Pinpointing oxidative modifications in proteins—recent advances in analytical methods
Ulrika
Törnvall
*
Department of Biotechnology, Center for Chemistry and Chemical Engineering, Lund University, P.O. Box 124, SE-221 00, Lund, Sweden. E-mail: ulrika.tornvall@biotek.lu.se; Fax: +46 (46)222 4713; Tel: +46 (46)222 7363
First published on 13th October 2010
Abstract
Oxidation is one of the key degradation pathways in proteins and is of relevance to analyze in a wide variety of research disciplines. The types of different modifications occurring during oxidation by reactive oxygen species (ROS) are almost as many as the number of available analytical methods. Protein oxidation in biological samples has traditionally been analyzed by various proteincarbonyl assays, sometimes combined with mass spectrometry (MS) for identification of the carbonylated proteins. MS is now increasingly used also for the determination of the exact position of the oxidation in the amino acid sequence, an approach that is aided by recent developments within the field of proteomics. The following review summarizes the effects of ROS on proteins, describes methods for labeling and separation of oxidized proteins in complex mixtures, and provides insight into various MS-based methods to localize the modifications within the protein primary structure. Pitfalls with the different techniques are given, as well as examples from various applications within biological studies, protein therapeutics, plant science, the food industry and industrial biotechnology.
 Ulrika Törnvall | Dr Ulrika Törnvall is a researcher at the Department of Biotechnology, Lund University, Sweden. Her research interests are within the development of more stable enzymes for industrial purposes, as well as bioanalytical and biophysical methods for the study of protein structure–function relationships. |
Introduction
Protein oxidation is of interest in many different disciplines, e.g. in studies of aging and age-related diseases such as Alzheimer's disease; in the field of therapeutic proteins; in studies of foodstorage; and in industrial biotechnology. The ability to detect amino acidoxidation and subsequent changes in protein activity and structure is therefore a highly important topic. However, oxidation is also an exceptionally complex issue since both the cause and the target, as well as the mechanisms and products, might vary considerably. The production of reactive oxygen species (ROS), e.g.superoxide, hydrogen peroxide, hydroxyl radicals and singlet oxygen, is an inevitable consequence of aerobic metabolism. ROS are thought to be involved in a number of important cellular signaling systems, but at elevated concentration (i.e. during stress), ROS can have deleterious effects, damaging biomolecules such as DNA, lipids and proteins.1,2 Oxidized proteins are known markers of oxidative stress in cells,3,4 but protein oxidation also takes place in many normal cellular regulatory processes such as modulation of enzyme activity5 and signalling.6–8 All of the different ROS mentioned above can oxidize proteins, but with different targets within the protein and with varying reaction rates.1,9–11 In addition, proteins do not all respond the same way upon oxidation; the oxidation of a particular amino acid can lead to full inactivation of a certain type of protein, whereas other types of proteins remain unaffected or even become activated by a similar modification.This review focuses on analytical techniques designed for evaluation of the effects caused by ROS on proteins. Different chemical modifications of amino acids occurring as a consequence of oxidation, as well as general methods for their detection, are first summarized. Tools for the evaluation of other main effects caused by ROS, e.g. aggregation, backbone cleavage, etc., are also briefly mentioned, since these effects might have important consequences for the analysis outcome. General methods for labeling and separation of oxidized proteins are then covered, followed by an in-depth description of methods aimed at localizing specific amino acids that have been oxidized. Finally, some examples of studies from each of the above-mentioned application areas are provided.
Types of oxidative modifications caused by reactive oxygen species
Oxidation of amino acid side chains by ROS can be reversible or irreversible, and might lead to further effects such as unfolding/misfolding, aggregation and/or increased susceptibility to proteolytic degradation (Fig. 1). Some ROS, such as radicals, might even cause backbone cleavage.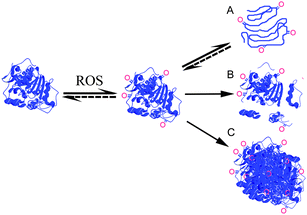 | ||
| Fig. 1 The effect of reactive oxygen species (ROS) on proteins can be oxidation of amino acid side chains, which may further cause: (A) unfolding (partial or total); (B) backbone cleavage; and/or (C) aggregation. Amino acid side chain oxidation and unfolding are in some cases reversible. Backbone cleavage can be either directly due to oxidation by radicals or indirectly due to oxidized proteins displaying increased susceptibility to proteolysis. Oxidations are schematically visualized and the shown pathways are not intended to be all-inclusive. | ||
Chemical modifications of amino acids
Due to the ease of oxidation of sulfur centers, the side chains of methionine and cysteine are the main oxidation sites within proteins under mild oxidative conditions. Depending on the oxidant used, histidine, tryptophan and tyrosine are also relatively susceptible to oxidation.9,10 Under more extreme conditions, such as those that exist during metal-catalyzed oxidation in e.g. the Fenton reaction (Fe2+ and H2O2 react to give HO˙) or γ-irradiation, protein carbonyls (aldehydes and ketones) are formed, especially in Lys, Arg, Pro and Thr residues. Hydroxyl radicals react with all amino acid side chains with varying rates,11 as well as cause extensive backbone cleavage. The products formed, along with the corresponding mass changes, have been summarized by Xu and Chance.12 The most commonly reported modifications resulting from amino acid side chain oxidations are given in Table 1.| Amino acid | Oxidized product | Mass shift |
|---|---|---|
| a +96 if both cysteines appear in the same peptide. b See Domingues et al.15 for other products of tryptophanoxidation. c Only for oxidation by hypochlorous acid, which is generated in vivo by the phagocyteenzymemyeloperoxidase.16 | ||
| Methionine | Methionine sulfoxide | 16 |
| Methionine sulfone | 32 | |
| Cysteine | Cysteine sulfonic acid | 48 |
| Cystine (disulfide) | — | |
| Cystine | Cleavage of disulfide bond and conversion to sulfonic acid | 48a |
| Tryptophan b | Hydroxytryptophan | 16 |
| N -Formylkynurenine | 32 | |
| Kynurenine | 4 | |
| 3-Hydroxykynurenine | 16 + 4 | |
| Histidine | Dehydro-2-imidazolone | 16 − 1 |
| Oxo-histidine | 16 | |
| 2-Imidazolone | 16 + 1 | |
| 5-Hydroxy-2-imidazalone | 32 + 1 | |
| Tyrosine | Dityrosine | (2M + H)+ − 2 |
| 3-Chlorotyrosine c | +35 | |
| Proline | Hydroxyproline/γ-glutamyl semialdehyde | +16 |
| Pyroglutamic acid | +14 | |
| Arginine | γ-Glutamyl semialdehyde | −43 |
| Lysine | Amino-adipicsemialdehyde | −1 |
Oxidation of Cys residues can result in a range of possible oxidation products, e.g. sulfenic, sulfinic and sulfonic acid, intra- and inter-disulfide bonds and mixed disulfide bonds (reaction of Cys with low molecular weight compounds).17 The formation of disulfide bonds is reversible and is known to be of regulatory importance, linking protein activity to redox-active processes and cellsignaling,6–8 and similar results have been found for Cys oxidation to sulfenic and sulfinic acid.18,19 The formation of sulfonic acid, on the other hand, seems to be irreversible and has been detected as the result of cystineoxidation under severely oxidizing conditions, leading to breakage of the disulfide bond.12,20–22
Met residues are sensitive to most ROS and the primary oxidation product is methionine sulfoxide (MetO), which can be further irreversibly oxidized to methionine sulfone under strongly oxidizing conditions. Recently, the formation of homocysteic acid in heavily oxidized protein samples was studied by tandem mass spectrometry (MS/MS, mass shift of 34 Da).23 The exact function of Met in proteins has not yet been fully elucidated, but it has been suggested that Met residues provide an antioxidant defense mechanism in organisms.24,25 This is supported by the susceptibility of Met to oxidation, by the existence of methionine sulfoxide reductases which catalyze the reduction of MetO to Met in a wide range of organisms, and by the fact that Metoxidation, especially for surface-exposed residues, does not necessarily lead to inactivation.24,25 However, since MetO is more polar than Met, its formation can have dramatic effects on enzyme conformation, even turning a previously hydrophobic pocket inside out.9,26 Furthermore, such conformational changes upon MetO formation have been suggested to precede proteincarbonylation.27 The position of MetO has traditionally been studied by chemical backbone cleavage with CNBr (Table 2), since CNBr specifically cleaves proteins on the C-terminal side of Met except at oxidized Met residues.
| Protease/chemical | Quality (purity) | Origin | Suppliera | Cleavage position (C-terminal) |
|---|---|---|---|---|
| a Promega (Madison, USA), Sigma-Aldrich (Steinheim, Germany), Novozymes A/S (Bagsvaerd, Denmark), Princeton Separations (Adelphia, USA), Seikagaku Corporation (Tokyo, Japan). b The CNBr method entails relatively harsh hydrolysis conditions and CNBr is highly toxic. c N -Terminal side. d Preference for F, L and E, does not cleave at V, A or G. Only at pH < 6. e Preference for aromatic and uncharged amino acids. | ||||
| Chymotrypsin | Sequencing grade | Bovine | Princeton Separations | F, Y, W (not before P) |
| Chymotrypsin | For molecular biology | Bovine | Sigma-Aldrich | F, Y, W, M, L (not before P) |
| CNBr b | — | — | Sigma-Aldrich | M (not if oxidized) |
| Endoproteinase Asp-N | Suitable for protein sequencing | Pseudomonas fragi | Sigma-Aldrich | Dc |
| Endoproteinase Lys-C | Sequencing grade | Lysobacter enzymogenes | Promega | K |
| Formic acid | — | — | Sigma-Aldrich | D |
| Metalloendopeptidase Lys-N | — | Grifola frondosa | Seikagaku Corporation | K c |
| Microwave digestion 106 | — | — | — | D |
| Pepsin | For molecular biology | Porcine | Sigma-Aldrich | Non-specificd |
| Proteinase K | For molecular biology | Tritirachium album | Sigma-Aldrich | Non-specifice |
| Savinase | Detergent | Bacillus clausii | Novozymes A/S | Non-specifice |
| Trypsin | Sequencing grade | Porcine | Promega | K, R (not before P) |
| Trypsin | For molecular biology | Porcine | Sigma-Aldrich | K, R + chymotrypsin activity |
| Trypsin | Detergent | Porcine | Novozymes A/S | K, R + chymotrypsin activity + other |
Tryptophan and tyrosine might form fluorescent products upon oxidation; e.g.kynurenin (λex = 365 nm, λem = 480 nm), and N-formylkynurenine (λex = 325 nm, λem = 434 nm),28 for Trp, and dityrosine, (λem = 415 nm, λex = 325 nm) for Tyr. Trp itself emits fluorescence at λex = 295 nm, and a decrease in fluorescence has been used as a marker for Trp oxidation.29–32 However, interpretation of such a decrease is risky, since the Trp fluorescence also depends on the local environment of the Trp residue(s). Any oxidation that affects the protein conformation might alter, or even totally quench, the intensity of Trp fluorescence, even if few or no Trp residues are oxidized.33–35 It has been proposed that surface-exposed Trp residues may act as antioxidants,36 similar to Met residues, even if oxidation of Trp is also known to cause inactivation of enzymes.
