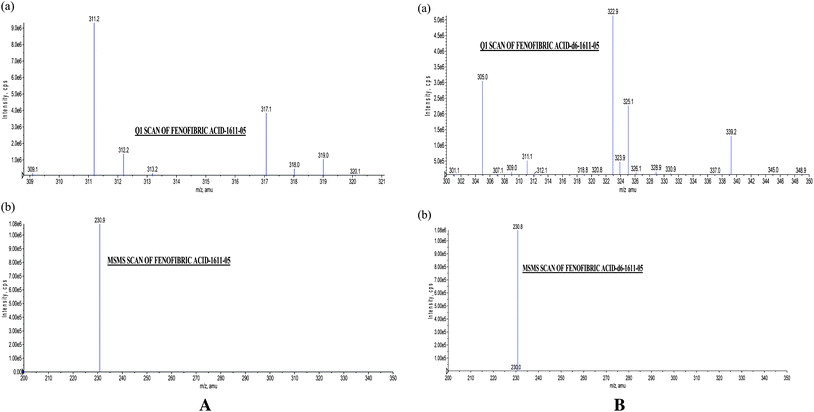 | ||
| Fig. 2 . (a) Q1 Scan of fenofibric acid (317.1), (b) MS/MS Scan of fenofibric acid (230.9) (b). (a) Q1 Scan of fenofibric acid-d6 (322.9), (b) MS/MS Scan of fenofibric acid-d6 (230.8). | ||
The mobile phase was optimized to be 0.2% formic acid solution/acetonitrile (35:65, v/v). The compounds were eluted at retention times of 2.30, 1.60, and 2.50 for fenofibric acid, fenofibric acid acyl-β-D-glucuronide and fenofibric acid-d6, respectively, and produced well resolved peak shapes. Refer to Fig. 3 for glucuronide resolution.
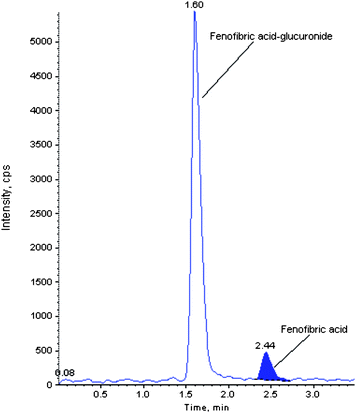 | ||
| Fig. 3 Representative chromatogram showing resolution of fenofibric acid glucuronide. | ||
3.2. Sample preparation
Due to its high plasma protein binding (>99%) and weak acidic14 properties (pKa = 4.5) it is difficult to extract fenofibric acid using conventional extraction procedures like protein precipitation, liquid–liquid extraction etc. Solid phase extraction was therefore used. This technique provided for improved recoveries and produced cleaner samples, thereby avoiding the introduction of non-volatile materials onto the column and MS system. During sample processing, ortho phosphoric acid was used as an inorganic acid to produce better stability. All plasma samples that were screened during the selectivity exercise, showed no significant interference (above 20% of LOQ response) at the retention times of analyte and IS. All samples were processed under a low light condition maintained in an ice cold water bath as a precaution to minimize potential back conversion of fenofibric acid acyl-β-D-glucuronide.3.3. Results of method validation
No interference from endogenous substance was observed in the selectivity exercise at the retention time of fenofibric acid and fenofibric acid-d6. This is explicit from the chromatogram of blank sample in Fig. 4. LOQ plasma samples spiked with IS are shown in Fig. 5. The R-square was consistently 0.99 or greater during the course of validation. The best fit for calibration curve of chromatographic response versus concentration were determined by weighted least square regression analysis with weighting factor of 1/concentration.2 The data of intra- and inter-day precision and accuracy for fenofibric acid from QC samples