Sample preparation of tropical and subtropical fruit biowastes to determine antioxidant phytochemicals
Mónica
González
*a and
Venerando
González
b
aPost-harvest and Food Technology Laboratory, Department of Tropical Fruit Crops, Instituto Canario de Investigaciones Agrarias, Apdo. 60, 38200, La Laguna, Spain. E-mail: mgonzal@icia.es; Fax: +34 922 476303; Tel: +34 922 476310
bDepartment of Analytical Chemistry, Nutrition and Food Science, University of La Laguna, Campus de Anchieta, Astrofísico Francisco Sánchez s/n, 38205, La Laguna, Spain
First published on 2nd November 2010
Abstract
This work is a review of the analytical strategies dealing with antioxidant phytochemicals in tropical and subtropical fruit by-products. The determination of bioactive compounds encompasses a number of different aspects and the analytical strategy employed depends on the biowaste, analyte and nature of the problem. In general, an analytical strategy involves recovering antioxidant phytochemicals from the sample matrix followed by separation, identification and measurement. For most phytochemicals, the recovery step typically involves extraction using a range of solvents. However, sample handling is often an ignored feature of the analysis. This review highlights the importance of sample preparation in the analysis of phytochemicals from tropical and subtropical fruit biowastes and the problems that can arise during this step. The various procedures are summarized and some typical “case studies” are presented.
1. Introduction
The main objective of fruit and vegetable processing is to supply wholesome, safe, nutritious and acceptable food to consumers throughout the year. However, this processing generates high amounts of biowaste materials such as peels, seeds and stones. Due to increasing production, disposal of these materials represents a growing problem, because they contain large quantities of nitrogen and phosphorus and their high water content makes them susceptible to modification by microorganisms. From an environmental perspective it is vital that plant by-products produced by the agro-food industry be reused. For example, the European Union requires that Member States reduce organic biodegradable waste in landfills by 65% (compared to 1995 levels) by no later than 20161 and that they must take the necessary measures to ensure that biowaste undergoes recovery operations.2Agro-industry waste is often utilized as feed or fertilizer. However, the cost of drying, storing and shipping biowastes limits the economic value of such products. One economically viable use for these wastes that is gaining interest is as food additives or supplements with high nutraceutical value.3
Multiple epidemiological studies have pointed out that consumption of fruits and vegetables imparts health benefits, such as reduced risk of coronary heart disease and stroke, as well as certain types of cancer. Apart from dietary fiber, these health benefits are mainly attributed to organic phytochemicals, such as polyphenolic compounds, carotenoids, tocopherols and sterols, among others, contained in fruits and vegetables. These antioxidant phytochemicals are also contained in the peels, seeds and stones of fruits and vegetables; in the majority of cases in greater quantities than in the edible part. These antioxidants are capable of slowing or preventing oxidation reactions that can produce free radicals by removing radical intermediates and inhibiting other oxidation reactions, but in order to do so they themselves must oxidize. Since oxidative stress might be an important part of many human diseases, the use of antioxidants is intensively studied, particularly in the prevention of stroke and neurodegenerative diseases. Other possible mechanisms of action by which polyphenols and other antioxidants may help to prevent disease include cell anti-proliferation, induction of cell cycle arrest and apoptosis, regulation of the host immune system, changes in cellular signalling, receptor sensitivity, enzyme activity, and gene regulation, etc., and they may have an anti-inflammatory effect.4,5
Furthermore, growing concern about food safety on the part of consumers, authorities and food industry producers has created a need to identify alternative natural and probably safer sources of food antioxidants. Replacing synthetic antioxidants with natural ones, such as phytochemicals, may have health benefits and increase functionality due to solubility in both oil and water, which could be useful for emulsions in food systems.
Unlike temperate fruits, tropical and subtropical fruits can be broadly defined as meeting all of the following criteria: crops that have their origin and commercial growing areas in the tropics or subtropics, plants that are evergreen and perennial, crops with a limited degree of frost resistance, and plants whose growth is practically nonexistent below 10 °C, with some exceptions according to species and individual age.6
Hundreds of tropical and subtropical fruits exist, but only around fifty are well known throughout most of the world. Production and trade figures allow the division of tropicals and subtropicals into three main categories with some overlapping: (i) major fruits, cultivated in most tropical (and subtropical) countries that are well known in both local and export-import markets (such as banana and plantain, coconut, mango, and pineapple); (ii) minor fruits, not so extensively cultivated, and of more limited consumption, both geographically and quantitatively; however, many are of considerable economic importance in their respective regional markets; examples of minor fruits are abiu, atemoya, avocado, breadfruit, carambola, cashew nut, cherimoya, durian, guava, jaboticaba, jackfruit, langsat, litchi, longan, macadamia, mangosteen, papaya, passion fruit, pulusan, rambutan, sapodilla, soursop, and white sapote and (iii) wild fruits belonging to diverse botanical families.6 Because wild fruits are not cultivated commercially in any country they are not used for processing and, therefore, not included in this review.
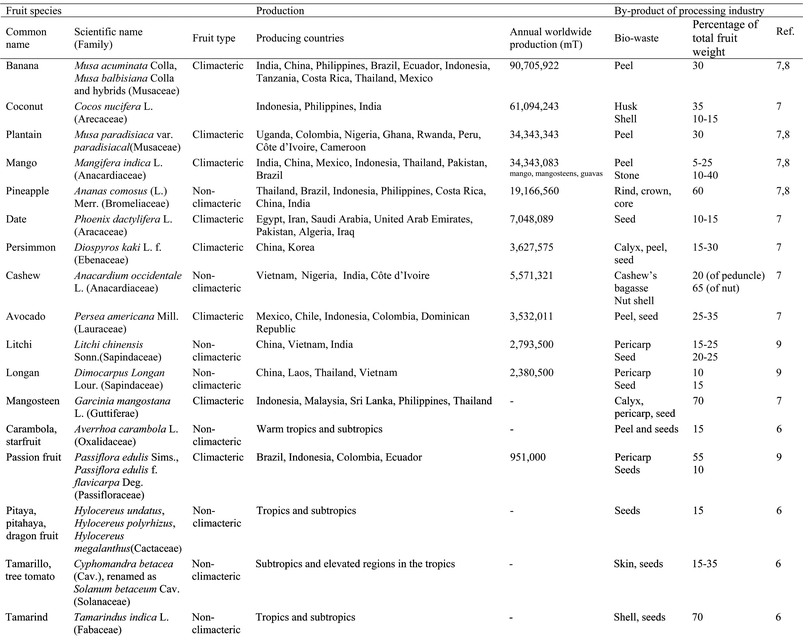
Fruits from the temperate zone are usually characterized by a large edible portion and moderate amounts of bio-waste material; in contrast, considerably higher ratios of by-products arise from tropical and subtropical fruit processing. Some data on worldwide production of the different tropical and subtropical fruits included in this review are presented in Table 1, alongside the different biowastes obtained from these fruits and the percentage of the total weight of the fruit that they represent.7–9
|
Developing rapid, rugged, robust and accurate analytical procedures is critical for the success of most of the steps necessary for designing, developing, and marketing value-added functional foods.10 Accurately identifying bioactive compounds is essential to finding relationships between different food components and health benefits. It is also critical to precisely quantify bioactive compounds to determine dietary intake levels and safety guidelines. Furthermore, assay procedures should be harmonized at the international level to analyze these value-added functional foods and facilitate their commerce in the global market.10
Analyzing phytochemicals from biowastes encompasses a number of distinct aspects and the analytical strategy will depend on the sample, analyte and nature of the problem. Although it is impossible for a single, global strategy to be valid for every by-product, given the diversity of analytes and plant materials, it is possible for a number of generalizations to be made. On the whole, the analytical procedure for determining phytochemicals from biowastes involves four common steps: sampling, sample storage, sample preparation (extraction from the plant material, hydrolysis and/or purification) and analytical determination (quantification and identification).
Most of the development made over the past few years in the analysis of phytochemicals from tropical and subtropical fruit biowastes has focused on the final analysis step. Remarkable innovations in instrumentation, spectroscopy, and chromatography have resulted in the rapid advancement of methods for high-throughput separation and detection of complex multi-component mixtures containing trace quantities of the analytes of interest. However, there has been limited research in the first three steps (sampling, sample storage, and sample preparation), which are the foundation for developing a quality, accurate, robust and rugged analytical procedure. In general, most uncertainty associated with analytic determination is usually related to these stages.10
This paper reviews analytical strategies dealing with antioxidant phytochemicals in tropical and subtropical fruit by-products: recovery of the phytochemicals from the sample matrix followed by separation, identification and quantification. It is focused on work published between 1990 and 2010, although most of the development made in the analysis of phytochemicals from tropical and subtropical fruit biowastes has been done over the past few years. The review emphasizes the importance of sample preparation in the determination of phytochemicals in these fruit by-products, as a critical step in the analysis of samples in which matrix components are biologically active and the analytes represent a diverse spectrum of numerous compounds, many of unknown identity.