Carbonylation is an irreversible and non-enzymatic modification of proteins that can lead to inactivation, crosslinking or breakdown,9,17 and it has been established that elevated levels of carbonyls are formed as a result of oxidative stress.3 However, a clear relationship between carbonylation and loss of function has not been found, possibly due to the high content of amino acids on the protein surface that are sensitive to oxidation but do not affect the activity, i.e. so-called sacrificial protection similar to that offered by Met residues.37,38 Nevertheless, proteincarbonyl groups are the most predominantly studied marker of protein oxidation and can be induced by almost all types of ROS on Arg, His, Lys, Pro and Thr residues.4,39 The most widely used method for determination of proteincarbonyl content (PCC) is to tag carbonyls with 2,4-dinitrophenylhydrazine (DNPH), as shown in Fig. 2. The dinitrophenylhydrazone product formed is stable and absorbs light at 370 nm (molar extinction coefficient of 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1), and the PCC can be determined by comparison with total protein content measured at 280 nm. The method, and suggested improvements thereof, have been extensively covered in the literature.40–43
000 M−1 cm−1), and the PCC can be determined by comparison with total protein content measured at 280 nm. The method, and suggested improvements thereof, have been extensively covered in the literature.40–43
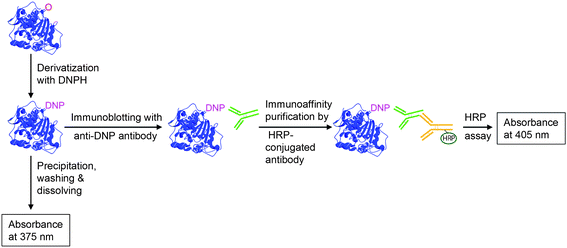 | ||
| Fig. 2 Measurement of protein carbonyls, derivatized with 2,4-dinitrophenylhydrazine (DNPH), either by a spectrophotometric or an immunodetection assay. As an alternative to horseradish peroxidase (HRP)-conjugated antibodies, antibodies conjugated with a chemiluminescent compound are also commonly used. | ||
Protein hydroperoxides are other major products of proteins exposed to ROS, but due to their reactivity they could rather be considered as reaction intermediates and have even been regarded as ROS themselves.44 Quantification of hydroperoxides can be performed with an iodometric assay under strictly anaerobic conditions,45 or with the so-called FOX assay,46 the latter of which can be combined with LC-MS/MS.47 However, in the following text, hydroperoxides will not be further discussed due to their labile nature and tendency to react further to form other more stable oxidation products.
Conformational changes, aggregation and backbone cleavage
Even for studies aimed at more specific identification of amino acidoxidation, it is often important to determine changes in the secondary and/or tertiary structure to identify possible interference with the method of choice. For example, aggregation or degradation might cause loss of protein molecules before analysis, which would be a major problem especially for quantitative evaluations.Cross-linking and aggregation due to oxidation can be evaluated using traditional SDS PAGE and/or native PAGE, size exclusion chromatography,48 or dynamic light scattering.22,49 Conformational changes are traditionally studied by circular dichroism (CD) but can also be evaluated using Fourier transform infrared FT-IR50 or MS.22,51–53 Increased hydrophobicity seems to be a consequence of such structural changes,54 which can be evaluated by hydrophobic interaction chromatography.55 A shift in the wavelength optimum for Trp fluorescence, due to changes in solvent-accessibility of Trp residues, is another indication of conformational changes.33,56
Oxidation by radicals is known to cause backbone cleavage; for an extensive compilation of the mechanisms and products formed, see Xu and Chance.12 However, fragmentation of oxidized proteins is not always a direct effect of backbone oxidation; it can also be indirectly due to conformational changes caused by oxidation of certain residues, leading to increased susceptibility to proteases.54,57 If the fragments are too small to be observed with SDS-PAGE, they might still be detectable by 2DGE,58 or by LC-MS/MS (as in Fig. 3, but leaving out the analytical protease digestion step).22 Quantification of both aggregation and fragmentation can be performed using a Bio-Rad densitometer.34
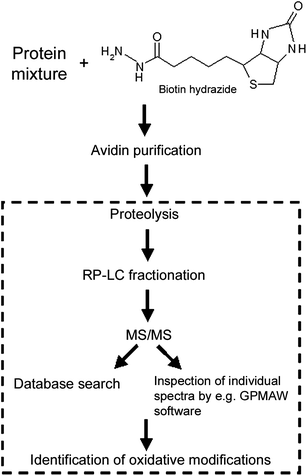 | ||
| Fig. 3 Schematic illustration of a strategy for the identification of oxidized residues in protein mixtures using biotin hydrazide labeling of carbonyls. Within the square is shown the part of the procedure which is general for detecting oxidative modifications and would be used for less complex samples such as purified proteins. | ||
Sample preparation for analysis of oxidative modifications
In principal, MS can be used for direct identification of all relatively stable amino acid side chain modifications caused by ROS, as further described below. However, for less stable modifications or for oxidized proteins in complex biological samples, a chemical labeling procedure might be necessary. In addition, for complex biological samples, different separation or enrichment techniques are sometimes needed prior to MS analysis.Separation techniques
A commonly employed method for separation of oxidized proteins in complex mixtures is gel electrophoresis, either one- or two-dimensional (2DGE), where the latter generally uses a first separation based on pI differences followed by a second separation based on differences in molecular weight.59 Drawbacks with gel-based separation include the limited sample loading capacity as well as difficulty in handling very small, very large, very acidic or very basic proteins. Non-gel approaches based on one- or two-dimensional LC methods are commonly used alternatives;60,61 these methods are especially well suited for subsequent MS/MS analysis. For the 2DLC approach, a strong cation exchange column normally constitutes the first dimension and reversed-phase (RP) chromatography the second. On-line multidimensional LC for peptide separation prior to MS/MS analysis has recently been reviewed,62 however, not in the context of protein oxidation analysis. Separation of undigested oxidized and non-oxidized proteins has been performed by anion-exchange,49RP-HPLC,63 or by using an Agilent 2100 bioanalyzer.64 The bioanalyzer separates proteins based on the gel electrophoresis principle, however, in a microfluidic chip format and using a gel that is not cross-linked.64Labeling of protein carbonyls
Visualization of proteins containing carbonyls can be accomplished by derivatization with DNPH and separation by gel electrophoresis, followed by immunodetection using antibodies specific to DNP (Fig. 2). This Western blotting method is available as commercial kits, including the Oxyblot™ kit provided by Intergen Inc (Chemicon, CA, USA), and the OxiSelect™ kit from Cell Biolabs, Inc. (San Diego, CA, USA). OxiSelect™ is available by detection with either Western blot, ELISA or spectrophotometry. These methods are all rather semiquantitative since they involve protein gel staining or antibody binding techniques. If desired, simultaneous identification and quantification of DNPH-derivatized protein carbonyls can instead be performed by ratiometric Raman spectroscopy.65Alternatively, labeling of protein carbonyls has been achieved by reaction with biotin hydrazide under mild pH conditions.66–68 The biotinylation method has been further developed for large-scale oxidative stress studies,48,60,69–71 with the general procedure as shown in Fig. 3. Biotin hydrazide labeling has also been combined with iTRAQ reagent labeling for improved identification and quantification using MS.70
Affinity selection might give false positives since some proteins may be merely associated with proteins possessing the modification under study without being oxidized themselves. Both DNPH and biotin hydrazide labeling suffer from the drawbacks of not distinguishing between carbonyls from different sources, e.g. primary and secondary carbonyl products as well as carbonyls from glycation, or conjugation with products of lipid peroxidation,4,72 and general concerns about overestimations with the DNPH assay have been reported.42,57,73 In addition, these chemicals do not react with all kinds of oxidized amino acids and probably do not even react with all different carbonylated amino acids such as N-formylkynurenine.38 Methods utilizing antibodies are quite frequently found to be rather non-specific.74 It has been noted that in oxidation studies, after enrichment of proteins with a certain modification such as carbonyls, these carbonylated proteins often also contain other oxidative modifications,48,69 which might complicate identification.