are summarized in Table 1. The precision and accuracy of this method conform to the FDA guidance document, which states that the accuracy determined at each concentration level must not exceed 15% (20% for LOQ) and precision must be within ±15% (±20% for LOQ) of the nominal value. The extraction recoveries from QC samples at low, middle and high concentrations were 75.4%, 74.6%, 73.8% for fenofibric acid, whereas it was 85.9% for IS. Recoveries were good and it was consistent, precise and reproducible with this proposed extraction method (Table 2). The results from stability test are presented in Table 3, which demonstrated a good stability of fenofibric acid under the conditions evaluated. Bench top stability in matrix (6.58 h), bench top stability during extraction (6.83 h), freeze-thaw stability (3 cycles), autosampler stability (49.13 h), long term stability (63 days, stored below -50 °C) were found to be acceptable. There was negligible or null matrix factor and matrix effect as demonstrated in Table 4. In the back conversion exercise shown in Table 5, it was observed that the ice bath condition resulted in a 44% lower back conversion of the acyl glucuronide to fenofibric acid. | ||
| Fig. 4 Chromatograms of extracted blanks (a) fenofibric acid, (b) fenofibric acid-d6. | ||
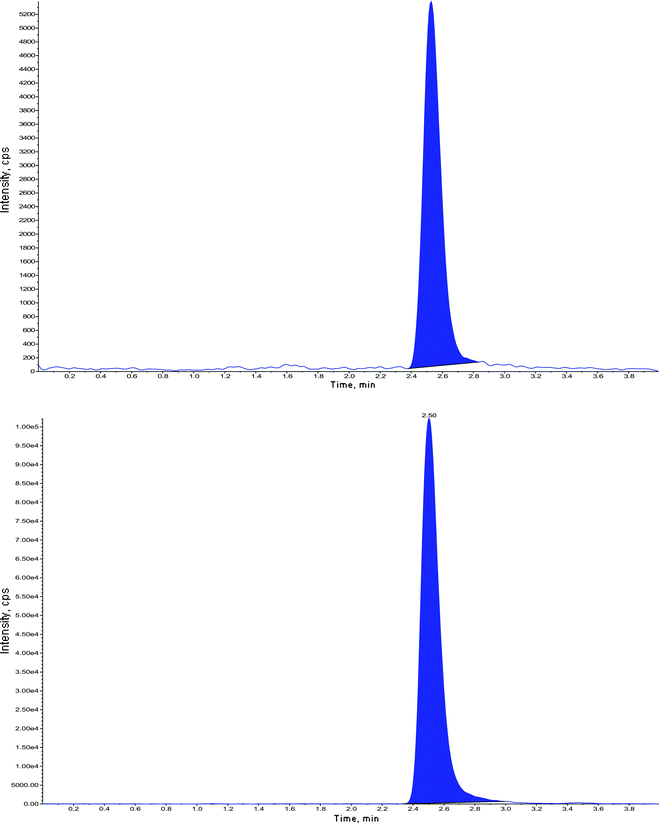 | ||
| Fig. 5 Chromatogram of extracted LOQ with internal standard – fenofibric acid-d6. | ||
| Nominal concentration (μg/mL) | Within batch precision (n = 6) | Between batch precision (n = 18) | ||||
|---|---|---|---|---|---|---|
| Concentration obtained (μg/mL) | Precision | Accuracy | Concentration obtained (ng/mL) | Precision | Accuracy | |
| (Mean ± SD) | (%) | (%) | (Mean ± SD) | (%) | (%) | |
| 0.151 (LOQ) | 0.1520 ± 0.00329 | 2.2 | 100.7 | 0.1503 ± 0.00382 | 2.5 | 99.6 |
| 0.403 (LQC) | 0.4047 ± 0.00437 | 1.1 | 100.4 | 0.4032 ± 0.00735 | 1.8 | 100.1 |
| 9.381 (MQC) | 9.6393 ± 0.14383 | 1.5 | 102.8 | 9.5985 ± 0.13437 | 1.4 | 102.3 |
| 15.635 (HQC) | 15.6175 ± 0.15667 | 1.0 | 99.9 | 15.6919 ± 0.22477 | 1.4 | 100.4 |
| Analyte | QC Level | Area response of unextracted samplea | Area response of extracted samplea | Recoveryb | Mean recovery of fenofibric acid |
|---|---|---|---|---|---|
| (Mean ± S. D.) | (Mean ± S. D.) | (%) | (± S. D.) | ||
a n = 4.