2. Sampling and storing plant material
The initial step in any analysis is sampling, in which the biowaste must be obtained in such a way that it truly represents the species to be analyzed. However, in published manuscripts details about the sampled biowastes are frequently poorly documented. In many instances, collection and storage procedures are not established and remain controversial. For example, a very controversial aspect is the choice of freeze-dried or fresh samples to extract phytochemicals from biowastes.11In many cases tropical fruit biowastes are obtained directly from agro-industries.12–21 However, most residues that will be analyzed are obtained from different kinds of fruit in the laboratory. These tropical and subtropical fruits can be obtained from fields belonging to the research institutes themselves or from commercial orchards,22–76 or local markets.77–100 However, when purchasing from local markets the researcher has no prior knowledge about the origins, variety and/or postharvest conditions (time elapsed after harvest and storage temperature of the fruit) of the samples. These factors must be characterized as they have an enormous impact on the quantity and even the type of bioactive compounds contained in plant material. The composition of bioactive compounds in tropical and subtropical fruit peel and seeds can vary depending on climate (temperatures, rainfalls and light hours), soil type and fertilization. Different varieties of tropical fruits, grown in different regions, in different years, can result in measurable differences in composition. The variety12,15,18,19,22–33,35–43,51–53,55,57–64,66,67,69–72,78–84,93,94,101–103 used in the analysis of tropical and subtropical fruit biowastes or the harvest index22,23,27 has been reported in some works, but rarely. The banana harvest index has been established as the caliber, measured in the middle finger of the outer whorl of the second hand from the distal end of the bunch.22,23 In mango, fruit maturity at harvest can be based on the days after full bloom, fruit shape and maturity of seed stone during destructive opening.27 Another aspect to keep in mind, especially when analyzing residues from climacteric fruit, is the ripening stage of the fruit at the moment the biowastes are obtained. Slight variability in the ripening stage could contribute to wide modifications in the bioactive compounds of climacteric fruit peel and seeds. For example, during the ripening process, there is a significant increase in the tocopherol content in the ripened banana peel.104 During ripening, the color of mango peel gradually changes from green to greenish-yellow, red, violet or yellow; these changes are related to an increase in carotenoids during ripening, ranging from 74 to 436 μg g−1 peel dry weight (DW).25 The carotenoid content was found to be greater in ripe mango peels (1400–4000 μg g−1 DW) compared to unripe peels (365–550 μg g−1 DW).26 The anthocyanin content (360–565 mg/100 g DW) was higher in ripe mango peel than in unripe peels (203–326 mg/100 g DW).25 Unripe mango peel had higher polyphenol content than ripe peels. Therefore, the total polyphenol content of unripe mango peels ranged from 90 to 110 mg g−1 peel DW, whereas it ranged from 55 to 100 mg g−1 DW in ripe peels, depending on the variety.25,26 Vitamin C and E contents ranged from 188 to 392 and 205 to 509 μg g−1 dry peel, respectively; and they were greater in ripe peels.26 The skin of “Hass” avocados changes color from green to purple and to black as the fruit ripens. This is the result of an initial decrease in chlorophyll content, followed by a dramatic increase in the level of the anthocyanin cyanidin 3-O-glucoside.58 The young mangosteen rind contains a higher content of phenolic compounds and tannins, promoting better free radical scavenging activity than the rind from the mature fruit. In contrast, the mature fruit rind contains higher amounts of total flavonoids and α-mangostin than the rind from the young mangosteen.105 The red colour of litchi fruit peel is mainly attributed to anthocyanins. The monomeric anthocyanin concentration of litchi pericarp (46 mg/100 g fresh weight (FW)) at ripeness makes them a potentially good source of anthocyanins.106 The individual anthocyanins identified were cyanidin-3-glucoside, cyanidin-3-rutinoside and malvidin-3-acetylglucoside. Only malvidin-3-acetylglucoside was detected in the unripe fruit. However, in the ripe fruit, cyanidin-3-rutinoside became the main anthocyanin (>75%),106–108 while cyanidin-3-glucoside represented less than 17%, and malvidin-3-acetylglucoside less than 9%.106 Therefore, it is essential that the ripening stage of the fruit be characterized when the fruit is analyzed. Climacteric fruits are usually harvested at physiological maturity,22,23,25,26,57,58,92,109 and then left to ripen at room temperature25,26,53,109 or in controlled conditions of temperature and relative humidity22,23,57,58,92 until full-ripening is reached, the stage at which the fruit biowaste is analyzed. Sometimes, biowastes are analyzed in fruits at physiological maturity24,25,84 or at different ripening stages,27,73,103,105 but normally antioxidant phytochemicals in biowastes are analyzed in ripe fruit.12,14,19,22–26,32,33,35–41,43,46–49,52,53,57,58,69,82,83,87,90,92,96,97,108–110 The ripening stage of banana is usually characterized in the middle finger of the outer whorl of each banana hand by using peel and pulp color, firmness, total soluble solid content and titratable acidity.22,23 The ripening stage of mangos and avocados has been characterized by measuring color and determining firmness.24,57,58 The characterization of cashew apples has been done on the basis of visual color, dry matter, total soluble solids, pH and titratable acidity.32,33 In tamarillo, the total soluble solids and moisture content were analyzed prior to obtaining the biowastes that were analyzed.49
Sample storage is an important step as there is often some delay between obtaining the biowaste and its analysis. For example, when biowastes are obtained from agro-industries, the storage conditions (temperature, time), from the time of collection to the time of analysis, are not usually specified. Improperly stored biowastes can rapidly lose their antioxidant properties, making them useless for extraction purposes. For example, pericarp browning, a common and important defect of harvested litchi fruit, is caused by anthocyanin degradation. The (−)-epicatechin is oxidized and oxidative products of (−)-epicatechin (o-quinones) catalyze other litchi flavanols and anthocyanin degradation, resulting in the pericarp browning of postharvest litchi fruit.107,108 Moreover, fresh biowaste has high moisture content (usually not reported) and therefore is at risk of microorganism growth unless stored in appropriate conditions. It is essential to inactivate all enzymatic and chemical reactions during storage so that the sample's identity does not change during this period.11 It is essential to dry tropical and subtropical fruit by-products to inactivate the enzymes responsible for degrading many active compounds and to decrease the rate of microbial growth. Drying time and temperature affect the activity and stability of bioactive compounds due to chemical and enzymatic degradation, losses by volatilization and/or thermal decomposition. Under natural drying conditions, biowastes are exposed to either direct sunlight for 3–7 days,14,15,18,102,111 or air-dried at room temperature for 1–2 weeks.47,78,112 Good air circulation is mandatory to reduce humidity as the biowastes dry. With artificial drying, the biowastes are oven-dried (with static or forced-air or under vacuum) at temperatures between 40–80 °C for 4–72 h12,13,15–17,30,35–36,38–41,45,52,53,71,72,85,88,90,97,105,110,113 or freeze-dried.22–24,27,28,32,33,37,42,51,55,68,80–83,86,87,89,96,100,101,114 Tamarind seeds have been dry heated along with acid washed sea sand on an open hot plate at 135 °C for 25 min.50
Some authors have described an increase (1.4 times) in the phenolic content of mango, longan and tamarind seeds when they are heated at temperatures between 105 and 160 °C prior to extraction.86,87 Coconut shell powder has been subjected to different toasting temperatures, from 100 to 200 °C, and toasting times, from 20 to 60 min, prior to the extraction of phenolic compounds.115 Thermal treatment, at 100 °C for 60 min, enhanced phenolic extraction from the coconut shell.111,115 The total phenol content in persimmon peel extracts (70% ethanol or water) increased between 4.2–6.9 times when the peel was heated at 150 °C for 20 min.112 This could be attributed to the formation of phenolic substances under milder heating temperature, probably due to the conversion of phenolic molecule precursors in this type of compound. This behavior may also be due to the fact that a large percentage of phenolic compounds are bound to cellular structures and heating treatments release bound phytochemicals from the matrix to make them more accessible in extraction.116
Reducing the particle size favors solvent extraction of phytochemicals,22,23 therefore, before extraction mechanical grinding of the biowastes is carried out to increase their surface area. Dried biowastes are usually ground with a ball, knife or hammer mills,12,13,16,18,22,23,27,28,30,55,80,82,83,85,88,90,111,115 blenders,35,36,38–41,53,78,110,112 grinders,20,21,42,52,86,87,96,117 mortars32,33,51 or processors.17,114 The particle size of the ground material is generally between 0.25 and 1 mm.13,14,17,18,22,23,32,50,52,53,55,78,87,90,91,105,111,113
Undoubtedly, the influence of storage temperature on the analyte of interest needs to be studied in detail in order to store samples under appropriate conditions. Dehydrated and ground samples are normally stored at room temperature,17,50,90,111 at 4 °C13,52 or below freezing at −20 °C12,22,23,27,37,47,51,67,81,86,87,96 or −80 °C.32,33,68 Generally, when anthocyanins are determined in plant residues, the samples do not dehydrate and are stored frozen at −20–−30 °C until extraction.62,106,107,118–120 Moreover, samples are usually stored protected from light,13,17,47,51,90,97 under nitrogen32,33 or in vacuum-packaged polyethylene pouches.51,52,81,86,87,90,96,97
3. Preparing tropical fruit biowastes for phytochemical analysis
The ultimate goal behind sample preparation is to obtain a sample extract uniformly enriched in all components of interest and free from interfering matrix components. This may include some of the following steps: extraction, hydrolysis, purification and/or pre-concentration. Fig. 1 summarizes the different techniques used to prepare tropical and subtropical fruit biowastes for phytochemical analysis.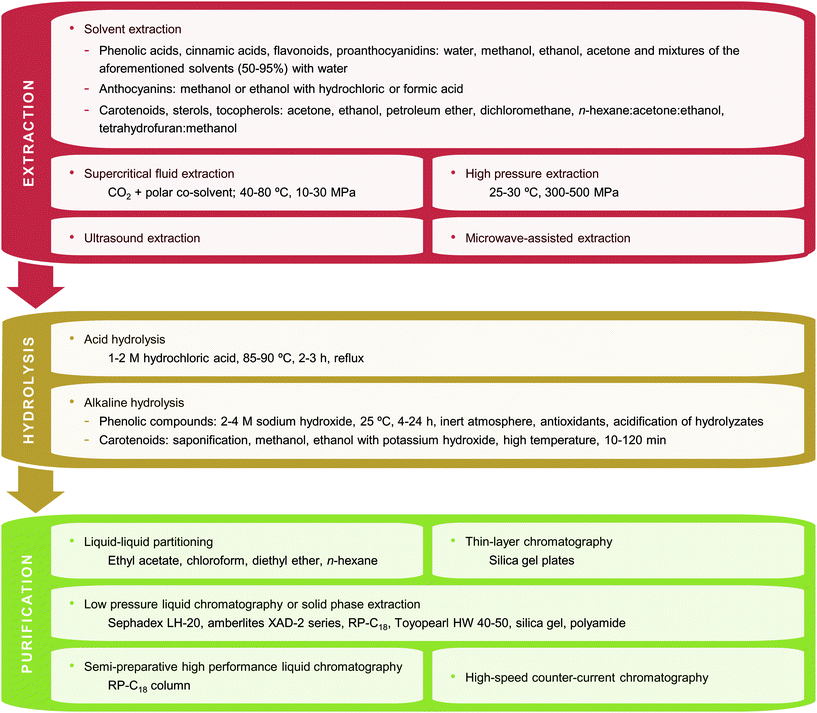 | ||
| Fig. 1 Techniques (extraction, hydrolysis and/or purification) used to prepare tropical and subtropical fruit biowastes for phytochemical analysis. | ||
3.1 Solvent extraction
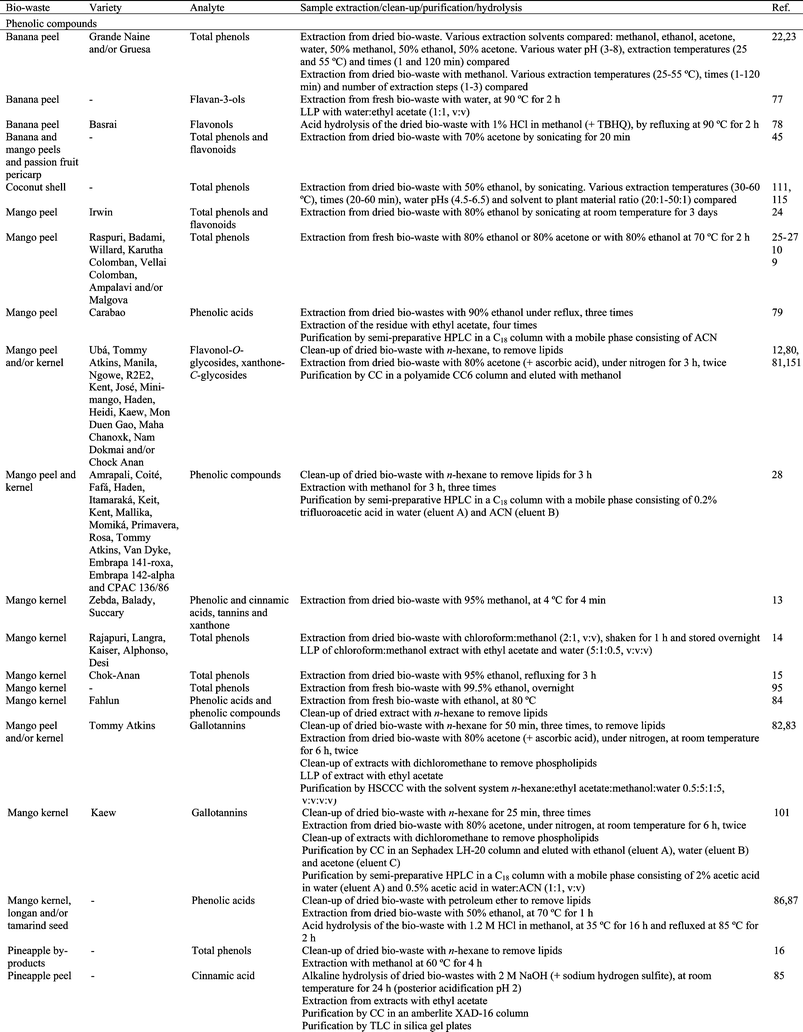
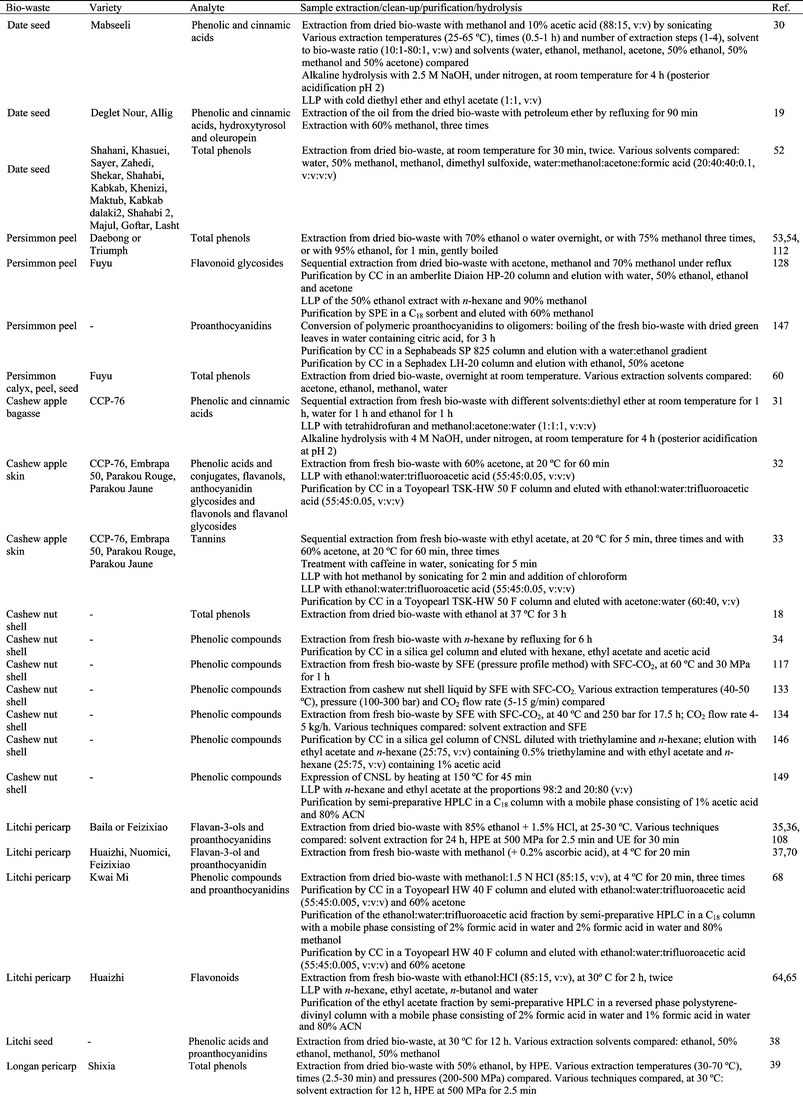