Labeling of cysteine residues
For assessment of Cys oxidation, labeling of free SHgroups can be performed with a reagent based on e.g. DTNB (Ellman's reagent), iodoacetamide or maleimide.75–77 A decrease in the amount of labeling is then used as a measure of increased Cys oxidation. In order to achieve direct monitoring of oxidized Cys and not merely the decrease in unmodified residues, Baty et al.78 blocked reduced thiols present in the protein, before reduction of oxidized thiols with dithiothreitol (DTT) and labeling with iodoacetamidofluorescein (followed by 2DGE separation). Proteins containing reversibly oxidized cysteines have been enriched in yeast cells exposed to oxidative stress by first blocking free thiols with N-ethylmalemide, followed by thiol-specific biotin–HPDP labeling.79 The proteins were then digested with trypsin, captured on streptavidin beads and finally eluted with β-mercaptoethanol and analyzed by LC-MS/MS. The method allowed identification of oxidized peptides in complex mixtures; however, it does not show which type of thioloxidation is present (e.g.sulfinic acid or disulfide bond formation), nor does it give any quantitative information. Formation of disulfide bridges upon oxidation has been specifically analyzed by 35S-Cys-labeling,80 or by using a non-sulfhydryl-containing phosphine-based disulfide-reducing agent instead of DTT or β-mercaptoethanol for reduction.81Cysteine sulfenic acids are highly unstable and therefore difficult to study, even if they have been reported to be stabilized if undergoing favorable electrostatic interaction with a neighboring amino acid, e.g.Arg, as reported for a synthetic peptide which was studied using MS/MS.82 Trapping procedures selective for sulfenic acid have also been reported using dimedone chemical reagent,83 dimedone–biotin,84 or reduction with arsenite to enable biotin–maleimidelabeling.19 Dimedone–biotin is too large to enter intact cells, whereas biotin–maleimide destroys protein structure. Therefore, Reddie et al.85 instead used a bifunctional chemical probe (an azide analog of dimedone) for selective and irreversible labeling of sulfenic-acid-containing proteins within living cells.
Labeling with so-called isotope-coded affinity tags is a technique for quantitative analysis of complex protein mixtures, and it has been employed for site-specific identification of carbonyls in yeast cells stressed with H2O2 using heavy and light isotope labeled Girard's P reagent,48 and with heavy and light isotopes of an iodoacetamide analog for evaluation of the relative sensitivity to oxidation for the Cys residues in human p21ras protein.86 However, also with this method, only the decrease in labeling of a certain residue can be measured, i.e. the modified Cys residues as such are not tagged and hence are not specifically examined.
Mass spectrometry for identification of oxidation sites
Since the development of the mild ionization techniques MALDI (matrix-assisted laser desorption/ionization) and ESI (electrospray ionization) during the 1980s, mass spectrometry has evolved as the predominant technique for the identification of proteins as well as for the characterization of protein primary structure. Accordingly, MS has been increasingly used in the field of protein oxidation, since any oxidative modification to a protein will lead to a change in mass. This mass shift can be observed using ESI-MS on the undigested protein,22,87 but more detailed information is commonly sought by performing MS and MS/MS studies on peptides (peptide mass fingerprinting) obtained from the protein (Fig. 4).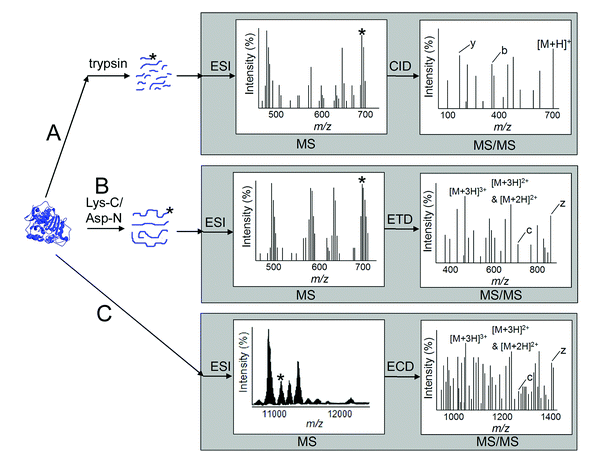 | ||
| Fig. 4 MS-based approaches for studying protein oxidation. (A) Bottom-up approach with tryptic proteolysis followed by MS and CID-MS/MS analysis of relatively small +1 and +2 charged peptides. (B) Middle-down approach with a more specific proteolysis followed by MS and ETD-MS/MS of larger +2, +3 and +4 charged peptides. (C) A top-down approach involving ESI-MS on the intact protein to determine total protein mass followed by ECD-MS/MS of a selected protein species with a modification of interest; very large and highly charged peptide fragments result, putting a premium on MS resolution and mass accuracy in interpretation. The grey boxes symbolize what takes place inside the MS equipment, the stars denote a certain peptide or protein (from an oxidized sample), chosen for CID, ETD or ECD. | ||
The most commonly used procedure for peptide mass fingerprinting is the bottom-up approach, i.e.proteasedigestion to generate peptides of mass <3 kDa, followed by separation of the peptides by LC and subsequent peptide sequencing by MALDI or ESI MS/MS (Fig. 3, lower part, and Fig. 4A), using collision induced dissociation (CID). The generated MS and MS/MSspectra are then typically exploited by comparison with peptidespectra from known proteins in bioinformatic databases such as those available at NCBI,88 and Swiss-Prot from the ExPASy proteomics server.89 For complex protein mixtures, various labeling and separation techniques are employed prior to MS analysis, e.g.derivatization of protein carbonyls with DNPH or biotinylation followed by separation by either 2DGE,60,90 or 2DLC.71,91 When the complex protein mixture is subjected to proteolysis prior to the separation and MS analysis, the technique is generally referred to as shotgun proteomics. Most often, positive MS mode is employed, but combining this with negative mode measurements has been shown to increase the number of identified oxidized peptides.23
Various search softwares, e.g. Mascot,92 and Sequest,93 are available to aid in the peptide identification. Mascot is freely available in a somewhat less extensive version than the commercial one.94 The combined technique of proteolysis, 2DLC, MS/MS and Sequest database searching is known as the multidimensional protein identification technology (MudPIT).95 Manual comparison of spectra is also possible with software programs such as GPMAW,96,97 especially advisable for verification of amino acid modifications and in ambiguous cases, since database search software has been shown to give false positive peptide identifications. An example of this is given by e.g. Chen et al., who found that the presence of unidentified peaks of high intensities in a spectrum could be an indication of a false positive.98
Trypsin is the predominant protease used in proteomic studies and is also a good choice for identification of proteins with posttranslational modifications (PTMs) such as oxidation (with only a few peptides needed from each protein). Though the use of high-specificity trypsin often suffices, the benefit of including low-specificity proteases in proteome research aimed at complete mapping of oxidation sites21 as well as phosporylation sites99,100 has been reported. Various proteases and chemicals available for protein digestion are summarized in Table 2. For a more complete list, and to obtain cleavage positions for proteins available in the Swiss-prot database, see Expasy's PeptideCutter.101 Addition of a reducing agent, e.g.DTT and β-mercaptoethanol, prior to protease digestion is often a prerequisite for achieving satisfactory sequence coverage in proteomic studies. However, the importance of running oxidation studies also without the addition of such reagents to enable the detection ofdisulfideoxidation to sulfonic acid has been highlighted.21
One outstanding advantage with MS-based techniques is of course the possibility to locate the exact position of the oxidative modification. The Mascot and Sequest programs can take into account a large number of different possible oxidative modifications (for a list, see the UNIMOD database13), but scientific guesses of which modifications to expect are often needed to a certain extent to limit the processing time. Detection of low-abundance PTMs might be facilitated by multiple rounds of MS/MS utilizing addition of already identified precursor ions to the exclusion list of the MS software.102 The development of MS/MS search tools specialized for identification of PTMs has recently been reviewed.103 Most of these tools are based on multiple round and/or tag extraction, the latter to reduce the number of candidates in the database for a certain spectrum, see for example the software InsPecT.104 Another recently developed tool is the ProtMapMS software package, aimed at protein structure studies and hence suitable for localizing the exact site of oxidative modifications.105 The verification takes into account information achieved from retention time analysis (i.e. that the oxidized peptide elutes prior to the unmodified version), and quantification is performed based on areas from the corresponding selected ion chromatograms.
Neutral loss scan
There are some peculiarities occurring during CID-MS/MS analysis that are not taken into account by most search engines such as Mascot, among which the neutral loss of sulfoxide, CH3SOH (64 Da), and sulfone, CH3SO2H (80 Da), from the side chain of Met sulfoxide or sulfone,107 respectively, are especially worth mentioning. Loss of sulfoxide is strongly diagnostic of Metoxidation; it occurs for the protonated precursor ion (Fig. 5) and also for all product ions containing the MetO. Extra peaks will appear in the MS/MSspectra at the corresponding mass-to-charge (m/z) values, and these non-sequence ions provide useful information regarding the presence and exact position of an oxidized amino acid. Some other covalent modifications similarly result in specific fragment ions (also referred to as reporter or marker ions), and a list of such useful fragment ions for simplified identification of covalent modifications has recently been published.108,109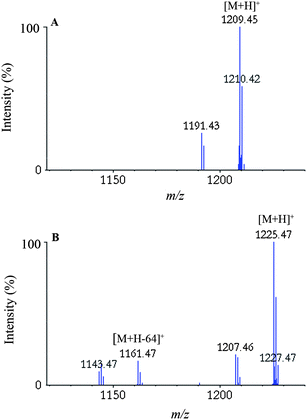 | ||
| Fig. 5 A close-up of the MS/MSspectra for the P.antarctica lipase B peptide QNCEPDLMPY: (A) without oxidation and (B) with oxidation of the Met residue, obtained after treatment with water and H2O2, respectively. The peak at m/z 1161.5 in (B) corresponds to the characteristic loss of CH3SOH occurring during MS/MS analysis of peptides containing oxidized Met. The additional peaks in the spectra correspond to the loss of water (−18 Da) from the respective ions. Reprinted from ref. 21 with permission. | ||
If peaks originating from neutral losses are present in high relative abundance, they can hamper peptide identification. This challenge might be solved by further analysis by MS3, as reported by Froelich and Reid and references therein.110 The identification of 3-chlorotyrosine, hydroxytyrosine and hydroxytryptophan after oxidation with HOCl has been performed by using diagnostic MS3 ions to map the oxidation products in a mixture of nine proteins.111 Limitations with such precursor ion scanning for the detection ofamino acidoxidation seem to be the relatively large number of false positives and the need for MS equipment with MSn capabilities. Fragmentation using the recently developed electron capture dissociation (ECD) or electron transfer dissociation (ETD) techniques provides another solution to the problem of high abundance peaks from neutral losses, as further described in the next section.