b

|
|||||
| Fenofibric acid | LQC | 81910.8 ± 5863.54 | 61006.0 ± 3921.97 | 75.4 | 74.60 ± 0.801 |
| MQC | 2389187.3 ± 83228.75 | 1787100.3 ± 56641.76 | 74.6 | ||
| HQC | 3821106.7 ± 60373.36 | 2827030.8 ± 143364.17 | 73.8 | ||
| Fenofibric acid-d6 | IS | 659498.8 ± 35754.71 | 566479 ± 16777.79 | 85.9 | |
| Exercise | QC(s) | Sample concentration (μg/mL) (mean ± S. D) | Concentration found (μg/mL) (mean ± S. D) | Precision | Accuracy | ||
|---|---|---|---|---|---|---|---|
| (Stability samples) | (Comparison samples) | (%) | (%) | ||||
| Bench top stability in matrix | LQC | 0.4038 ± 0.00929 | 0.4018 ± 0.00797 | 2.3 | 2.0 | 100.2 | 102.0 |
| HQC | 15.4728 ± 0.10089 | 15.6268 ± 0.12228 | 0.7 | 0.8 | 99.0 | 99.1 | |
| Bench top stability during extraction | LQC | 0.3808 ± 0.00310 | 0.3840 ± 0.01236 | 0.8 | 3.2 | 94.5 | 94.6 |
| HQC | 15.8225 ± 0.13337 | 15.9753 ± 0.07862 | 0.8 | 0.5 | 101.2 | 102.3 | |
| 3 Freeze/thaw stability | LQC | 0.4000 ± 0.00770 | 0.3965 ± 0.00311 | 1.9 | 0.8 | 99.3 | 97.7 |
| HQC | 15.1108 ± 0.24329 | 15.3768 ± 0.06962 | 1.6 | 0.5 | 96.6 | 98.5 | |
| Autosampler stability (49.13 h) | LQC | 0.4140 ± 0.01849 | 0.4040 ± 0.00698 | 4.5 | 1.7 | 102.7 | 99.5 |
| HQC | 15.4503 ± 0.14341 | 15.7195 ± 0.08913 | 0.9 | 0.6 | 98.8 | 100.7 | |
| LTS (stored below −50 °C) | LQC | 0.4003 ± 0.00250 | 0.4078 ± 0.00550 | 0.6 | 1.3 | 99.3 | 104.0 |
| HQC | 15.4030 ± 0.01657 | 15.4610 ± 0.08558 | 0.1 | 0.6 | 98.5 | 98.7 | |
| Lot | LQC (μg mL−1) | HQC (μg mL−1) |
|---|---|---|
| 0.393 | 15.705 | |
| Lot#1-1 | 0.401 | 15.454 |
| Lot#1-2 | 0.403 | 15.659 |
| Lot#2-1 | 0.405 | 15.801 |
| Lot#2-2 | 0.401 | 15.389 |
| Lot#3-1 | 0.398 | 16.234 |
| Lot#3-2 | 0.404 | 16.654 |
| Lot#4-1 | 0.399 | 15.554 |
| Lot#4-2 | 0.398 | 15.405 |
| Lot#5-1 | 0.398 | 15.638 |
| Lot#5-2 | 0.390 | 15.252 |
| Lot#6-1 | 0.407 | 15.753 |
| Lot#6-2 | 0.399 | 15.501 |
| Mean | 0.4003 | 15.6912 |
| S.D. (±) | 0.00441 | 0.39529 |
| C.V. (%) | 1.1 | 2.5 |
| % Nominal | 101.8 | 99.9 |
| N | 12 | 12 |
| Matrix factor | ||
|---|---|---|
| Acceptance criteria: The %CV of matrix factor between low, middle and high QC level should be ±15% (Mean value 1 ± 0.05) | QC | % Matrix Factor |
| LQC | 1.01 | |
| MQC | 0.99 | |
| HQC | 1.01 | |
| Mean | 1.001 | |
| S D (±) | 0.0121 | |
| C.V. (%) | 1.2 | |
| N | 3 | |
| Extracted blank spiked with glucuronide at 8 μg mL−1 | Storage condition | Mean area response of fenofibric acid to show its extent of formation from glucuronide |
|---|---|---|
| a Percent reduction in back conversion in ice water bath = 1229.5/2738.5 × 100% = 44.9%. | ||
| Blank + Glucuronide | Room temp | 2738.5 |
| Blank + Glucuronide | Ice cold water bath | 1229.5 |
Values for all method validation parameters indicate that the methods can be applied for routine bioanalysis.