It is practically impossible to develop an efficient and uniform method for the extraction of all phytochemical compounds from tropical and subtropical fruit biowastes with a single solvent system, as the polarities of different compounds vary significantly and the manner in which they associate with the sample matrix is also variable. Therefore, in order to accurately estimate the diverse bioactive compounds present in different biowaste matrices it is essential to optimize sample preparation.Phytochemical compounds in plants can be distributed in various forms. For example, phenolic compounds exist as free aglycons or as conjugates with sugars or esters, or as polymers with multiple monomeric units. Moreover, phenolic compounds are not uniformly distributed and may be associated with cell walls, carbohydrates, or proteins.11,121 In addition, the task of phytochemical recovery is complicated as plant material is a natural matrix with high enzyme activity and the stability of bioactive compounds varies significantly, some are relatively stable while others are highly reactive;11,121 this complicates their extraction and quantitative recovery becomes particularly problematic. Some precautions are usually taken during extraction in order to prevent oxidation of bioactives and other deteriorative processes. Drying the plant by-product before extraction, immediately immersing the biowaste in methanol and using an acid extraction medium protects the material from oxidation.11 Moreover, the extractions can be carried out under reduced light22,23,57,58,82,92,95 or under an inert atmosphere.57,82,83,101 Some antioxidants can be used for this purpose as well.37,43,51,57,69,70,78,80,82,92
Extraction is the first and most important step in recovering and purifying active ingredients from plant biowastes. The objective when extracting phytochemical antioxidants from plant sources is to liberate these compounds from the structures where they are found. Solvent choice, contact time and temperature, number of extraction cycles, solvent to plant material ratio and extraction technique significantly influence the extraction efficiency. The role of each factor in the extraction process is not always obvious; the chemical characteristics of the solvent and the diverse structure and composition of natural products ensures that each biowaste-solvent system behaves differently and in an unpredictable manner.
|
|

|
|
|
In general, no single solvent will provide optimum recovery of all phenols or even a limited range of phenols. Plant phenols are ionizable with typical pKa values ranging from 8 to 12.11,121 Thus, they exhibit considerable diversity in terms of acidity and polarity,and they also range from hydrophobic to hydrophilic. The polarity of the solvent and that of the different phenolic compounds affect extraction efficiency and the activity of the obtained extracts. In general, highly hydroxylated aglycone forms of phenolic compounds are soluble in alcohols. Less polar solvents such as ethyl acetate, acetone and chloroform are used for the less polar and highly methoxylated aglycone forms that are very common in fruit peel. The most polar phytochemicals can be extracted using water. Moreover, there are some important distinctions between fresh and dried samples. In the case of extractants using aqueous mixtures, the required proportion of water in the extractant is lower with fresh samples. Furthermore, with dried materials, low polarity solvents and ethyl acetate will simply leach the sample whereas alcoholic solvents presumably rupture cell membranes and enhance the extraction of endocellular materials.11,121
Solvent extraction of anthocyanins warrants special consideration. The fact that their chemistry is complicated by various pH-dependent equilibria11,121 has been exploited in traditional anthocyanin recovery strategies by extracting the flavylium cation form with methanol or ethanol containing hydrochloric (HCl) or formic acid (Table 2). The low pH value of the extraction solvent prevents the degradation of these pigments.122 However, pigment degradation occurs as the acids are concentrated during the evaporation of the alcohol-HCl or alcohol-formic acid mixtures. Small amounts of dilute acid may also cause partial or total hydrolysis of acylated anthocyanins that are present in plant tissues.122
In addition to the solvent, many other factors contribute to the efficiency of the extraction process. Extraction of phenolic compounds is typically conducted at room temperature, 20 to 30 °C (Table 2). However, increasing temperature improves extraction efficiency due to the enhanced diffusion rate and solubility of phytochemicals in solvents. Therefore, in some cases hot solvents (at temperatures between 37 and 90 °C) or heat reflux are used to extract bioactive compounds from tropical and subtropical fruit biowastes. However, it must be taken into account that the degradation rate of antioxidant compounds is time and temperature dependent. It has been widely reported that extraction temperature affects the stability of bioactive compounds. In addition, at high temperatures bioactive compounds can react with other components of the plant material, thus impeding extraction. In some cases cold extraction, at 4 °C, has been reported (Table 2). The mass transfer rate and the chemical solubility of the phytochemicals in the solvent can be significantly improved with agitation during the extraction procedure.14,18,27,30,32,33,37,38,75,78,83,90,95,101,114,123
The recovery of bioactive compounds from biowastes is also influenced by the extraction time and the number of extraction processes. Extraction times of a few minutes (between 1 and 30 min), hours (between 1 and 12 h) or days (between 1 and 3 days) have been used (Table 2). Usually a single extraction step is used, but sometimes from 2 to 8 extraction steps have been used (Table 2). Another aspect to take into account when extracting antioxidant compounds from biowastes is the solvent to plant material ratio. The most commonly reported ratios are between 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 10:1 (v:w).13–15,18,22,23,31,33,37,38,43,46,49,50,55,69,76–78,83,84,90,95,96,99,101,114,124–126 However, higher ratios between 20:1 and 100 :1 (v:w)12,27,30,32,55,59,61,64,65,67,75,80,82,89,127 or between 125:1 and 250:1 (v:w)49,52,86,87 have also been used.
:1 and 10:1 (v:w).13–15,18,22,23,31,33,37,38,43,46,49,50,55,69,76–78,83,84,90,95,96,99,101,114,124–126 However, higher ratios between 20:1 and 100 :1 (v:w)12,27,30,32,55,59,61,64,65,67,75,80,82,89,127 or between 125:1 and 250:1 (v:w)49,52,86,87 have also been used.
In the majority of cases, the procedures employed to extract phytochemicals from tropical and subtropical fruit biowaste have not been specifically optimized for them, but have been optimized for the extraction of other types of plant material. Extraction methods differ between different biowastes because of their different matrices, with unique properties in terms of structure and composition (related to specie, variety, ripening stage). Therefore, considerable caution should be exercised when using methods that have been developed to analyze specific plant tissue types and phytochemical extraction should be optimized for each biowaste.
The effect of solvents on the extraction of phenolic compounds from banana peel has been evaluated (Table 2).22 Acetone (50%) was the most effective solvent for the extraction of phenolic compounds from this biowaste, extracting 1.5–3.5 times more than methanol, 50% methanol or 50% ethanol. Moreover, experimental design and response surface methodology was used in order to optimize the number of extraction steps, extraction time and temperature when extracting phenolics from banana peel (Table 2).23 The factor that had the greatest impact was the number of extraction steps: the extraction was improved by increasing the number of extractions. Optimum extraction of phenolic compounds was obtained with 3 extraction steps, with homogenization for 1 min at 25 °C and further centrifugation. The extraction of phenolics from mango kernel has been optimized by using 95% ethanol for 4.5 h or refluxing with ethanol for 3 h.15 The phenolic compounds extracted using reflux were 2.5 times higher than those extracted using shaking. The effects of solvent to sample ratio, temperature, extraction time, number of extractions and solvent type on the efficiency of phenolic extraction from date seeds has been also studied (Table 2).30,52 The optimum conditions were considered to be a two-stage extraction, each stage lasting 1 h at 45 °C with a solvent to sample ratio of 60:1 (v:w).30 Acetone (50%) was selected as a very efficient solvent for extraction.30 Extraction by dimethyl sulfoxide also gave a high total phenolic content in the extracts obtained from seeds of 14 date varieties.52 How the solvent (acetone, ethanol, methanol and water) affected the extraction of phenolic compounds from three different biowastes obtained from persimmon was evaluated.55 Acetone was the most effective solvent for the extraction of phenolics from persimmon calyx, seed and peel. Temperature and pH significantly influenced the extraction yield of anthocyanins from litchi fruit pericarp and antioxidant activity.67 Temperatures from 45–60 °C and pH from 3.0 to 4.0 exhibited a relatively high antioxidant activity. Five different solvents were used to evaluate total phenolic content, antioxidant capabilities and antityrosinase activity in litchi seeds (Table 2).38 There were no significant differences in the phenolic content among all the extracts except for those obtained using water. The effect of different solvents on phenolic extraction from longan pericarp has also been studied (Table 2).40 Extracting with methanol or ethanol obtained the highest total phenolic content, followed by water and ethyl acetate. Dried mangosteen pericarp was extracted with 95% ethanol by maceration, percolation, Soxhlet extraction, ultrasonic extraction and shaking.97 Soxhlet extraction promoted the maximum content of xanthones in the extracts and it was also recommended because of its low reagent consumption and its low extraction time. Moreover, the extraction solvent was also optimized.97 50% Ethanol was the appropriate solvent for extracting phenolic compounds and tannins from mangosteen pericarp, while 95% ethanol was recommended for α-mangostin extraction. The extraction of phenolics from carambola residues was carried out at different ratios of organic solvents and water, extraction temperatures and times (Table 2).89 The optimum solvent was 50% acetone. Moreover, the extraction efficiency increased with increased temperature and time. Considering the stability of antioxidants at high temperatures, 90 °C was chosen as a suitable extraction temperature and 45 min as the optimum extraction time. The effect of acetone concentration, extraction temperature and extraction time (Table 2) on phenolic content extracted from carambola residue was also studied by using response surface methodology.90 Acetone concentration was statistically the most significant factor and the optimal extraction conditions obtained were: 65% acetone concentration, 43 °C extraction temperature, and 234 min extraction time.