ETD/ECD and top-down approaches
The development of ECD for Fourier-transform-ion cyclotron resonance (FT-ICR) MS,112 and ETD for quadrupole ion trap MS,113 has provided an alternative to CID for MS/MS. While CID produces predominantly b- and y-ions (Fig. 4), ECD and ETD generate c- and z-type sequence ions from N–Cα backbone cleavage. In addition, ECD and ETD tend to be independent of peptide size and sequence, as well as of precursor ion proton mobility of the peptide and modified residues, and give increased backbone fragmentation without loss of the side chain of modified residues.110 Hence, unambiguous identification of the modified residue is possible, as shown for oxidized Met and His residues by a few research groups.114–116 Additionally, problems with quantification of oxidation, due to varying dissociation patterns caused by side-chain modifications when using CID, are also avoided. Since ECD breaks disulfide bonds, the implementation of this technique should simplify the determination of disulfides. Guan et al. could accordingly locate the position of MetO within a 28-amino-acid-long peptide containing an intra-disulfide linkage using ECD.115 Mixed integer linear optimization combined with ETD has been proposed as a new computational method enabling identification of PTMs in highly modified proteins,117 but has to date not been used for oxidative modifications.Top-down approaches, as opposed to the bottom-up approaches described in the previous section, have also been utilized for characterization of protein oxidation (Fig. 4). In one exemplary approach, the protein is subjected to limited dissociation in the MS to give a set of peptides with masses that sum up to the protein total mass, in principle yielding total sequence coverage.118 Modified peptides can then be easily spotted and the determination of oxidized residues can be done on these peptides in an MS/MS experiment, using CID, ETD or ECD. The high mass accuracy and mass-resolving power of modern FT-ICR- and Orbitrap™-MS instruments make them especially well suited for the identification of oxidative modifications by the top-down approach,119,120 although their current high cost (especially the case for the FT-ICR) still restricts their availability for many researchers. The top-down approach has been used to detect oxidation in viral prolyl-4-hydroxylase,121 and compared with the bottom-up approach for characterization of oxidative damage to myoglobin.122ETD/ECD is especially suited for long and highly charged peptides with z > +2 (e.g. from Lys-C or Asp-Nproteolysis, or the recently reported microwave digestion, see Table 2), while CID has been shown to be better for peptides with z = +2 (e.g. from tryptic cleavage), and bottom-up with CID and top-down with ETD/ECD seem to be complementary methods rather than competitive.122,123 Particularly suitable for purified proteins, methods based on direct infusion (top-down) provide much faster analysis compared with chromatographic peptide separation (15 min instead of 1–2 hours) and the risk of contamination between samples is also diminished.124
Large-scale studies using the top-down approach are so far scarce due to the extensive amount of data that are generated, but development of improved data analysis software is ongoing.125 In the meanwhile, middle-down approaches have been used (Fig. 4), utilizing limited proteolysis with e.g. endoproteinase Lys-C or CNBr, followed by fragmentation using higher-energy collisions in an OrbitrapMS.126
Artifactual oxidation and other challenges
Sample handling is known to be responsible for amino acidoxidation artifacts and samples should preferably be stored cold, dark and in the absence of oxygen and metal ions. Artifactual oxidation of Trp and Met to hydroxytryptophan and methionine sulfoxide, respectively, has been reported to be high in proteins separated by gel electrophoresis (as compared with in-solution digestion) prior to MS analysis.110,127Ex vivo PTMs are also known to occur during electrospray ionization, which was shown by Chen and Cook to be especially pronounced for stainless steel electrospray emitters submitted to corrosion.128 Corrosion, which occurs gradually during normal use, gives irregularities in the surface, which can cause electrical discharge, promoting the production of reactive radicals. Routine monitoring of the electrospray current may promote the detection of discharge conditions; a current above 1 µA was suggested by the authors to indicate the need for polishing of the emitter tube, or for its replacement. The degree of oxidation during electrospray ionization has also been suggested to be dependent on the needle voltage. No oxidation of Met was detected at a spray voltage below 4.2 kV, whereas 8% oxidation occurred at 4.2–4.4 kV in a study by Guzzetta et al.,129 and similar values were observed in a study by Morand et al.130 In addition, the use of formic acid instead of TFA seems to cause less artifactual Metoxidation. An oxidative modification formed prior to MS analysis can be easily distinguished from one originating from electrospray conditions by the use of LC-MS/MS, since oxidized and non-oxidized peptides will have different chromatographic retention times.22,131,132
Since disulfide bridges can be formed, or broken, as a result of oxidative stress, the amount of intact disulfide bridges might be relevant to monitor. However, since disulfides often hamper proteolysis as well as fragmentation, peptides containing disulfide bridges can be difficult to identify with MS, and the traditional MS-based method for the evaluation of which disulfide bonds are present in a protein is to compare masses of non-reduced and reduced aliquots of the protein after pepsin proteolysis.133 A specific search for an MSspectrum with m/z corresponding to the sum of two (predefined) Cys containing peptides minus 2 Da (S–S instead of two S–H) and comparison of the corresponding MS/MSspectrum with calculated expected fragments from e.g. MS-Digest (from the Sequest Browser) has been suggested to be an efficient method for detecting disulfides.134 Fragmentation of intra- and inter-peptidedisulfide bonds has actually been observed during nanoESI CID,135 and an algorithm for automatic identification of disulfide bonds in proteins after proteolysis based on their MS/MS fragmentation patterns was recently reported.136 Fragmentation of Cys-containing peptides has been investigated by FT-ICR-MS and found to vary depending on which blocking reagent (e.g.iodoacetamide or maleimide-based) was used.137Cysteine sulfinic and sulfonic acid fragmentation patterns have also been described.138
Quantification of oxidized products by using extracted ion chromatogram areas created by the MS bottom-up approach has several benefits compared with UV detection, but interpretation should be performed with care. All products cannot be assumed to ionize identically, and oxidation may alter the fragmentation characteristics,139 even if the use of isotopically labeled peptides as internal standard might provide a solution.140 The MS signal generally decreases for peptides containing modifications compared to the unmodified peptide, e.g. a Cys residue oxidized to sulfonic acid will result in a more negatively charged peptide with lower ionization efficiency in positive mode. Decreased peptide retrieval can also be an effect of decreased proteolysis caused by an increased tendency of the oxidized protein to cross-link,55,57,141 which additionally complicates data analysis due to randomly linked peptides. If oxidation instead causes partial unfolding of the protein, the opposite effect (i.e. increased proteolysis) might be observed.54,57
The large number of possible amino acid modifications after ROS treatment, the potential for (unexpected) backbone cleavage, and cross-linking certainly complicate MS analysis as well as increase the risk of false identification. An additional problem, especially in complex mixtures and samples with heavily oxidized proteins, is the presence of peptides with similar m/z or peptide isomers. It has been estimated that 10–20% of the MS/MSspectra from complex biological samples contain more than one molecular species.142 Even in a study of a purified protein (the 33 kDa lipase B from P. antarctica), after tryptic digestion of a non-oxidized sample, two peptides were found with both nearly identical m/z (1087.53 and 1087.56 Da, respectively) and elution time (U. Törnvall, unpublished data). Only one of them was detected by the database search using Mascot, whereas the other peptide present in the spectrum was conclusively identified by manual comparison of the b- and y-series ions using GPMAW. One of the two peptides was of extra interest since it contained oxidative-sensitive residues (one Met and one Trp). Clearly, the risk of co-elution increases with increased complexity of the mixture. Moreover, peptide isomers with the same sequence, differentiated only by having the same modification positioned at different amino acid residues, might be present. This complicates identification further since many fragmentation peaks of the two peptides may be the same. An algorithm for the identification of spectra containing two isomers has been presented by Chen and Bern et al.142 Site-directed mutagenesis has also been successfully used to identify oxidation sites in peptides containing more than one oxidation-sensitive amino acid,132 though this is a rather labor-intensive method. The use of ECD/ETD might, at least partly, provide a solution to the problem.
For unknown systems (e.g. if the protein sequence is not present in any database) other methods are needed for the detection of oxidized residues. De novo sequencing, in combination with a mass-based alignment algorithm, has been used by Searle et al.,143 who detected several oxidized Met and Trp residues in crystallin. A c-type ion ladder de novo sequencing method utilizing metalloendopeptidaseLys-N (Table 2) and MS/MS with ETD has recently been presented.144 A non-MS-based alternative would be total amino acid analysis, e.g. by acid hydrolysis followed by amino acid evaluation using HPLC (with e.g. UV145 or fluorescence146 detection), or GC-MS (after derivatization to volatile esters).147 Total proteinhydrolysis by enzymes offers gentler conditions and hence lowered risk of reduction/decomposition of oxidized residues, or artifactual oxidation.148–150 Drawbacks of an enzyme digestion approach might be a failure to give quantitative digestion, and potential problems caused by enzymeautolysis.