4. Conclusion
To our best knowledge, this is the first publication for determination of fenofibric acid in human plasma that reports a chromatographic resolution of fenofibric acid acyl-β-glucuronide. Special conditions (viz. low-light and ice bath) however, needed to be maintained at the clinic during sample collection as well as during sample analysis. Results of matrix suppression/enhancement showed no matrix effect existed in this method. The method was furthermore applied successfully for clinical sample analysis. Finally conducting bioavailability study on Indian volunteers would act as authentic reference for fenofibric acid disposition in Asian or other ethnic populations. Thus all the above factors contribute uniqueness and improvement on previous available methods and for the first time complete method validation details with exhaustive stability exercises presented.5. Acknowledgments
The authors thank Ranbaxy Laboratories Ltd. for giving permission to publish in-house data.6. References
- A. D. Edgar, Curr. Ther. Res. Clin. Exp., 1990, 47, 952–961.
- R. H. Knopp, C. E. Walden and G. R. Warnik, Am. J. Med., 1987, 83, 75–84 CrossRef CAS.
- D. R. Guay, Ann. Pharmacother., 1999, 33, 1083–1103 CrossRef CAS.
- J. A. Balfour, D. Mac Tavish and R. C. Heel, Drugs, 1990, 40, 260–290 CrossRef CAS.
- John Caldwell, Cardiology, 1989, 76(Suppl. 1), 33–44 CrossRef.
- http://www.cipladoc.com/therapeutic/pdf_cipla/fenolip.pdf .
- R. K. Trivedi, R. R. Kallem, R. Mullangi and N. R. Srinivas, J. Pharm. Biomed. Anal., 2005, 39(3–4), 661–9 CrossRef CAS.
- B. Mertens, B. Cahay, R. Klinkenberg and B. streel, J. Chromatogr., A, 2008, 1189, 493–502 CrossRef CAS.
- D. Bhavesh, S. Shah and Shivprakash, Biomed. Chromatogr., 2009, 23(9), 922–928 CrossRef CAS.
- S. K. Dubey, M. S. Tomar, A. K. Patni, A. Khuroo, S. Reyar and T. Monif, E–J. Chem., 2010, 7(1), 25–36 Search PubMed.
- Guidance for Industry: Bioanalytical Method Validation, U.S. Food and Drug Administration, Center for Drug Evaluation and Research and Center for Veterinary Medicine, Rockville, MD, 2001 Search PubMed.
- The 2nd calibration and validation group workshop on recent issues in Good Laboratory Practice bioanalysis, Bioanalysis ( 2000), 1(1), pp. 19–30 Search PubMed.
- C. Harvengt and J. P. Desager, Eur. J. Clin. Pharmacol., 1980, 17, 459–463 CrossRef CAS.
- http://www.drugbank.ca/drugs/DB01039 .
| This journal is © The Royal Society of Chemistry 2010 |