Once extracts have been obtained they are usually concentrated at low temperature (35–45 °C) or lyophilized. The residues are then redissolved in an adequate solvent for subsequent purification and analysis.13–16,20,21,24,31,38,43,44,50,54,62,64,65,69,70,72,75,77–87,89,93,94,96,98,99,101,105,123,124,128
:acetone:ethanol (2:1:1, v:v:v)49,92 or tetrahydrofuran:methanol (1:1, v:v),51 followed by saponification and purification. Dichloromethane is sometimes added to improve the extraction efficiency which, however, increases the risk of altering the sample composition.104 Extraction can be achieved by refluxing with a Soxhlet extractor for 6–8 h.47,104
Sometimes mango kernels,81,82 pineapple by-products,16 longan seeds44 and tamarind seed coats50,100 and tamarind pericarp100 are defatted (Table 2), prior to extracting antioxidant compounds from them, with n-hexane16,81,82,100 or petroleum ether.37,43,44,50 Other times, the extract obtained from mango kernel83,84 or mangosteen pericarp114 is extracted with n-hexane,84 dichloromethane83 or diethyl ether114 to remove residual lipid material.
Carotenoids exhibit pronounced photo- and thermal sensitivity. These phytochemicals, in their natural environment, are incorporated in lipoproteins or membranes, and are relatively well protected. However, if they are isolated in extractants, they readily undergo trans-cis isomerization catalyzed by light, acids and bases, oxygen, heat, traces of metal ions, etc.129 Therefore, some measures must be taken during the sample pre-treatment: antioxidants must be used, laboratory operations must be carried out in dimmed, yellow or red light, evaporation should be carried out under a protective nitrogen or argon atmosphere at temperatures below 40 °C and the samples should be stored in darkness, under nitrogen or argon, at temperatures around −20 °C.129
3.2 Other extraction techniques
The traditional liquid–liquid methods used to extract phytochemicals have several drawbacks: they are time consuming, laborious and have low selectivity and/or low extraction yields. Knowledge of phytochemical extraction is desirable beyond a purely analytical context, as it also has practical applications in the food industry. The use of large amounts of toxic solvents is strictly regulated in food ingredient manufacturing. Therefore, safer, more environmentally friendly and more effective and selective extraction technologies are being developed to obtain bioactive compounds from tropical and subtropical fruit biowastes, including supercritical fluid extraction (SFE), ultrasonic extraction (UE), microwave-assisted extraction (MAE) and high pressure (HPE) extraction.Extraction of cashew nut shell liquid (CNSL) from split cashew shell with SFC-CO2 is an interesting approach because it could allow for much more of the CNSL to be used by the food industry.117,133,134 The composition of the supercritical carbon dioxide and solvent extracted CNSL from the raw cashew nut shells were comparable.134 Philip et al.133 evaluated the isolation of anacardic acid from natural CNSL by SFC-CO2 under a range of operating conditions of pressure, temperature and CO2 flow rate. The best working conditions were found to be 50 °C and 300 bar at a flow rate of 5 g min−1.133 Under these conditions it was possible to quantitatively isolate high purity anacardic acid from crude natural CNSL within 150 min. Two extraction methods were also used to extract anacardic acid compounds from the cashew shell:117 (i) the typical extraction method, in which CO2 flows through the extractor at constant temperature, and (ii) the pressure profile method, in which the extractor content is subjected to pressurization with CO2 and depressurization steps before beginning the extraction. The pressure profile method promoted CO2 dissolution into the shell material because of the changes produced in its structure, giving yields 10 times greater than those of the typical extraction method for the same amount of CO2. Under these conditions, the composition of the extracted oil was virtually the same as that obtained by expressed CNSL, without chemical degradation of the anacardic acid.117
The xanthones from mangosteen pericarp have been extracted without ethanol and with 2–3% ethanol at 30 MPa and 50 °C in SFC-CO2.88 SFE using ethanol as a modifier significantly increased the extraction yields of xanthones. The performance of SFE with pure CO2 when extracting phytochemicals from tamarind seed coat was very low.91 However, adding an ethanol co-solvent increased the extraction of (−)-epicatechin substantially (500 times) and the antioxidant activity of the extracts. Luegthanaphol et al. optimized the extraction temperature and pressure over the range 35–80 °C and 10–30 MPa, respectively, and highlighted that the amount of (−)-epicatechin extracted decreased when temperature and pressure increased.91 SFC-CO2 extraction was studied as a method to extract antioxidant compounds from tamarind seed coat.21 Different combinations of pressure and temperature were used with and without ethanol as modifier. As temperature and pressure were increased (80 °C and 30 MPa optimum conditions), more phenolic compounds were extracted.21
The extraction of carotenoids from persimmon peels using SFE at 60 °C and 30 MPa, was carried out.132 CO2 was used as the SCF and 5–20% of ethanol was added as a co-solvent. When increasing the ethanol amount from 0 to 10%, the carotenoid yield in the extraction was improved, although the selectivity of the extracted carotenoids was drastically depressed.
The use of ultrasound (at an intensity of 4,870 W m−2) with 50% ethanol has been evaluated to extract phenolic compounds from coconut shell.111,115 The effect of extraction temperature and time, solvent to biowaste ratio and water pH was evaluated using response surface methodology. Phenolic extraction was maximized when pH values of 6.5, high solvent to solid ratio (50:1, v:w) and low temperature (30 °C) were employed. Increasing extraction time did not increase the extraction of phenolics (optimum 15 min). At optimum operating conditions, sonication enhanced the phenolic extraction yield 3 fold. Flavanols have been extracted from fresh litchi pericarp using acidified methanol in an ultrasonic bath at room temperature for 0.5 h (Table 2).108 Longan pericarp or seed in 70% acetone (solvent to solid ratio 125:1, v:w) was also subjected to ultrasonic treatment (input power 700 W) for 20 min.42
HPE has been used to extract bioactive compounds from litchi and longan pericarp. Prasad et al. studied the effect of extraction conditions on the extraction yield, including type of solvent and concentration, solvent to plant material ratio, acidic medium, extraction pressure, time and temperature.35,36,39–41,110 These authors did not find significant differences in the phenolic content when the litchi pericarp was extracted using HPE, UE or conventional extraction.35,36 However, the HPE technique increased the flavonoid extraction yield from litchi pericarp up to 2.6 times in comparison with UE and up to 10 times compared with conventional extraction. In longan pericarp, the optimum HPE conditions for phenolic compounds39 and corilagin41 were determined to be 500 MPa for 2.5 min at 30 °C, 50% ethanol and a solvent:solid ratio of 50:1 (v:w). The HPE technique resulted in the highest extraction of total phenolic and corilagin content (1.2–1.4 times) and was faster than either UE or conventional extraction.39,41 Xanthones have also been extracted from mangosteen pericarp with ethanol using HPE at 100 °C for 15 min.137 The extraction yield using HPE was twice that obtained by UE; moreover, both solvent consumption and extraction time were reduced.
The phenolic compounds from longan pericarp were extracted with 95% ethanol (solvent:plant material ratio 10:1, v:w) employing MAE, at 80 °C for 30 min (Table 2).113 The phenolic content of extracts obtained from longan pericarp was similar when using either MAE or Soxhlet extraction. However, the antioxidant activity of microwave-assisted extracts was superior to that of Soxhlet extracts, had faster extraction times (2 h for Soxhlet and 30 min for MAE) and required less solvent. All of which are clear advantages to using this technique.113
3.3 Hydrolysis
Most phenolic compounds in plant peels and seeds primarily occur in the bound form as conjugates with sugars, fatty acids, or proteins. Therefore, it is important that a hydrolysis process is adopted in order to obtain maximum yield during phenolic extraction. Sometimes hydrolysis is carried out simultaneously with extraction and sometimes after the extraction process. Acid hydrolysis15,49,78,87 has been the traditional approach to measure aglycones and phenolic acids from flavonoids glycosides and phenolic acid esters, respectively. Hydrolysis of flavonoid glycosides requires relatively high concentrations (1–2 M) of acids under refluxing conditions (Table 2). Moreover, acid hydrolysis is seen to more closely reflect dietary intakes although it is evident that absorption, metabolism and bioavailability of plant phenols are complex and that understanding how they work is still very limited.11,121 The hydrolysis of flavonols from banana peel was carried out with acidified methanol containing 1.2 M HCl at 90 °C under reflux for 2 h, to obtain aglycons of flavonol glycosides.78 Dried mango seed kernel was refluxed with 1.2 M HCl in ethanol for 3 h, after which the acid solution was neutralized with 1% sodium carbonate.15 An acid-catalyzed hydrolysis with aqueous methanol and HCl, at a final concentration of 1.2 M, was also employed to liberate phenolic acids from their bound forms in longan seed and mango kernel.87 The solution was stirred at 35 °C for 16 h and refluxed at 85 °C for 2 h. Tamarillo pomace was hydrolyzed with 1% HCl in ethanol at room temperature for 60 min.49Alkaline hydrolysis conditions have been employed to isolate phenolic acids from the biowastes obtained from tropical and subtropical fruits in order to determine bound phenols.30,31,85,114 Hydrolysis of esterified ferulic acid from pineapple peel with sodium hydroxide (NaOH) has been described by Tilay et al.,85 who performed alkaline treatment with 2 M NaOH for 24 h at 22 °C. The residue of date seeds obtained after extraction was hydrolyzed with 2.5 M NaOH and stirred at room temperature for 4 h.30 The residue obtained after extracting the soluble phenolic compounds from cashew apple bagasse31 or mangosteen pericarp114 was hydrolyzed with 4 M NaOH for 4 h at room temperature. Next, the residue obtained after hydrolysis of mangosteen pericarp was heated with 2 M HCl for 30 min at 95 °C to liberate the phenolic acids from glycosidic bonds.114 Sometimes the use of an inert atmosphere,30,31,114 antioxidant,85 or the acidification of the hydrolyzates30,31,85,114 are essential precautions due to the poor stability of some phenolic acids in alkaline conditions. The hydrolysis method of choice is always a compromise between efficiently producing aglycones from plant material and degrading aglycones. Enzyme-assisted hydrolysis of plant material is a very interesting option due to its high specificity, the fact that it uses mild reactive conditions and because it achieves highly stable extracts with antioxidant activity and without organic solvent residues.141,142 However, its use has not been reported in the hydrolysis of tropical and subtropical biowastes.
Saponification is usually recommended to analyze the carotenoids, sterols or tocopherols of plant tissues129 as it simplifies chromatographic profiles by removing potentially interfering compounds such as chlorophyll degradation products and chlorophyll-esters. The disadvantage of this approach is that it involves extra handling steps including partitioning out of the saponified analytes, evaporation of the extracts to dryness and making up to solution before injection into the chromatograph, thereby increasing both the time and the expense of the analysis. To saponify lipid extract or oil prior to carotenoid, sterol or tocopherol determination, an aliquot of the sample is stirred with methanol25,26,104,109 or ethanol13,47,102,143 containing potassium hydroxide (KOH). Saponification is usually carried out at high temperature for 10–120 min (Table 2).13,47,51,102,104,143
Unlike most C–C single bonds that are often chemically resistant, the inter-flavanol C–C bonds in the B-type linkage of the proanthocyanidins can be readily broken under mild acidic conditions such as 1% HCl in ethanol118,119 or methanol,44,49,144 at room temperature for 2 h44,49 or at 4 °C for more than 6 h.118,119,144 This procedure has been carried out to hydrolyze anthocyanins from litchi pericarp,44,118,119 mangosteen pericarp144 and tamarillo pomace.49 The depolymerization of proanthocyanidins from litchi68 or mangosteen145 pericarp was carried out with thiol reagents.68,145 Tannins from litchi pericarp have been mixed with toluene-a-thiol 5% in 0.2 N HCl in methanol, stirred and heated for 2 min at 90 °C.68 Thiolytic degradation, followed by mass spectrometric and nuclear magnetic resonance analysis indicated the presence of catechin, epicatechin and proanthocyanidin A-type constitutive units.The mangosteen pericarp was mixed with methanol, 36% HCl and mercaptoacetic acid and heated at 65 °C for 12 h.145 This depolymerization process gave rise to 4β-(carboxymethyl)sulfanyl-(−)-epicatechin methyl ester, with a yield (calculated as epicatechin) 6 times higher than the proanthocyanidins extracted from mangosteen pericarp (without hydrolysis).