Applications
Biological samples
The identification of protein oxidation in biological samples is referred to as redox proteomics. In such studies, modifications might be missed due to the protein degradation or repair occurring in vivo, by decomposition of the modification during sample preparation, or if the method of choice detects only a limited number of the different PTMs that have formed. As an example of the latter case, in a study of metal-catalyzed oxidation of bovine serum albumin (used as a model protein of biological interest), as many as 106 different oxidized residues (including Cys, Met, Lys, Arg, Pro, Tyr, His, Phe, Trp, Asp, Asn) were detected using MALDI MS and Mascot.151 An assay aimed at measuring only PCC would miss a large number of these oxidations. In the study, total PCC analyzed by the DNPH method was found to correlate well with LC-MS/MS data on carbonyl formation on Arg, Lys and Pro.151Even though increased carbonylation has been shown to be a result of aging, it apparently occurs only on a few specific proteins.90 After selection of proteins containing carbonyls in human plasma,69 or in yeast cells exposed to oxidative stress,48 several other oxidatively modified amino acids were detected, such as Cys, Met, Trp and Tyr. Certain domains in the proteins seemed to be more susceptible to oxidation than others.48 In the study of human plasma, the specific locations of oxidations were determined by both MALDI and ESI MS/MS; more peptides were identified with the latter method.69
2DGE is widely used for protein separation in biological samples, based on isoelectric point and mass. Staining with derivatives or antibodies specific for the modifications can be used for the selection of proteins to be further studied by MS/MS. Using biotin hydrazide and 2DGE, Oh-Ishi et al. thus detected increased levels of protein carbonyls in muscles of a diabetes model rat, relative to the control.68 Tsaytler et al. used a combined method with biotinylation of both carbonyls (biotin hydrazide) and oxidized thiols (biotin maleimide), after blocking with N-ethylmaleimide.152 By affinity purification with neutravidin agarose followed by in-gel tryptic digestion and nanoLC-MS/MS, they showed that untreated cancer cells contained both carbonyls and oxidized thiols. Slade et al. used three different techniques to evaluate carbonyl formation in breast cancer cells (hydroperoxide of linoleic acid as oxidant and not ROS); two based on biotinlabeling and 2DLC-MS/MS (strong cation exchange and LC) and the third based on 2DGE, Western blot, in-gel digestion and LC-MS/MS.153 Only proteins detected by all three techniques were counted as oxidized, hence the number of false positives was presumed to be decreased. Other human diseases associated with carbonylated proteins include Alzheimer's disease, cystic fibrosis, Parkinson's disease, psoriasis and arthritis.17,39
Analysis of the Cys/CysS ratio performed by LC-FT-ICR MS and stable isotopic dilution for quantification has indicated that the level of cysteineoxidation in human plasma seems to increase with age.154 The method is proposed as suitable for routine analysis with the Cys/CysS ratio used as a biomarker of oxidative stress, instead of carbonyls. Fluorescent oxidation products with λex around 360 and λem around 430 have also been proposed as a more sensitive and reliable biomarker for oxidative stress than PCC in e.g. plasma.155 The exact nature of these products is not known and the fluorescence is thought to stem from a mixture of oxidation products, such as various oxidized aromatic residues, but also from non-protein substances in the case of complex biological samples.28,141,155
Along with the above-mentioned markers, glutamic and aminoadipic semialdehydes have also been proposed as suitable markers of oxidative stress since they generally constitute the main part of the protein carbonyls formed, and they can be more specifically assayed.150 A general conclusion that can be drawn from the studies of protein oxidation in biological samples is that the optimal marker, as well as the best method for its analysis, remains to be conclusively identified.
Protein-based pharmaceuticals
Oxidation of therapeutic proteins during production, formulation or storage can lead to variations in effective dose and hence compromise the quality of the product due to loss of biological activity, or evoke an undesirable immunogenic response or other unwanted side effects.10,156 Conversion of Met to MetO has commonly been reported for proteinpharmaceuticals during manufacture and storage, even under refrigeration.10,156–158 Determination of the amino acid position of the oxidation and/or mechanism behind the oxidative damage might shed light on the best strategy for stabilization, e.g. site-directed mutagenesis, chemical modification, physical methods (solid vs. liquid formulation), altered storage and package conditions or the use of a chemical additive such as antioxidants.In a study of metal-catalyzed oxidation of the hormone relaxin, used as a therapeutic agent, one His residue was oxidized mainly to 2-oxohistidine, and both Met residues were converted to MetO, with one being more easily oxidized than the other.159 Ji et al. used trypsin digestion and LC-MS/MS with Sequest database searching to identify oxidized residues in a parathyroid hormone as model protein.160 Different residues were oxidized depending on the ROS system used; i.e. H2O2 and tert-butyl hydroperoxide (two Met residues), Fenton reaction with iron (Met and Trp) or Fenton reaction with copper (Met, Trp and His). An MS method for peptide mapping of Metoxidation in pharmaceuticals, as well as possibilities for transfer of the method to quality control, have been proposed by Houde et al.161
Since oxidation of some amino acid residues can be very slow under normal storage conditions, forced oxidation is commonly applied to study the stability of protein therapeutic formulations, which might give valuable indications during formulation screening.160 However, the exact position and degree of a certain modification under model treatment are not necessarily the same as after long-term storage, as shown for a human recombinant monoclonal antibody.162
Plant science
In line with the situation for animals, an elevated level of oxidized proteins in plants is an indicator of stress conditions, even if oxidation of proteins has also been shown to play a key role in normal cell physiology.36 ROS are mainly formed in the chloroplast and peroxisome of photosynthesizing plant cells. In non-photosynthesizing plant cells, ROS are probably produced mainly in the mitochondrion, see Møller et al. and references therein.73N-Formylkynurenine has been detected in 29 peptides from 17 different proteins in plant mitochondria using LC-MS/MS combined with the Mascot search engine for data analysis.163There is an extensive range of enzymes involved in the removal of ROS in plants.164 The relationship between amount of oxidized protein and age seems to be more complicated in plants than in animals. An accumulation of oxidized proteins (detected by DNPHderivatization of carbonyls and the OxyBlot kit) has been shown to occur in Arabidopsis seeds both during artificial and natural aging, and a direct correlation with loss of germination viability was seen.165 However, in a study of PCC in Arabidopsis leaves by Johansson et al.,166 it was found that even if the carbonyl content increased during the first 20 days, this was followed by a drastic reduction prior to bolting and flower development (reproductive phase). One possible explanation could be the benefit of limiting the transmission of oxidatively damaged components to the offspring, and as stated by the authors, reproduction in animals also takes place at a stage when oxidative damage is low, i.e. early to middle stage of the life cycle (see also Møller et al.73).
Food industry
Similar to the situation in the health sector, protein oxidation has been less studied in food than other markers of oxidative stress (e.g.oxidation of lipids). Nevertheless, protein oxidation has been shown to lead to reduced tenderness and juiciness and hence to decreased eating quality.167 The analytical methods employed to study protein oxidation in food are mainly adapted from biomedical research, and include measurements of loss of tryptophan fluorescence, gain in fluorescence due to dityrosine formation, spectrophotometric measurements of PCC, or SDS-PAGE to study disulfide bond formation. However, a few reports on novel techniques are available, e.g. a method based on derivatization with p-aminobenzoic acid and NaCNBH3, followed by LC-ESI MSn analysis, for the detection of semialdehydes in various foodproteins.168Oxidation of proteins in milk has been reported to cause off-flavor as well as inhibit enzymatic processes important to many dairy processes, e.g.fermentation and cheese ripening.33 Upon photo-oxidation of milk proteins, LC-MS and UV detection after DNPHderivatization showed oxidation of His, Met and Trp residues.33 Structural changes were also observed. Dityrosine formation and changes in tertiary structure were detected by HPLC-fluorescence, changes in secondary structure were investigated by CD, and changes in quaternary structure were monitored by gel filtration. Determination of oxidized milk proteins has also been performed by enzymehydrolysis (pronase, leucineaminopeptidase and prolidase) followed by HPLC/UV.148
Industrial biotechnology
Despite the many favorable characteristics of enzymes as catalysts for industrial production of chemicals, such as their high specificity and selectivity, the broad front implementation of biocatalysis in industry is as yet far from reality. One major reason for this is the often low stability of enzymes under the conditions required for the industrial process. Thermostability has been in focus, but the presence of an oxidant in the reaction system might in fact hamper many promising industrial enzymes, e.g. proteases as bleaching agents due to the release of H2O2;169D-amino acid oxidase, lipases and chloroperoxidases in various reactions utilizing H2O2 as a substrate;49,170,171 and lipases used for biodiesel production due to the presence of secondary lipid peroxidation products.172The oxidation of industrial enzymes has mainly been studied using the bottom-up MS approach as described above. Obviously, when studying industrial enzymes, the conditions during oxidation are easier to mimic than for biological systems. On the other hand, the concentration of the oxidant is generally higher than during physiological conditions, and more effort is probably needed to remove the oxidant after treatment if it interferes with the analytical method, as e.g. H2O2 does in CD experiments.22 In a study of the industrially important lipase B from Pseudozyma (formerly Candida) antarctica, which is exposed to H2O2 as a reagent in the chemo-enzymatic epoxidation of vegetable oils, Törnvall et al. detected sulfoxide and sulfone formation on Met residues, Trp oxidation to hydroxytryptophan and Cys oxidation to sulfonic acid by using protease digestion directly on the immobilized enzyme followed by ESI-MS/MS and Mascot/GPMAW evaluation.21 Both trypsin and savinase were utilized for proteolysis, in order to maximize sequence coverage, and in total 11 oxidative modifications were observed after treatment with H2O2.
The enzymeD-amino acid oxidase is utilized industrially for conversion of cephalosporin C.49 However, denaturation due to protein oxidation by O2 or H2O2, which are both present during the process, constitutes a serious obstacle for cost-effective use of this enzyme. In a study by de la Mata et al.,173 the deactivation of D-amino acid oxidase by H2O2 was correlated with oxidation of Trp (measured as disappearance of fluorescence) and Cys (detected by Ellman's reagent DTNB) residues, and these modifications were shown to hamper proper dimerization of the enzyme. Alkylation of Met residues by 14C-labeled iodoacetic acid revealed that no Metoxidation had occurred. Slavica et al. showed by ESI-MS/MS that the amino acid oxidase produced by Trigonopsis variabilis contained a fraction with the surface-exposed Cys108 oxidized to sulfinic acid.49 The oxidized fraction comprised about one fourth of the biocatalyst preparation and could be separated from the non-oxidized form using anion-exchange chromatography.
The inactivation of a liquid detergent subtilisin enzyme by H2O2 has been shown to be due to the oxidation of one of its five Met residues and more stable variants have been produced by saturated mutagenesis of this residue.174 A solid formulation of the detergentenzyme savinase has been studied using CNBr cleavage for the detection of MetO after exposure to H2O2. The most exposed but partially buried Met222 was found to be the only oxidized Met (the other two Met residues are more structurally hindered and inaccessible to solvent).169
The chloroperoxidase from Caldariomyces fumago can catalyze a wide range of industrially important oxygeninsertion reactions. Unfortunately, it displays low operational stability, especially when H2O2 is used as the oxidant. Grey et al. used on-line tryptic digestion of the enzyme followed by LC-MS/MS and observed that in the presence of moderate concentrations of H2O2, two Met residues (and one Trp) were oxidized without loss of enzyme activity.170 However, H2O2 in combination with the chloroperoxidase substrate indole inactivated the enzyme and the cause for this was found to be the oxidation of the Cys50 residue to sulfonic acid (+48 Da). Cys50 functions as the ligand for the iron in the hemeprosthetic group of the chloroperoxidase, and after oxidative inactivation, the hemegroup was reported to be absent.
Conclusion
Protein oxidation is intensively studied, as illustrated by the growing number of publications available on this subject. This research has been enabled mainly due to new improvements within the field of proteomics. However, in the area of redox proteomics, the optimal marker of oxidative stress has not yet been identified. Indeed, if it is widely found that different diseases affect different targets, such a marker may not even exist. The question of whether the modifications cause a disease to progress or rather simply accompany ongoing pathological processes remains to be conclusively answered. Nevertheless, protein carbonyls, dityrosine, disulfide bonds, products of tryptophanoxidation and others have all been proposed as suitable for assessment of oxidative stress and human diseases, and the debate is still ongoing. More research is needed to validate the different markers and their relationship with oxidative stress, as well as to find the best analytical technique to identify and quantify them. Clearly, an important aid in this research will be the development of more sophisticated MS equipment and methods, such as ECD and ETD, which are MS fragmentation methods complementary to CID. This development will also be beneficial in studies of oxidative inactivation of industrially important enzymes, and can hopefully shed some light on enzyme stability as well as provide clues on how to create biocatalysts more optimized to withstand harsh industrial conditions.Abbreviations
| CD | circular dichroism |
| CID | collision induced dissociation |
| DNP | 2,4-dinitrophenol |
| DNPH | 2,4-dinitrophenylhydrazine |
| DTT | dithiothreitol |
| ECD | electron capture dissociation |
| ETD | electron transfer dissociation |
| ESI | electrospray ionization |
| FT-IR | fourier transform infrared |
| FT-ICR | fourier-transform-ion cyclotron resonance |
| MALDI | matrix-assisted laser desorption/ionization |
| MetO | methionine sulfoxide |
| MudPIT | multidimensional protein identification technology |
| MS | mass spectrometry |
| MS/MS | tandem mass spectrometry |
| m/z | mass-to-charge |
| PCC | protein carbonyl content |
| PTM | posttranslational modification |
| ROS | reactive oxygen species |
| RP | reversed-phase |
| 2DGE | two-dimensional gel electrophoresis |
| 2DLC | two-dimensional liquid chromatography |
Acknowledgements
The author would like to thank the Swedish Research Council for financial support. I am grateful to Dr Curt Reimann and Dr Martin Hedström, Lund University, for critical discussion and proofreading.References
- B. Halliwell and J. M. C. Gutteridge, Free Radicals in Biology and Medicine, Clarendon Press, Oxford, 1989, pp. 96–98 Search PubMed.
- B. D′Autréaux and M. B. Toledano, Nat. Rev. Mol. Cell Biol., 2007, 8, 813–824 CrossRef CAS.
- B. S. Berlett and E. R. Stadtman, J. Biol. Chem., 1997, 272, 20313–20316 CrossRef CAS.
- E. Shacter, Drug Metab. Rev., 2000, 32, 307–326 CrossRef CAS.
- A. Claiborne, H. Miller, D. Parsonage and R. P. Ross, FASEB J., 1993, 7, 1483–1490 CAS.
- T. Finkel, FEBS Lett., 2000, 476, 52–54 CrossRef CAS.
- I. M. Møller and L. J. Sweetlove, Trends Plant Sci., 2010, 15, 370–374 CrossRef CAS.
- S. G. Rhee, Y. S. Bae, S. Lee and J. Kwon, Sci. STKE, 2000, 2000, PE1 Search PubMed.
- M. J. Davies, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1703, 93–109 CrossRef CAS.
- S. Li, C. Schöneich and R. T. Borchardt, Biotechnol. Bioeng., 1995, 48, 490–500 CrossRef CAS.
- G. V. Buxton, C. L. Greeenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CAS.
- G. Xu and M. R. Chance, Chem. Rev., 2007, 107, 3514–3543 CrossRef CAS.
- UNIMOD, http://www.unimod.org/, retrieved March 2010.
- K. L. Schey and E. L. Finley, Acc. Chem. Res., 2000, 33, 299–306 CrossRef CAS.
- M. R. M. Domingues, P. Domingues, A. Reis, C. Fonseca, F. M. L. Amado and A. J. V. Ferrer-Correia, J. Am. Soc. Mass Spectrom., 2003, 14, 406–416 CrossRef CAS.
- D. I. Pattison and M. J. Davies, Curr. Med. Chem., 2006, 13, 3271–3290 CrossRef CAS.
- I. Dalle-Donne, D. Giustarini, R. Colombo, R. Rossi and A. Milzani, Trends Mol. Med., 2003, 9, 169–176 CrossRef.
- C. Jacob, A. L. Holme and F. H. Fry, Org. Biomol. Chem., 2004, 2, 1953–1956 RSC.
- A. T. Saurin, H. Neubert, J. P. Brennan and P. Eaton, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17982–17987 CrossRef CAS.
- C. R. Robbins and M. K. Bahl, J. Soc. Cosmet. Chem., 1984, 35, 379–390 Search PubMed.
- U. Törnvall, C. M. Fürst, R. Hatti-Kaul and M. Hedström, Rapid Commun. Mass Spectrom., 2009, 23, 2959–2964 CrossRef.
- U. Törnvall, M. Hedström, K. Schillén and R. Hatti-Kaul, Biochimie, 2010 DOI:10.1016/j.biochi.2010.1007.1008.
- M. Bern, J. Saladino and J. S. Sharp, Rapid Commun. Mass Spectrom., 2010, 24, 768–772 CrossRef CAS.
- R. L. Levine, L. Mosoni, B. S. Berlett and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 15036–15040 CrossRef CAS.
- J. Moskovitz, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1703, 213–219 CrossRef CAS.
- W. Vogt, Free Radical Biol. Med., 1995, 18, 93–105 CrossRef CAS.
- J. Moskovitz and D. B. Oien, Antioxid. Redox Signaling, 2010, 12, 405–415 Search PubMed.
- Y. Fukunaga, Y. Katsuragi, T. Izumi and F. Sakiyama, J. Biochem., 1982, 92, 129–141 CAS.
- E. E. Dubinina, S. V. Gavrovskaya, E. V. Kuzmich, N. V. Leonova, M. G. Morozova, S. V. Kovrugina and T. A. Smirnova, Biochemistry (Moscow), 2002, 67, 343–350 CrossRef CAS.
- M. Estévez, P. Kylli, E. Puolanne, R. Kivikari and M. Heinonen, Meat Sci., 2008, 80, 1290–1296 CrossRef CAS.
- A. Giessauf, E. Steiner and H. Esterbauer, Biochim. Biophys. Acta, 1995, 1256, 221–232.
- M. Heinonen, D. Rein, M. Satué-Gracia, S. Huang, J. German and E. Frankel, J. Agric. Food Chem., 1998, 46, 917–922 CrossRef CAS.
- T. K. Dalsgaard, D. Otzen, J. H. Nielsen and L. B. Larsen, J. Agric. Food Chem., 2007, 55, 10968–10976 CrossRef CAS.
- K. J. A. Davies, M. E. Delsignore and S. W. Lin, J. Biol. Chem., 1987, 262, 9902–9907 CAS.
- J. P. Privat, R. Lotan, P. Bouchard, N. Sharon and M. Monsigny, Eur. J. Biochem., 1976, 68, 563–572 CrossRef CAS.
- S. Rinalducci, L. Murgiano and L. Zolla, J. Exp. Bot., 2008, 59, 3781–3801 CrossRef CAS.
- A. T. Nguyen and R. P. Donaldson, Arch. Biochem. Biophys., 2005, 439, 25–31 CrossRef CAS.
- L. J. Sweetlove and I. M. Møller, Adv. Bot. Res., 2009, 52, 1–23 Search PubMed.
- M. F. Beal, Free Radical Biol. Med., 2002, 32, 797–803 CrossRef CAS.
- J. M. Fagan, B. G. Sleczka and I. Sohar, Int. J. Biochem. Cell Biol., 1999, 31, 751–757 CrossRef CAS.
- R. L. Levine, D. Garland, C. N. Oliver, A. Amici, I. Climent, A. G. Lenz, B. W. Ahn, S. Shaltiel and E. R. Stadtman, Methods Enzymol., 1990, 186, 464–478 CAS.
- S. Luo and N. B. Wehr, Redox Rep., 2009, 14, 159–167 Search PubMed.
- A. Z. Reznick and L. Packer, Methods Enzymol., 1994, 233, 357–363 CAS.
- J. M. Gebicki, Redox Rep., 1997, 3, 99–110 Search PubMed.
- W. Jessup, R. T. Dean and J. M. Gebicki, Methods Enzymol., 1994, 233, 289–303 CAS.
- S. P. Wolff, Methods Enzymol., 1994, 233, 182–189 CAS.
- P. E. Morgan, D. I. Pattison, C. L. Hawkins and M. J. Davies, Free Radical Biol. Med., 2008, 45, 1279–1289 CrossRef CAS.
- H. Mirzaei and F. Regnier, J. Chromatogr., A, 2007, 1141, 22–31 CrossRef CAS.
- A. Slavica, I. Dib and B. Nidetzky, Appl. Environ. Microbiol., 2005, 71, 8061–8068 CrossRef CAS.
- P. Lasch, T. Petras, O. Ullrich, J. Backmann, D. Naumann and T. Grune, J. Biol. Chem., 2001, 276, 9492–9502 CrossRef CAS.
- R. Grandori, J. Mass Spectrom., 2003, 38, 11–15 CrossRef CAS.
- L. Konermann and D. J. Douglas, Biochemistry, 1997, 36, 12296–12302 CrossRef CAS.
- S. Venkatesh, K. B. Tomer and J. S. Sharp, Rapid Commun. Mass Spectrom., 2007, 21, 3927–3936 CrossRef CAS.
- T. Grune, T. Reinheckel and K. J. A. Davies, FASEB J., 1997, 11, 526–534 CAS.