3.4 Purification of extracts
In order to identify or quantify phytochemicals from crude extracts obtained from fruit biowastes, partitioning steps are usually carried out to reduce the number of antioxidant compounds and other substances contained in the extracts that could interfere with analytic measurements. For example, many extracts contain significant quantities of carbohydrates and/or lipids that can potentially interfere with subsequent quantification. Various strategies have been designed to cope with this situation including liquid–liquid partitioning, low pressure liquid chromatography (CC), solid phase extraction (SPE), thin-layer chromatography (TLC), semi-preparative high performance liquid chromatography (HPLC) and/or high-speed counter-current chromatography (HSCCC).:water:trifluoroacetic acid mixture (55:45:0.05, v:v:v), were passed through a methacrylic size-exclusion Toyopearl TSK-HW 50 F (12 × 1 cm i.d.) column at 1 ml min−1 and monomeric phenolics were eluted with the above solvent mixture. Tannins from cashew apple skin were also purified from 60% acetone extracts.33 Prior to purification by adsorption chromatography, tannins were flocculated with caffeine, a process that eliminates monomeric phenols, including anthocyanins, but not flavan-3-ols. The crude caffeine-tannin powder, dispersed in an ethanol:water:trifluoroacetic acid mixture (55:45:0.05, v:v:v), was passed through a column packed with Toyopearl TSK-HW 50 F at 1 ml min−1. Tannins were eluted with 60% acetone.33 The hexanic fraction obtained from cashew nut shell has been purified on a silica gel column with a gradient of hexane, ethyl acetate and acetic acid.34,146 The crude polyphenolic extract obtained from litchi pericarp was fractionated by CC on a Toyopearl HW 40 F (16 × 2.5 cm) column.68 The monomeric polyphenols were recovered in the fraction eluted with ethanol:water: trifluoroacetic acid (55:45: 0.005, v:v:v) and the proanthocyanidin fraction was then eluted by 60% acetone. Crude polyphenols from longan pericarp were fractionated on a Sephadex LH-20 (60 × 4.5 cm i.d.) column by using methanol as eluant at a flow rate of 1 ml min−1.42 Polyamide, Sephadex LH-20 and silica gel chromatography have also been tested for the purification of the phenolic crude extracts from longan pericarp.43,69,70 The extracts were passed through a polyamide (250 g, 45 × 2.5 cm, 60–80 mesh) column and then eluted with a water:methanol mixture at a gradient of 100, 80, 60, 40, 20 and 0% (v:v), at a flow rate of 2.5 ml min−1. The fractions containing phenols were loaded on a Sephadex LH-20 column (15 × 1.5 cm) and eluted with methanol at a flow rate of 3 ml min−1. To purify the substrates a fraction eluted from the Sephadex column was passed through a silica gel (4 g, 40 × 0.6 cm, 200–300 mesh) column using chloroform:methanol:formic acid (90:10:0.2, v:v:v) as the eluant at a flow rate of 0.2 ml min−1.43,70 The residue obtained from longan seed extraction was suspended in water and partitioned successively with petroleum ether, chloroform, ethyl acetate and n-butanol.44 The chloroform extract was passed through a silica gel column and eluted with chloroform:methanol mixtures with increasing polarities (94:1 to 4:1, v: v). The fraction obtained by eluting with chloroform:methanol (19:1, v:v) was further separated using polyamide (80–100 mesh) CC and eluted with 5–80% aqueous methanol. The subfraction obtained by eluting with 10% methanol was passed through a Sephadex LH-20 column and eluted with methanol to yield ethyl gallate. The ethyl acetate extract was passed through a polyamide column and the first fraction, eluted with water, was further separated on a Sephadex LH-20 column using methanol and yielded 1-β-O-galloyl-D-glucopyranose. Other fractions obtained with water, 10% methanol, 30% methanol and methanol yielded gallic acid, methyl brevifolin carboxylate, brevifolin, corilagin, 4-O-α-L-rhamnopyranosyl-ellagic acid and ellagic acid.44 Extraction of phenolic and cinnamic acid from pitaya seed oil was done using SPE on a diol-bonded phase cartridge. The sorbent was conditioned by consecutively passing methanol and then n-hexane through the cartridge.47 The oil dissolved in n-hexane was applied to the column and the analytes were eluted with methanol.47
Amberlite XAD-series or Sephadex LH-20 column chromatography is commonly used for anthocyanin purification. The concentrated extract obtained from litchi pericarp was purified with an XAD-7 (50 × 2.0 cm) column.118,119 Liu et al. purified the concentrated extract obtained from litchi pericarp on an Amberlite XAD-16 (30 × 2.5 cm i.d.) column and eluted the fraction with 40% ethanol; after the ethanol was removed, the fraction was extracted with ethyl acetate.107 Subsequently, the anthocyanin fraction, in water phase, was applied to a Toyopearl HW-40S (20 × 1.6 cm i.d.) column and the pigments were eluted using a methanol:water:formic acid mixture (50:48:2, v:v:v) at a flow rate of 0.8 ml min−1. PVPP was also used to purify the anthocyanin extracts obtained from litchi pericarp.106 The absorbed anthocyanins were eluted with methanol:HCl. The anthocyanin extracts from tamarillo peelings were applied to an XAD-7 (800 mg, 40 mm i.d.) column. The pigments were eluted with a mixture of methanol:acetic acid (19:1, v:v).48 Then, the XAD-7 isolates of peelings were fractionated by high-speed counter-current chromatography.
Fractionation of polymers and oligomers from proanthocyanidin of persimmon peel was carried out by column separation.147 Previously, a mixture of freshly crushed persimmon peel and dried green tea leaves in water containing citric acid was boiled for 3 h. At this stage, substitution at the C-4 positions of polymeric proanthocyanidin with monomeric tea catechins occurred, and consequently, the polymeric molecules were converted into oligomers. The filtrate was directly applied to a Sephabeads SP 825 (45 × 10 cm i.d.) column. Elution with water washed out non-phenolic compounds. Further elution with water containing increasing amounts of ethanol (20–80% ethanol) yielded a mixture of oligomeric proanthocyanidin and tea catechins. The mixture was subsequently subjected to Sephadex LH-20 CC with ethanol. The monomeric tea catechins were eluted out with ethanol, and further elution with 50% aqueous acetone yielded oligomers.147 To prepare polymeric proanthocyanidin from persimmon peel, an aqueous acetone extract was subjected to MCI-gel CHP 20P CC with water containing methanol (0–80%) to yield polymers.
Carotenoid extracts from banana peel were purified with an alumina column and eluted with acetone: n-hexane (2:1, v: v)77 or successively with petroleum ether, ether:petroleum ether (9:1, v: v), ether:petroleum ether (1:4, v :v), ether:petroleum ether (1:3, v:v) and methanol:ether (1:9, v:v).92 A silica gel column was used to purify sterols from mango seed kernel oil.143
:methanol:ethyl acetate :ethyl methyl ketone (6:1.6:2:2, v:v:v:v) as the mobile phase for gallic acid and methyl gallate and chloroform:ethanol:formic acid (3:5:1, v:v: v) for pentagalloylglucopyranose. The qualitative analysis of ferulic acid extracts from pineapple peel was performed by HPTLC on pre-coated silica gel 60 F254 (20 × 10 cm) plates.85 The plate was developed with the mobile phase benzene:dioxane :acetic acid (85:15:1, v:v:v) at room temperature. The xanthones and flavan-3-ols of the mangosteen pericarp were separated by TLC on silica gel 60 F254 using n-hexane:chloroform as the mobile phase.125,126,148 The C-glycosylflavonoids of the pericarp extracts from passion fruit were also purified by TLC.42 Plates coated with silica gel F254 were used as adsorbent and ethyl acetate:formic acid:water (80:10:10, v:v:v) and ethyl acetate:acetone:acetic acid:water (60:20:10:10, v:v: v:v) as the mobile phase. Unsaponifiable matter from mango143 or date seed oil19 was fractionated on silica gel plates. The plates were developed with n-hexane:diethyl ether (4:1, v:v)143 or (7:3, v:v).19 Visualization of TLC spots or bands is normally carried out with an ultraviolet (UV) lamp on a silica gel plate impregnated with an indicator84,85,125,126,148 or by spraying the plate with sulfuric acid in ethanol,125,126,148 diphenylboryloxyethylamine in methanol,46 0.01% rhodamine in ethanol143 or with 0.2% 2,7-dichlorofluorescein in ethanol.19
:v) formic acid in water (eluant A) and 60% (v:v) acetonitrile in water (eluant B), at a flow rate of 9 ml min−1. A preparative separation of individual rhamnetin glycosides from mango peel was performed with the same preparative stationary phase described above, but with the following mobile phase: 2% (v:v) acetic acid in water (eluant A) and of 0.5% acetic acid in water and acetonitrile (1:1, v:v; eluant B).81 The separation of rhamnetin glycosides was achieved isocratically (37% eluant B) at a flow rate of 17 ml min−1. For final purification, a Chromabond C18 (1 g, 6 ml bed volume) SPE column was used. The rhamnetin glycosides were eluted with acidified acetonitrile (pH 3).81 After low-pressure liquid chromatography on a Sephadex LH-20 sorbent column, gallotannins from mango kernel were purified by using semi-preparative HPLC on a Supelcosil LC-18-DB semi-preparative (25 × 1.0 cm, 5 μm) column, operated at room temperature.101 The gallotannins were eluted, in a first step, with a gradient system consisting of 2% (v:v) acetic acid in water (eluant A, pH 2.6) and 0.5% acetic acid in water:acetonitrile (1:1, v:v, eluant B, pH 3.6) at a flow rate of 4.0 ml min−1. The second purification step was performed using an isocratic solvent system (70% A).101 To separate individual compounds in extracts of cashew nut shell and the anacardic acid, cardanol and cardol fractions, semi-preparative HPLC was carried out with an RP-C18 column (25 × 1.0 cm).149 The mobile phase consisted of 1% acetic acid in 80% acetonitrile run in the isocratic mode at a flow rate of 3.0 ml min−1.149 Flavonoids from litchi pericarp were partitioned with n-hexane, ethyl acetate and n-butanol, sequentially.64,65 The ethyl acetate fraction was purified on a reversed phase polystyrene-divinyl (10 × 0.6 cm) column. 2% Formic acid in water (eluant A) and 1% formic acid in water:acetonitrile (1:4, v:v; eluant B) were used as mobile phase.64,65 The phenolic compounds from tamarind seed and seed coat extracts were purified using a Develosil ODS-10 (25 × 2.0 cm) column with 0.1% trifluoroacetic acid in 17% acetonitrile at a flow rate of 5.0 ml min−1.20 Sudjaroen et al.100 also purified the extracts obtained from tamarind pericarp and seeds by using a RP-C18 (1.0 cm i.d.) column with 2% acetic acid in water and methanol at 3.0 ml min−1.
:ethyl acetate:methanol:water (0.5:5:1:5, v:v:v:v). After equilibration, the upper phase was used as the stationary phase, while the lower phase served as the mobile phase. Once, the sample was injected, the mobile phase was pumped at a flow rate of 3 ml min−1 in head-to-tail mode. After a run time of 300 min, the upper phase was used as the mobile phase to facilitate the elution of higher galloylated tannins. The xanthones from mangosteen pericarp were separated at the semi-preparative scale by HSCCC with a biphasic solvent system consisting of heptane:ethyl acetate:methanol:water (2:1:2:1, v:v:v:v).137 The HSCCC column was filled with the upper organic and then equilibrated with the lower aqueous phase, in the descending mode.
The anthocyanins contained in the extracts obtained from tamarillo peel were also fractionated by HSCCC, with a solvent system consisting of n-butanol:tert-butyl methyl ether:acetonitrile:water (2:2:1:5, v:v:v:v) acidified with 0.1% trifluoroacetic acid.48 The organic phase was used as the stationary phase and elution mode was head-to-tail.