- C. Giulivi, R. E. Pacifici and K. J. A. Davies, Arch. Biochem. Biophys., 1994, 311, 329–341 CrossRef CAS.
- A. Milzani, R. Rossi, P. D. Simplicio, D. Giustarini, R. Colombo and I. Dalle-Donne, Protein Sci., 2000, 9, 1774–1782 CrossRef CAS.
- R. T. Dean, S. Fu, R. Stocker and M. J. Davies, Biochem. J., 1997, 324, 1–18 CAS.
- L. J. Sweetlove, J. L. Heazlewood, V. Herald, R. Holtzapffel, D. A. Day, C. J. Leaver and A. H. Millar, Plant J., 2002, 32, 891–904 CrossRef CAS.
- D. Sheehan, Biochem. Biophys. Res. Commun., 2006, 349, 455–462 CrossRef CAS.
- B.-S. Yoo and F. E. Regnier, Electrophoresis, 2004, 25, 1334–1341 CrossRef CAS.
- K. Zhu, M. T. Kachman, F. R. Miller, D. M. Lubman and R. Zand, J. Chromatogr., A, 2004, 1053, 133–142 CrossRef CAS.
- H. Malerod, E. Lundanes and T. Greibrokk, Anal. Methods, 2010, 2, 110–122 RSC.
- C. V. Olson, D. H. Reifsnyder, E. Canova-Davis, V. T. Ling and S. E. Builder, J. Chromatogr., A, 1994, 675, 101–112 CrossRef CAS.
- S. Somani, G. Mandal, S. Banerjee, K. S. Prasad and S. Padmanabhan, Anal. Lett., 2009, 42, 1070–1083 CrossRef CAS.
- D. Zhang, D. Jiang, M. Yanney, S. Zou and A. Sygula, Anal. Biochem., 2009, 391, 121–126 CrossRef CAS.
- W.-G. Chung, C. L. Miranda and C. S. Maier, Electrophoresis, 2008, 29, 1317–1324 CrossRef CAS.
- D. J. O'Shannessy, M. J. Dobersen and R. H. Quarles, Immunol. Lett., 1984, 8, 273–277 CrossRef CAS.
- M. Oh-Ishi, T. Ueno and T. Maeda, Free Radical Biol. Med., 2003, 34, 11–22 CrossRef CAS.
- A. G. Madian and F. E. Regnier, J. Proteome Res., 2010, 9, 1330–1343 CrossRef CAS.
- D. L. Meany, H. Xie, L. V. Thompson, E. A. Arriaga and T. J. Griffin, Proteomics, 2007, 7, 1150–1163 CrossRef CAS.
- B. A. Soreghan, F. Yang, S. N. Thomas, J. Hsu and A. J. Yang, Pharm. Res., 2003, 20, 1713–1720 CrossRef CAS.
- R. L. Levine, N. Wehr, J. A. Williams, E. R. Stadtman and E. Shacter, Methods Enzymol., 1994, 99, 15–24.
- I. M. Møller, P. E. Jensen and A. Hansson, Annu. Rev. Plant Biol., 2007, 58, 459–481 CrossRef.
- C. Schöneich and V. S. Sharov, Free Radical Biol. Med., 2006, 41, 1507–1520 CrossRef.
- S. L. Cuddihy, J. W. Baty, K. K. Brown, C. C. Winterbourn and M. B. Hampton, Methods Mol. Biol., 2009, 519, 363–375 CAS.
- P. Eaton, Free Radical Biol. Med., 2006, 40, 1889–1899 CrossRef CAS.
- J. P. Fabisiak, A. Sedlov and V. E. Kagan, Antioxid. Redox Signaling, 2002, 4, 855–865 Search PubMed.
- J. W. Baty, M. B. Hampton and C. C. Winterbourn, Proteomics, 2002, 2, 1261–1266 CrossRef CAS.
- B. McDonagh, S. Ogueta, G. Lasarte, C. A. Padilla and J. A. Bárcena, J. Proteomics, 2009, 72, 677–689 CrossRef CAS.
- C. Appenzeller-Herzog, J. Riemer, B. Christensen, E. S. Sorensen and L. Ellgaard, EMBO J., 2008, 27, 2977–2987 CrossRef CAS.
- L. M. Landino, T. E. Skreslet and J. A. Alston, J. Biol. Chem., 2004, 279, 35101–35105 CrossRef CAS.
- V. Shetty, D. S. Spellman and T. A. Neubert, J. Am. Soc. Mass Spectrom., 2007, 18, 1544–1551 CrossRef CAS.
- W. S. Willett and S. D. Copley, Chem. Biol., 1996, 3, 851–857 CrossRef CAS.
- R. L. Charles, E. Schröder, G. May, P. Free, P. R. J. Gaffney, R. Wait, S. Begum, R. J. Heads and P. Eaton, Mol. Cell. Proteomics, 2007, 6, 1473–1484 CrossRef CAS.
- K. G. Reddie, Y. H. Seo, W. B. Muse, III, S. E. Leonard and K. S. Carroll, Mol. BioSyst., 2008, 4, 521–531 RSC.
- M. Sethuraman, N. Clavreul, H. Huang, M. E. McComb, C. E. Costello and R. A. Cohen, Free Radical Biol. Med., 2007, 42, 823–829 CrossRef CAS.
- D. Manzanares, K. Rodriguez-Capote, S. Liu, T. Haines, Y. Ramos, L. Zhao, A. Doherty-Kirby, G. Lajoie and F. Possmayer, Biochemistry, 2007, 46, 5604–5615 CrossRef CAS.
- NCBI, http://www.ncbi.nlm.nih.gov/, retrieved August 2010.
- ExPASy Proteomics Server—Swiss-Prot, http://www.expasy.ch/sprot/, retrieved August 2010.
- K. England and T. Cotter, Biochem. Biophys. Res. Commun., 2004, 320, 123–130 CrossRef CAS.
- B. K. Kristensen, P. Askerlund, N. V. Bykova, H. Egsgaard and I. M. Møller, Phytochemistry, 2004, 65, 1839–1851 CrossRef CAS.
- D. N. Perkins, D. J. C. Pappin, D. M. Creasy and J. S. Cottrell, Electrophoresis, 1999, 20, 3551–3567 CrossRef CAS.
- J. K. Eng, A. L. McCormack and J. R. Yates, J. Am. Soc. Mass Spectrom., 1994, 5, 976–989 CrossRef.
- Mascot Science, http://www.matrixscience.com/, retrieved April 2010.
- M. P. Washburn, D. Wolters and J. R. Yates, Nat. Biotechnol., 2001, 19, 242–247 CrossRef CAS.
- GPMAW, http://www.gpmaw.com/, retrieved March 2010.
- J. Bunkenborg and R. Matthiesen, Interpretation of Collision-Induced Fragmentation Tandem Mass Spectra of Posttranslationally Modified Peptides Methods in Molecular Biology: Mass Spectrometry Data Analysis in Proteomics, Humana Press Inc., Totowa, 2007, pp. 169–194 Search PubMed.
- Y. Chen, J. Zhang, G. Xing and Y. Zhao, J. Proteome Res., 2009, 8, 3141–3147 CrossRef CAS.
- A. Schlosser, J. T. Vanselow and A. Kramer, Anal. Chem., 2005, 77, 5243–5250 CrossRef CAS.
- B. Wang, R. Malik, E. A. Nigg and R. Korner, Anal. Chem., 2008, 80, 9526–9533 CrossRef CAS.
- Expasy PeptideCutter, http://www.expasy.ch/tools/peptidecutter, 2009.
- J. Seo, J. Jeong, Y. M. Kim, N. Hwang, E. Paek and K. Lee, J. Proteome Res., 2008, 7, 587–602 CrossRef CAS.
- E. Ahrné, M. Müller and F. Lisacek, Proteomics, 2010, 10, 671–686 CrossRef CAS.
- S. Tanner, H. Shu, A. Frank, L. Wang, E. Zandi, M. Mumby, P. A. Pevzner and V. Bafna, Anal. Chem., 2005, 77, 4626–4639 CrossRef CAS.
- P. Kaur, J. G. Kiselar and M. R. Chance, Anal. Chem., 2009, 81, 8141–8149 CrossRef CAS.
- N. J. Hauser, H. Han, S. A. McLuckey and F. Basile, J. Proteome Res., 2008, 7, 1867–1872 CrossRef CAS.
- F. M. Lagerwerf, M. van de Weert, W. Heerma and J. Haverkamp, Rapid Commun. Mass Spectrom., 1996, 10, 1905–1910 CrossRef CAS.
- C. Hung, A. Schlosser, J. Wei and W. D. Lehmann, Anal. Bioanal. Chem., 2007, 389, 1003–1016 CrossRef CAS.
- J. Roeser, R. Bischoff, A. P. Bruins and H. P. Permentier, Anal. Bioanal. Chem., 2010, 397, 3441–3455 CrossRef CAS.
- J. M. Froelich and G. E. Reid, Proteomics, 2008, 8, 1334–1345 CrossRef CAS.
- L. Mouls, E. Silajdzic, N. Haroune, C. M. Spickett and A. R. Pitt, Proteomics, 2009, 9, 1617–1631 CrossRef CAS.
- R. A. Zubarev, N. L. Kelleher and F. W. McLafferty, J. Am. Chem. Soc., 1998, 120, 3265–3266 CrossRef CAS.
- J. E. P. Syka, J. J. Coon, M. J. Schroeder, J. Shabanowitz and D. F. Hunt, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9528–9533 CrossRef CAS.
- R. Bakhtiar and Z. Guan, Biotechnol. Lett., 2006, 28, 1047–1059 CrossRef CAS.
- Z. Guan, N. A. Yates and R. Bakhtiar, J. Am. Soc. Mass Spectrom., 2003, 14, 605–613 CrossRef CAS.
- R. Srikanth, J. Wilson and R. W. Vachet, J. Mass Spectrom., 2009, 44, 755–762 CrossRef CAS.
- P. A. DiMaggio, N. L. Young, R. C. Balibant, B. A. Garcia and C. A. Floudas, Mol. Cell. Proteomics, 2009, 8, 2527–2543 CrossRef CAS.
- N. L. Kelleher, H. Y. Lin, G. A. Valaskovic, D. J. Aaserud, E. K. Fridriksson and F. W. McLafferty, J. Am. Chem. Soc., 1999, 121, 806–812 CrossRef CAS.
- J. Reinders and A. Sickmann, Biomol. Eng., 2007, 24, 169–177 CrossRef CAS.
- C. Zhao, M. Sethuraman, N. Clavreul, P. Kaur, R. A. Cohen and P. B. O'Connor, Anal. Chem., 2006, 78, 5134–5142 CrossRef CAS.
- Y. Ge, B. G. Lawhorn, M. Elnaggar, S. K. Sze, T. P. Begley and F. W. McLafferty, Protein Sci., 2003, 12, 2320–2326 CAS.
- L. J. Deterding, S. Bhattacharjee, D. C. Ramirez, R. P. Mason and K. B. Tomer, Anal. Chem., 2007, 79, 6236–6248 CrossRef CAS.
- J. Wiesner, T. Premsler and A. Sickmann, Proteomics, 2008, 8, 4466–4483 CrossRef CAS.
- S. Barnes, E. M. Shonsey, S. M. Eliuk, D. Stella, K. Barrett, O. P. Srivastava, H. Kim and M. B. Renfrow, Biochem. Soc. Trans., 2008, 36, 1037–1044 CrossRef CAS.
- B. A. Garcia, J. Am. Soc. Mass Spectrom., 2010, 21, 193–202 CrossRef CAS.
- J. Zhang, H. Liu and V. Katta, J. Mass Spectrom., 2010, 45, 112–120 CAS.
- I. Perdivara, L. J. Deterding, M. Przybylski and K. B. Tomer, J. Am. Soc. Mass Spectrom., 2010, 21, 1114–1117 CrossRef CAS.
- M. Chen and K. D. Cook, Anal. Chem., 2007, 79, 2031–2036 CrossRef CAS.
- A. W. Guzzetta, R. A. Thakur and I. C. Mylchreest, Rapid Commun. Mass Spectrom., 2002, 16, 2067–2072 CrossRef CAS.
- K. Morand, G. Talbo and M. Mann, Rapid Commun. Mass Spectrom., 1993, 7, 738–743 CAS.
- K. Gevaert, J. Van Damme, M. Goethals, G. R. Thomas, B. Hoorelbeke, H. Demol, L. Martens, M. Puype, A. Staes and J. Vandekerckhove, Mol. Cell. Proteomics, 2002, 1, 896–903 CrossRef CAS.
- S. W. Griffiths and C. L. Cooney, J. Chromatogr., A, 2002, 942, 133–143 CrossRef CAS.
- J. J. Gorman, T. P. Wallis and J. J. Pitt, Mass Spectrom. Rev., 2002, 21, 183–216 CrossRef CAS.
- T. Yen and B. A. Macher, Methods Enzymol., 2006, 415, 103–113 CAS.
- M. Mormann, J. Eble, C. Schwöppe, R. M. Mesters, W. E. Berdel, J. Peter-Katalinic and G. Pohlentz, Anal. Bioanal. Chem., 2008, 392, 831–838 CrossRef CAS.
- S. Choi, J. Jeong, S. Na, H. S. Lee, H. Kim, K. Lee and E. Paek, J. Proteome Res., 2010, 9, 626–635 CrossRef CAS.
- S. M. Chowdhury, G. R. Munske, R. C. Ronald and J. E. Bruce, J. Am. Soc. Mass Spectrom., 2007, 18, 493–501 CrossRef CAS.
- Y. Wang, S. Vivekananda, L. Men and Q. Zhang, J. Am. Soc. Mass Spectrom., 2004, 15, 697–702 CrossRef CAS.
- T. Grunert, K. Pock, A. Buchacher and G. Allmaier, Rapid Commun. Mass Spectrom., 2003, 17, 1815–1824 CrossRef CAS.
- C. J. Silva, B. C. Onisko, I. Dynin, M. L. Erickson, W. H. Vensel, J. R. Requena, E. M. Antaki and J. M. Carter, Biochemistry, 2010, 49, 1854–1861 CrossRef CAS.
- A. J. Grant, W. Jessup and R. T. Dean, Free Radical Biol. Med., 1993, 18, 259–267 CAS.
- X. Chen, P. Drogaris and M. W. Bern, J. Proteome Res., 2010, 9, 3270–3279 CrossRef CAS.
- B. C. Searle, S. Dasari, P. A. Wilmarth, M. Turner, A. P. Reddy, L. L. David and S. R. Nagalla, J. Proteome Res., 2005, 4, 546–554 CrossRef CAS.
- N. Taouatas, M. M. Drugan, A. J. R. Heck and S. Mohammad, Nat. Methods, 2008, 5, 405–407 CrossRef CAS.
- W. J. M. Underberg, M. A. Hoitink, J. L. E. Reubsaet and J. C. M. Waterval, J. Chromatogr., B: Biomed. Sci. Appl., 2000, 742, 401–409 CrossRef CAS.
- T. Fox, G. Tsaprailis and A. M. English, Biochemistry, 1994, 33, 186–191 CrossRef CAS.
- J. W. Heinecke, F. F. Hsu, J. R. Crowley, S. L. Hazen, C. Leeuwenburgh, D. M. Mueller, J. E. Rasmussen and J. Turk, Methods Enzymol., 1999, 300, 124–144 CAS.
- J. H. Baxter, C.-S. Lai, R. R. Phillips, L. Dowlati, J. J. Chio, S. T. Luebbers, S. R. Dimler and P. W. Johns, J. Chromatogr., A, 2007, 1157, 10–16 CrossRef CAS.
- J. Pietzsch, Biochem. Biophys. Res. Commun., 2000, 270, 852–857 CrossRef CAS.
- J. R. Requena, C.-C. Chao, R. L. Levine and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 69–74 CrossRef CAS.
- S. Guedes, R. Vitorino, R. Domingues, F. Amado and P. Domingues, Rapid Commun. Mass Spectrom., 2009, 23, 2307–2315 CrossRef CAS.
- P. A. Tsaytler, M. C. O'Flaherty, D. V. Sakharov, J. Krijgsveld and M. R. Egmond, J. Proteome Res., 2008, 7, 3868–3878 CrossRef CAS.
- P. G. Slade, M. V. Williams, V. Brahmbhatt, A. Dash, J. S. Wishnok and S. R. Tannenbaum, Chem. Res. Toxicol., 2010, 23, 557–567 CrossRef CAS.
- J. M. Johnson, F. H. Strobel, M. Reed, J. Pohl and D. P. Jones, Clin. Chim. Acta, 2008, 396, 43–48 CrossRef CAS.
- T. Wu, W. C. Willett, N. Rifai and E. B. Rimm, Am. J. Epidemiol., 2007, 166, 552–560 CrossRef.
- J. L. Cleland, M. F. Powell and S. J. Shire, Crit. Rev. Ther. Drug Carrier Syst., 1993, 10, 307–377 CAS.
- D. Liu, D. Ren, H. Huang, J. Dankberg, R. Rosenfeld, M. J. Cocco, L. Li, D. N. Brems and R. L. Remmele, Biochemistry, 2008, 47, 5088–5100 CrossRef CAS.
- M. C. Manning, D. K. Chou, B. M. Murphy, R. W. Payne and D. S. Katayama, Pharm. Res., 2010, 27, 544–575 CrossRef.
- S. Li, T. H. Nguyen, C. Schöneich and R. T. Borchardt, Biochemistry, 1995, 34, 5762–5772 CrossRef CAS.
- J. A. Ji, B. Zhang, W. Cheng and Y. J. Wang, J. Pharm. Sci., 2009, 98, 4485–4500 CrossRef CAS.
- D. Houde, P. Kauppinen, R. Mhatre and Y. Lyubarskaya, J. Chromatogr., A, 2006, 1123, 189–198 CrossRef CAS.
- C. Chumsae, G. Gaza-Bulseco, J. Sun and H. Liu, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 850, 285–294 CrossRef CAS.
- I. M. Møller and B. K. Kristensen, Free Radical Biol. Med., 2006, 40, 430–435 CrossRef CAS.
- R. Mittler, S. Vanderauwera, M. Gollery and F. Van Breusegem, Trends Plant Sci., 2004, 9, 490–498 CrossRef CAS.
- L. Rajjou, Y. Lovigny, S. P. C. Groot, M. Belghazi, C. Job and D. Job, Plant Physiol., 2008, 148, 620–641 CrossRef CAS.
- E. Johansson, O. Olsson and T. Nyström, J. Biol. Chem., 2004, 279, 22204–22208 CrossRef CAS.
- M. N. Lund, R. Lametsch, M. S. Hviid, O. N. Jensen and L. H. Skibsted, Meat Sci., 2007, 77, 295–303 CrossRef CAS.
- M. Estévez, V. Ollilainen and M. Heinonen, J. Agric. Food Chem., 2009, 57, 3901–3910 CrossRef CAS.
- S. Biran, A. D. Jensen, S. Kiil, P. Bach and O. Simonsen, J. Biotechnol., 2009, 141, 73–79 CrossRef CAS.
- C. E. Grey, M. Hedström and P. Adlercreutz, ChemBioChem, 2007, 8, 1055–1062 CrossRef CAS.
- U. Törnvall, C. Orellana-Coca, R. Hatti-Kaul and D. Adlercreutz, Enzyme Microb. Technol., 2007, 40, 447–451 CrossRef CAS.
- D. Pirozzi, Eur. J. Lipid Sci. Technol., 2003, 105, 608–613 CrossRef CAS.
- I. de la Mata, F. Ramón, V. Obregón, M. P. Castillón and C. Acebal, Enzyme Microb. Technol., 2000, 27, 234–239 CrossRef CAS.
- D. A. Estell, T. P. Graycar and J. A. Wells, J. Biol. Chem., 1985, 260, 6518–6521 CAS.
| This journal is © The Royal Society of Chemistry 2010 |