4. Identification and quantification
In general, identification and quantification procedures are universally applicable to phytochemical extracts regardless of species or plant part. Remarkable developments in instrumentation have resulted in rapid advancement of methods for high-throughput separation and detection of complex multi-component mixtures containing trace quantities of the analytes of interest. However, the analysis of phytochemical compounds in biowastes obtained from tropical and subtropical fruits made over the past few years has focused on identification. There has been considerably less “interest” in quantifying the antioxidant phytochemicals, presumably due to the limited range of bioactive compounds commercially available as suitable reference compounds.4.1 Spectrophotometric methods
Traditional methods to determine components relied on colorimetric measurement of the total phenols using reagents of varying selectivity. Folin-Ciocalteau12,13,15,16,18,19,22–27,29–31,35,36,38,40,42,45,49,50,52–56,75,77,78,86,89,90,97,99,105,109–111,113–115 or Prussian blue42 are the classical assays recommended for total phenol measurement (Table 3). These assays, however, are not specific for phenols and hence other compounds, such as ascorbic acid, may interfere. Moreover, the diversity of phenolics means that selection of a reagent and/or absorbing wavelength will be a compromise. Results are expressed in terms of molar equivalents of a commonly occurring phenolic, such as gallic [e.g., gallic acid equivalents (GAE)],(+)-catechin, pyrogallol, caffeic acid or tannic acid (Table 3). In most cases the absorbance is measured around 760 nm, with the Folin-Ciocalteau reactive being used as the chromophore (Table 3).The determination of total phenols can be coupled with the use of an insoluble matrix, such as PVPP, to measure tannins. PVPP is added to extracts from fruit by-products and this agent binds tannins that precipitate.42,50 Other than tannins, the supernatant only has simple phenolics; therefore, tannins can be quantified by calculating the difference between total phenolic compounds and the phenols in the supernatant, both measured by the Folin-Ciocalteau assay.
|
Total flavonoids are usually determined by using a colorimetric method (with measurement at 510 nm) based on the formation of acid-stable complexes between aluminium chloride and the C-4 keto group and either the C-3 or C-5 hydroxyl group of flavones and flavonols. In addition, aluminium chloride forms acid-labile complexes with the orthodihydroxyl groups in the A- or B-ring of flavonoids. This method has been used to determine flavonoids in banana peel,45,78 mango peel,24,45 date seeds,30 litchi pericarp,64,68–70 mangosteen pericarp,105 passion fruit peel45 and tamarillo pomace.49 Results are usually expressed as quercetin,49,105 catechin30,78 and rutin24,45 equivalents. However, only flavones and flavonols are found to form stable complexes with aluminium chloride; none of the colorimetric methods can detect all types of flavonoids.
The analysis of anthocyanins is complex due to their ability to undergo structural transformations and complexation reactions. The total anthocyanin content in crude extracts containing other phenolic materials has been determined by measuring solution absorbance at a single wavelength, usually 520–535 nm.59,67,106,120,144 This is possible because the typical absorption band, in the 490 to 550 nm region of the visible spectra, of the anthocyanins is far from the absorption bands of other phenolics, which have spectral maxima in the UV range. In many instances, however, this simple method is inappropriate because of interference from anthocyanin degradation products from browning reactions. In those cases, the approach has been to use differential and/or subtractive methods to quantify anthocyanins and their degradation products. The monomeric anthocyanin concentration is widely evaluated by the pH differential method, based on the structural transformation of anthocyanins that occurs with a change in pH (colored at pH 1.0 and colorless at pH 4.5).22,25,48,49,61,63,66,99,106,109 Results are usually expressed as cyanidin 3-glucoside22,25,59–61,63,66,99,109,144 or as delphinidin 3-glucoside48 equivalents.
The determination of condensed tannins (proanthocyanidins) is based on oxidative depolymerization of condensed tannins in butanol-HCl reagent.42 The presence of iron is considered to increase the reproducibility and sensitivity of the assay. Vanillin-HCl is also used to determine condensed tannins.49,99
Due to the presence of a long system of conjugated double bonds, carotenoids are intensely colored and thus strongly absorb in the visible region, between 400 and 500 nm, usually exhibiting three absorption bands or two bands plus a shoulder, with vibrational fine structure, the middle band having the highest intensity.129 These absorption properties provide a simple and cheap possibility for direct analytical determination of the total carotenoid content by measuring the absorption of the extracts at 450 nm; the results are usually expressed as trans-β-carotene equivalents.51,53
Some phenolic compounds possess native fluorescence. This characteristic has been used to determine major phenolics in persimmon peel.54 Therefore, fluorescence emission has been measured at excitation (λexc) and emission (λem) wavelengths suitable for each phenolic acid that has been determined: gallic acid λexc = 260 nm, λem = 357 nm, pH = 4.63; protocatechuic acid λexc = 290 nm, λem = 363 nm, pH = 10.7; vanillic acid λexc = 305 nm, λem = 378 nm, pH = 9.3; p-coumaric acid λexc = 330 nm, λem = 443 nm, pH = 10.7; and ferulic acid λexc = 340 nm, λem = 460 nm, pH = 11.2.
4.2 Chromatographic methods
The need to profile and identify individual phytochemical compounds has seen traditional methods replaced by separation methods. Several techniques have been used to separate and determine the different types of antioxidant compounds. The limited volatility of most of the phytochemicals restricts the application of gas chromatography (GC) for their separation. However, with suitable derivatization they are adapted to GC coupled to a flame ionization detector (FID)31,114 or mass spectrometry (MS)34,100 determination (Table 4). Currently, the most common methods to determine plant sterols and tocopherols involve extraction of the lipid fraction from the biowaste followed by alkaline hydrolysis (saponification), extraction of the non-saponifiables, derivatization and separation and quantification of the derivatives by GC.13,47,104,143 Moreover, GC has also been used to confirm phenolic100 or carotenoid92 identity. To volatilize hydroxy-containing phytochemicals, enhance component resolution and stabilize thermally labile phenolic compounds, carotenoids, tocopherols and sterols, these compounds are more frequently analyzed as their trimethysilyl (TMS) derivatives.31,47,92,100,104,114 The derivatization process involves dissolving the dried non-saponifiable material of the fruit biowaste in dry pyridine, followed by treatment of the mixture with bis-(trimethylsilyl)-trifluoroacetamide with heating for 15–30 min at 60 °C. The mixture is then left to stand at room temperature overnight. Depending on the sample, matrices and the number of analyte species in samples, GC analyses of TMS derivatives of plant phenols, carotenoids, sterols and tocopherols are run by temperature programming at various gas flow rates (Table 4). The majority of the methods employ polysiloxane phases for antioxidant analysis, mainly 100% poly(dimethylsiloxane) or 5% phenyl and 95% poly(dimethylsiloxane).13,31,34,47,100,104,114,143
|
|
|
|
HPLC is currently the most popular and reliable technique for the analysis of phenolic compounds obtained from biowastes of tropical and subtropical fruits (Table 4). A typical system involves reversed phase liquid chromatography (RP-HPLC) comprising a C18 stationary phase, in a column ranging from 15 to 30 cm in length and usually with a 4.6 mm internal diameter (i.d.) and 5 μm packing. In some instances, isocratic elution has provided adequate resolution due to selectivity effects of one or more components of the mobile phase, although gradient elution has usually been mandatory because of the complexity of the phenolic profile of most samples (Table 4). Elution systems are usually binary, with an aqueous acidified polar solvent and a less polar organic solvent such as acetonitrile or methanol. Occasionally tetrahydrofuran and 2-propanol have also been used as the organic solvent. The greatest alteration observed in the mobile phases is the acid type used as modifier to minimize peak tailing. Most often acetic or formic acid are employed; however, phosphoric acid and trifluoroacetic acid have also been used. Flow rates are usually 1.0 ml min−1 and columns are kept at ambient or slightly above ambient temperature. Phenolic elution is typical of RP-HPLC, that is, polar compounds elute first, followed by those of decreasing polarity. Hence, an elution order can be developed as phenolic acids < cinnamic acids < flavonoids although overlap of the individual members of different classes is inevitable because of the diversity of compounds.121 In phenolic and cinnamic acids, polarity is increased most by hydroxyl groups at the 4-position, followed by those at the 3- and 2-positions. The elution pattern for flavonoids containing equivalent substitution patterns is flavanone glycoside followed by flavonol and flavone glycosides and then the free aglycones in the same order.121
HPLC has been shown to be the most convenient and reliable technique for the analysis of carotenoids. Both normal phase and reversed phase HPLC methods, operating with isocratic or gradient elution and a wide variety of mixtures of different organic solvents as mobile phases, have been used to separate and quantify carotenoids in fruit biowastes. Separation has been more effective using a RP-C30 stationary phase instead of RP-C18, because the resolution of geometrical isomers with this column is outstanding (Table 4). However, for plant material with a relatively simple carotenoid profile there seems to be little advantage gained by using RP-C30 columns rather than conventional RP-C18 columns. The high price of RP-C30 columns combined with a limited column lifetime for this type of analysis significantly increases overall costs.51 In reversed-phase systems, non-aqueous mobile phases are recommended because the pronounced hydrophobicity of carotenoids makes it difficult or impossible to separate them in mobile phases containing water. Various mixtures of solvents, mostly of methanol, acetonitrile and tetrahydrofuran, have been successfully used for this purpose.129
Carotenoids from banana peel were separated in an ODS-A column with different isocratic solvent systems: methanol:ethyl acetate (62:38, v:v), methanol:ethyl acetate (88:12, v:v) or methanol:water:ethyl acetate (80:10:10, v:v:v).92 The analysis of these analytes in banana peel was carried out using a RP-C18 column with acetonitrile and methanol:ethyl acetate (1:1, v:v), containing triethylamine and BHT, as mobile phase (Table 4).51 Carotenoids in persimmon peel have been analyzed using a RP-C30 column with a mobile phase consisting of methanol:methyl tert-butyl ether:water (81:15:4, v:v::v) (eluant A) and methanol:methyl tert-butyl ether (10:90, v:v) (eluant B), both containing ammonium acetate.102 Tocopherols in pitaya seeds have been analyzed by HPLC with fluorescence detection.47 The separation of α- and γ-tocopherols was performed in a LichroCart Purospher STAR Si column with n-hexane:2-propanol (99:1, v:v) as mobile phase (Table 4).
Other separation techniques, such as capillary electrophoresis, have not been used to detect antioxidant phytochemicals in tropical and subtropical biowastes.
4.3 Detection
Routine detection in HPLC is typically based on measuring UV absorption or, less commonly, visible radiation in the case of anthocyanins, often involving a photo-diode array (PDA) detector. No single wavelength is ideal for all classes of phenols since they display absorbance maxima at distinctly different wavelengths. The most commonly used wavelength for routine detection is 280 nm which represents a suitable compromise, although detection at other wavelengths has been applied (Table 4). Two absorption bands are characteristic of flavonoids; band II, with a maximum in the 240–285 nm range, is believed to arise from the A-ring and band I with a maximum in the 320–385 nm range, representing the B-ring absorption. Anthocyanins show band II and band I absorption maxima in the 265–275 and 465–560 nm regions, respectively.The complementary nature of fluorescence detection has been demonstrated and used in series with a UV detector to detect tannins. Michodjehoun-Mestres et al. used a fluorescence detector (275 nm for excitation and 322 nm for emission) connected in series to a PDA detector (280 nm) to detect tannins in cashew apple skin.33 However, this technique, and others such as electrochemistry, have not been commonly used to detect antioxidant phytochemicals in this type of biowaste.
Mass spectrometry (MS) is a powerful tool for elucidating phenolic structures. MS can be carried out either on-line in combination with chromatographic techniques or off-line. The on-line coupling of chromatography with MS has been the single-most important advance in the analysis of bioactive compounds in biowastes obtained from tropical and subtropical fruits (Table 4). Moreover, the number of applications involving direct inlet introduction of phytochemicals to the mass spectrometer has increased significantly in the last years.
In coupled mode, the mass spectrometer may function simply as a highly selective detector but it is in qualitative analysis that it excels providing unsurpassed opportunities for compound “identification”. GC-MS has been used, with electron impact ionization (EI), to identify phenolic compounds in cashew nut shell34 or phenolic and cinnamic acids in mangosteen pericarp114 previously derivatizing the phenolic compounds. Moreover, GC-MS has been used to identify sterols and tocopherols from banana peel104 or sterols from mango seed kernel oil.143 However, it is the hyphenation of LC with MS that has revolutionized the analysis of non-volatile species (Table 4). The development of soft ionization techniques, such as atmospheric pressure ionization (API), to analyze polar, non-volatile, thermolabile molecules has facilitated the analysis of high molecular mass molecules such as plant phenols by LC-MS. API-based interfacing systems are mainly based on electrospray ionization (ESI). API mass spectra typically comprise protonated molecular ions [M + H]+ in positive ion mode or deprotonated molecular ions [M − H]− in negative ion mode with few fragment ions and thus have a low structure information content. An acid (acetic or formic) is often added to mobile phases in positive ion ESI as a source of protons to assist ionization.
On rare occasions, MS can provide sufficient data for full structure analysis. Generally, the analyte fragmentation in mass spectra is used to determine molecular mass, elemental formula and to establish the distribution of substituents on the phenolic rings. In less favorable situations, the mass spectrum will assist in structural elucidation although other techniques such as nuclear magnetic resonance (NMR) are used for a definitive structural assignment.28,34,43,44,48,69,70,73,77,79,93,94,124–126,128 NMR spectra of phenols are frequently complex and identifying isolated compounds is complicated by the absence of suitable reference standards. In many instances, the combination of UV, MS and 1H NMR will provide adequate information for structural elucidation.20,74,100,148 In other cases, information on the 13C NMR20,34,43,44,69,70,73,74,77,100,124,125,128,149 signals is necessary along with 2D correlation experiments involving 1H–1H correlations such as COSY (Correlation Spectroscopy)20,48,100 or 1H–13C correlation experiments such as HMBC (Heteronuclear Multiple Quantum Coherence)20,73 and HSQC (Heteronuclear Multiple Bond Coherence)48 as applied in the structural identification of anthocyanins isolated from tamarillo peel,48 xanthones from mangosteen pericarp73 or phenolic compounds isolated from tamarind seeds and tamarind seed coats.20
5. Future trends
The exploitation of biowastes generated from fruit processing as a source of functional compounds and their application in food is a promising field which requires interdisciplinary research by food technologists, food chemists, nutritionists and toxicologists. The design of functional foods (their complex matrix and their composition of bioactive principles) requires careful assessment of potential risks which might arise from isolated compounds recovered from by-products. Therefore, the bioactivity, bioavailability and toxicology of phytochemicals need to be carefully assessed by in vitro and in vivo studies. Minimization of potentially unsafe constituents and optimization of valuable compounds may be achieved by plant breeding. Moreover, research on the stability of phytochemicals and how they interact with other food ingredients during processing and storage needs to be carried out.From this global perspective, it is vital to develop analytical methods designed specifically to quantify phytochemicals. One of the current difficulties with phytochemical analysis is lack of confidence in methodology. There is no discussion on how the recovery of bioactive compounds from tropical and subtropical fruit biowastes may be influenced by many factors, e.g., specie, variety, ripening stage, preharvest and postharvest conditions and extraction conditions. Moreover, there are no standard procedures to isolate phytochemicals and, in the majority of cases, the procedures employed to extract phytochemicals from tropical and subtropical fruits biowaste have not been specifically optimized for them. Therefore, any quantitative data on phytochemical concentration must be evaluated in this light. In this sense, developing more effective and selective extraction techniques will significantly improve the process of obtaining bioactive compounds from tropical and subtropical fruit biowastes. Intense work needs to be carried out to increase the commercial availability of bioactive compounds as suitable reference compounds and of reference materials. In addition, it is necessary to carry out validation studies on the analytical methods developed specifically to analyze bioactive compounds from tropical and subtropical fruit biowastes.
Acknowledgements
M. González would like to thank the Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA) for awarding the contract within the framework of the “Recursos y Tecnologías Agrarias del Plan Nacional de Investigación Científica, Desarrollo e Innovación Tecnológica 2000–2003” strategic action, financed with the involvement of the European Social Fund. This work has been supported by the projects RTA2006-00187 and MAC/I/C054 BIOMUSA (financed by FEDER).References
- European Union, Off. J. Eur. Commun., L182, European Community, Brussels, 1999 Search PubMed.
- European Union, Off. J. Eur. Commun., L312/3, European Community, Brussels, 2008 Search PubMed.
- V. Oreopoulou and C. Tazia, in Utilization of by-products and treatment of waste in the food industry, ed. V. Oreopoulou and W. Russ, Springer, London, 2007, ch. 11, pp. 209–232 Search PubMed.
- A. J. Parr and G. P. Bolwell, J. Sci. Food Agric., 2000, 80, 985–1012 CrossRef CAS.
- I. F. F. Benzie, Eur. J. Nutr., 2000, 39, 53–61 CrossRef CAS.
- V. Galán-Saúco, in Encyclopedia of food and culture, ed. S. H. Katz and W. W. Weaver, Charles Scribners's Sons, New York, 2003, vol. 2, pp. 70–78 Search PubMed.
- FAO, Tropical Fruits Network, 2008, accessed April 2010, http://www.itfnet.org/ Search PubMed.
- FAOSTAT, Food and Agriculture Organization of the United Nations Statistical Database, 2009, accessed April 2010, http://faostat.fao.org Search PubMed.
- FAO, Tropical fruits compendium, Joint meeting of the fourth session of the Sub-Group on Bananas and the fifth session of the Sub-Group on Tropical Fruits, Rome, 2009 Search PubMed.
- D. L. Luthria, J. Sci. Food Agric., 2006, 86, 2266–2272 CrossRef CAS.
- D. Tura and K. Robards, J. Chromatogr., A, 2002, 975, 71–93 CrossRef CAS.
- S. M. R. Ribeiro, L. C. A. Barbosa, J. H. Queiroz, M. Knödler and A. Schieber, Food Chem., 2008, 110, 620–626 CrossRef CAS.
- A. E. M. Abdalla, S. M. Darwish, E. H. E. Ayad and R. M. El-Hamahmy, Food Chem., 2007, 103, 1134–1140 CrossRef CAS.
- D. Puravankara, V. Boghra and R. S. Sharma, J. Sci. Food Agric., 2000, 80, 522–526 CrossRef CAS.
- P. Maisuthisakul and M. H. Gordon, Food Chem., 2009, 117, 332–341 CrossRef CAS.
- A. C. de Oliveira, I. B. Valentim, C. A. Silva, E. J. H. Bechara, M. P. de Barros, C. M. Mano and M. O. F. Goulart, Food Chem., 2009, 115, 469–475 CrossRef.
- J. M. C. da Costa, E. M. F. Felipe, G. A. Maia, I. M. Brasil and F. F. H. Hernández, Rev. Cienc. Agron., 2007, 38, 228–232 Search PubMed.
- V. Kamath and P. S. Rajini, Food Chem., 2007, 103, 428–433 CrossRef CAS.
- S. Besbes, C. Blecker, C. Deroanne, N. Bahloul, G. Lognay, N. E. Drira and H. Attia, J. Food Lipids, 2004, 11, 251–265 CrossRef CAS.
- T. Tsuda, M. Watanabe, K. Ohshima, A. Yamamoto, S. Kawakishi and T. Osawa, J. Agric. Food Chem., 1994, 42, 2671–2674 CrossRef CAS.
- T. Tsuda, K. Mizuno, K. Ohshima, S. Kawakishi and T. Osawa, J. Agric. Food Chem., 1995, 43, 2803–2806 CrossRef CAS.
- R. González-Montelongo, M. G. Lobo and M. González, Food Chem., 2010, 119, 1030–1039 CrossRef CAS.
- R. González-Montelongo, M. G. Lobo and M. González, Sep. Purif. Technol., 2010, 71, 347–355 CrossRef CAS.
- H. Kim, J. Y. Moon, H. Kim, D. S. Lee, M. Cho, H. K. Choi, Y. S. Kim, A. Mosaddik and S. K. Cho, Food Chem., 2010, 121, 429–436 CrossRef CAS.
- C. M. Ajila, K. A. Naidu, S. G. Bhat and U. J. S. Prasada Rao, Food Chem., 2007, 105, 982–988 CrossRef CAS.
- C. M. Ajila, S. G. Bhat and U. J. S. Prasada Rao, Food Chem., 2007, 102, 1006–1011 CrossRef CAS.
- T. Thanaraj, L. A. Terry and C. Bessant, Food Chem., 2009, 112, 786–794 CrossRef CAS.
- J. C. Barreto, M. T. S. Trevisan, W. E. Hull, G. Erben, E. S. de Brito, B. Pfundstein, G. Würtele, B. Spiegelhalder and R. W. Owen, J. Agric. Food Chem., 2008, 56, 5599–5610 CrossRef CAS.
- M. Al-Farsi, C. Alasalvar, M. Al-Abid, K. Al-Shoaily, M. Al-Amry and F. Al-Rawahy, Food Chem., 2007, 104, 943–947 CrossRef CAS.
- M. A. Al-Farsi and C. Y. Lee, Food Chem., 2008, 108, 977–985 CrossRef CAS.
- P. R. B. Broinizi, E. R. S. Andrade-Wartha, A. M. O. Silva, A. J. V. Novoa, R. P. Torres, H. M. C. Azeredo, R. E. Alves and J. Mancini-Filho, Cienc. Tecnol. Aliment., 2007, 27, 902–908 Search PubMed.
- L. Michodjehoun-Mestres, J. M. Souquet, H. Fulcrand, C. Bouchut, M. Reynes and J. M. Brillouet, Food Chem., 2009, 112, 851–857 CrossRef CAS.
- L. Michodjehoun-Mestres, J. M. Souquet, H. Fulcrand, E. Meudec, M. Reynes and J. M. Brillouet, Food Chem., 2009, 114, 989–995 CrossRef CAS.
- S. G. De Lima, C. M. Feitosa, A. M. G. L. Cito, J. M. Moita-Neto, J. A. D. Lopes, A. S. Leite, M. C. Brito, S. M. M. Dantas and A. A. C. Melo-Cavalcante, Genetics and Molecular Research, 2008, 7, 806–818 Search PubMed.
- K. N. Prasad, B. Yang, M. Zhao, N. Ruenroengklin and Y. Jiang, J. Food Process Eng., 2009, 32, 828–843 Search PubMed.
- K. N. Prasad, B. Yang, M. Zhao, B. S. Wang, F. Chen and Y. Jiang, Int. J. Food Sci. Technol., 2009, 44, 960–966 CrossRef.
- J. Sun, Y. Jiang, J. Shi, X. Wei, S. J. Xue, J. Shi and C. Yi, Food Chem., 2010, 119, 753–757 CrossRef CAS.
- K. N. Prasad, B. Yang, S. Yang, Y. Chen, M. Zhao, M. Ashraf and Y. Jiang, Food Chem., 2009, 116, 1–7 CrossRef.
- K. N. Prasad, E. Yang, C. Yi, M. Zhao and Y. Jiang, Innovative Food Sci. Emerging Technol., 2009, 10, 155–159 CrossRef CAS.
- K. N. Prasad, J. Hao, J. Shi, T. Liu, J. Li, X. Wei, S. Qju, S. Xue and Y. Jiang, Innovative Food Sci. Emerging Technol., 2009, 10, 413–419 CrossRef CAS.
- K. N. Prasad, B. Yang, M. Zhao, X. Wei, Y. Jiang and F. Chen, Sep. Purif. Technol., 2009, 70, 41–45 CrossRef CAS.
- N. He, Z. Wang, C. Yang, Y. Lu, D. Sun, Y. Wang, W. Shao and Q. Li, Sep. Purif. Technol., 2009, 70, 219–224 CrossRef CAS.
- J. Shi, J. Sun, X. Wei, J. Shi, G. Cheng, M. Zhao, J. Wang, B. Yang and Y. Jiang, LWT–Food Sci. Technol., 2008, 41, 1742–1747 CrossRef CAS.
- G. Zheng, L. Xu, P. Wu, H. Xie, Y. Jiang, F. Chen and X. Wei, Food Chem., 2009, 116, 433–436 CrossRef CAS.
- G. P. P. Lima, S. A. da Rocha, M. Takaki, P. R. R. Ramos and E. O. Ono, Int. J. Food Sci. Technol., 2008, 43, 1838–1843 CrossRef CAS.
- L. M. Sena, S. M. Zucolotto, F. H. Reginatto, E. P. Schenkel and T. C. Monteiro-De-Lima, Exp. Biol. Med., 2009, 234, 967–975 Search PubMed.
- H. K. Lim, C. P. Tan, R. Karim, A. A. Ariffin and J. Bakar, Food Chem., 2010, 119, 1326–1331 CrossRef CAS.
- N. H. Hurtado, A. L. Morales, M. L. González-Miret, M. L. Escudero-Gilete and F. J. Heredia, Food Chem., 2009, 117, 88–93 CrossRef CAS.
- R. M. Ordóñez, M. L. Cardozo, I. C. Zampini and M. I. Isla, J. Agric. Food Chem., 2010, 58, 331–337 CrossRef CAS.
- P. Siddhuraju, LWT–Food Sci. Technol., 2007, 40, 982–990 CrossRef CAS.
- M. W. Davey, J. Keulemans and R. Swennen, J. Chromatogr., A, 2006, 1136, 176–184 CrossRef CAS.
- M. R. S. Ardekani, M. Khanavi, M. Hajimahmoodi, M. Jahangiri and A. Hadjiakhoondi, Iranian J. Pharm. Res., 2010, 9, 141–146 Search PubMed.
- M. S. Akter, M. Ahmed and J. B. Eun, Int. J. Food Sci. Technol., 2010, 45, 1464–1471 CrossRef CAS.
- S. Gorinstein, Z. Zachwieja, M. Folta, H. Barton, J. Piotrowicz, M. Zemser, M. Weisz, S. Trakhtenberb and O. Martín-Belloso, J. Agric. Food Chem., 2001, 49, 952–957 CrossRef CAS.
- I. C. Jang, E. K. Jo, M. S. Bae, H. J. Lee, G. I. Jeon, E. Park, H. G. Yuk, G. H. Ahn and S. C. Lee, J. Med. Plants. Res., 2010, 4, 155–160 Search PubMed.
- M. Suksomtip and S. Pongsamart, LWT–Food Sci. Technol., 2008, 41, 2002–2007 CrossRef CAS.
- O. B. O. Ashton, M. Wong, T. K. McGhie, R. Vather, Y. Wang, C. Requejo-Jackman, P. Ramankutty and A. B. Woolf, J. Agric. Food Chem., 2006, 54, 10151–10158 CrossRef CAS.
- K. A. Cox, T. K. McGhie, A. White and A. B. Woolf, Postharvest Biol. Technol., 2004, 31, 287–294 CrossRef CAS.
- S. Underhill and C. Critchley, Aust. J. Exp. Agric., 1994, 34, 115–122 CrossRef CAS.
- Y. Jiang, J. Sci. Food Agric., 2000, 80, 305–310 CrossRef CAS.
- Z. Zhang, X. Pang, Z. Ji and Y. Jiang, Food Chem., 2001, 75, 217–221 CrossRef CAS.
- Z. Zhang, P. Xuequn, C. Yang, Z. Ji and Y. Jiang, Food Chem., 2004, 84, 601–604 CrossRef CAS.
- Z. Zhang, X. Pang, D. Xuewu, Z. Ji and Y. Jiang, Food Chem., 2005, 90, 47–52 CrossRef CAS.
- M. Zhao, B. Yang, J. Wang, B. Li and Y. Jiang, Food Chem., 2006, 98, 539–544 CrossRef CAS.
- M. Zhao, B. Yang, J. Wang, Y. Li, L. Yu and Y. Jiang, Int. Immunopharmacol., 2007, 7, 162–166 CrossRef CAS.
- X. Duan, Y. Jiang, X. Su, Z. Zhang and J. Shi, Food Chem., 2007, 101, 1365–1371 CrossRef CAS.
- N. Ruenroengklin, J. Zhong, X. Duan, B. Yang, J. Li and Y. Jiang, Int. J. Mol. Sci., 2008, 9, 1333–1341 Search PubMed.
- P. Sarni-Manchado, E. Le Roux, C. Le Guernevé, Y. Lozano and V. Cheynier, J. Agric. Food Chem., 2000, 48, 5995–6002 CrossRef CAS.
- J. Sun, Y. Jiang, X. Wei, J. Shi, Y. You, H. Liu, Y. Kakuda and M. Zhao, Food Res. Int., 2006, 39, 864–870 CrossRef CAS.
- J. Sun, J. Shi, Y. Jiang, S. J. Xue and X. Wei, J. Agric. Food Chem., 2007, 55, 5864–5868 CrossRef CAS.
- N. Rangkadilok, L. Worasuttayangkurn, R. N. Bennett and J. Satayavivad, J. Agric. Food Chem., 2005, 53, 1387–1392 CrossRef CAS.
- N. Rangkadilok, S. Sitthimonchai, L. Worasuttayangkurn, C. Mahidol, M. Ruchirawat and J. Satayavivad, Food Chem. Toxicol., 2007, 45, 328–336 CrossRef CAS.
- S. Suksamrarn, N. Suwannapoch, R. Ratananukul, N. Aroonlerk and A. Suksamrarn, J. Nat. Prod., 2002, 65, 761–763 CrossRef CAS.
- S. Suksamrarn, N. Suwannapoch, W. Phakhodee, J. Thanuhiranlert, P. Ratananukul, N. Chimnoi and A. Suksamrarn, Chem. Pharm. Bull., 2003, 51, 857–859 CrossRef CAS.
- S. Dangcham, J. Bowen, I. B. Ferguson and S. Ketsa, Postharvest Biol. Technol., 2008, 50, 37–44 CrossRef CAS.
- T. Ichimura, A. Yamanaka, T. Ichiba, T. Toyokawa, Y. Kamada, T. Tamamura and S. Maruyama, Biosci., Biotechnol., Biochem., 2006, 70, 718–721 CrossRef CAS.
- S. Someya, Y. Yoshiki and K. Okubo, Food Chem., 2002, 79, 351–354 CrossRef CAS.
- B. Sultana, F. Anwar, M. R. Asi and S. A. S. Chatha, Grasas Aceites, 2008, 59, 205–217 Search PubMed.
- L. Y. Jiang, S. He, Y. J. Pan and C. R. Sun, Food Chem., 2010, 119, 1285–1292 CrossRef CAS.
- A. Schieber, N. Berardini and R. Carle, J. Agric. Food Chem., 2003, 51, 5006–5011 CrossRef CAS.
- N. Berardini, R. Fezer, J. Conrad, U. Beifuss, R. Carle and A. Schieber, J. Agric. Food Chem., 2005, 53, 1563–1570 CrossRef CAS.
- N. Berardini, R. Carle and A. Schieber, Rapid Commun. Mass Spectrom., 2004, 18, 2208–2216 CrossRef CAS.
- C. Engels, M. G. Gänzle and A. Schieber, J. Agric. Food Chem., 2010, 58, 775–780 CrossRef CAS.
- S. Nithitanakool, P. Pithayanukul, R. Bavovada and P. Saparpakorn, Molecules, 2009, 14, 257–265 Search PubMed.
- A. Tilay, M. Bule, J. Kishenkumar and U. Annapure, J. Agric. Food Chem., 2008, 56, 7644–7648 CrossRef CAS.
- Y. Y. Soong and P. J. Barlow, Food Chem., 2004, 88, 411–417 CrossRef CAS.
- Y. Y. Soong and P. J. Barlow, Food Chem., 2006, 97, 524–530 CrossRef CAS.
- A. S. Zarena and K. Udaya-Sankar, J. Supercrit. Fluids, 2009, 49, 330–337 CrossRef CAS.
- G. Shui and L. P. Leong, Food Chem., 2006, 97, 277–284 CrossRef CAS.
- C. F. Yap, C. W. Ho, W. M. W. Aida, S. W. Chan, C. Y. Lee and Y. S. Leong, Sains Malaysiana, 2009, 38, 511–520 Search PubMed.
- S. Luegthanaphol, D. Mongkholkhajornsilp, S. Douglas, P. L. Douglas, L. Pengsopa and S. Pongamphai, J. Food Eng., 2004, 63, 247–252 CrossRef.
- A. Subagio, N. Morita and S. Sawada, J. Nutr. Sci. Vitaminol., 1996, 42, 553–566 Search PubMed.
- F. Yamaguchi, T. Ariga, Y. Yoshimura and H. Nakazawa, J. Agric. Food Chem., 2000, 48, 180–185 CrossRef CAS.
- F. Yamaguchi, M. Saito, T. Ariga, Y. Yoshimura and H. Nakazawa, J. Agric. Food Chem., 2000, 48, 2320–2325 CrossRef CAS.
- T. Kabuki, H. Nakajima, M. Arai, S. Ueda, Y. Kuwabara and S. Dosako, Food Chem., 2000, 71, 61–66 CrossRef CAS.
- Y. Y. Soong and P. J. Barlow, J. Chromatogr., A, 2005, 1085, 270–277 CrossRef CAS.
- W. Pothitirat, M. T. Chomnawang, R. Supabphol and W. Gritsanapan, Pharm. Biol., 2010, 48, 182–186 CrossRef CAS.
- G. Shui and L. P. Leong, J. Chromatogr., A, 2004, 1022, 67–75 CrossRef CAS.
- L. C. Wu, H. W. Hsu, Y. C. Chen, C. C. Chiu, Y. I. Lin and J. A. Ho, Food Chem., 2006, 95, 319–327 CrossRef CAS.
- Y. Sudjaroen, R. Haubner, G. Würtele, W. E. Hull, G. Erben, B. Spiegelhalder, S. Changbumrung, H. Bartsch and R. W. Owen, Food Chem. Toxicol., 2005, 43, 1673–1682 CrossRef CAS.
- C. Engels, M. Knödler, Y. Y. Zhao, R. Carle, M. G. Gänzle and A. Schieber, J. Agric. Food Chem., 2009, 57, 7712–7718 CrossRef CAS.
- R. Izuchi, H. Takahashi and Y. Inada, Biosci., Biotechnol., Biochem., 2009, 73, 2793–2795 CrossRef CAS.
- J. Sun, X. Xiang, C. Yu, J. Shi, H. Peng, B. Yang, S. Yang, E. Yang and Y. Jiang, Sci. Hortic., 2009, 120, 555–559 CrossRef CAS.
- L. Oliveira, C. S. R. Freire, A. J. D. Silvestre and N. Cordeiro, J. Agric. Food Chem., 2008, 56, 9520–9524 CrossRef CAS.
- W. Pothitirat, M. T. Chomnawang, R. Supabphol and W. Gritsanapan, Fitoterapia, 2009, 80, 442–447 CrossRef CAS.
- J. Rivera-López, C. Ondorica-Falomir and P. Wesche-Ebeling, Food Chem., 1999, 65, 195–200 CrossRef.
- L. Liu, S. Cao, B. Xie, Z. Sun and J. Wu, J. Agric. Food Chem., 2007, 55, 9074–9078 CrossRef CAS.
- L. Liu, S. Cao, Y. Xu, M. Zhang, G. Xiao, Q. Deng and B. Xie, Food Chem., 2010, 118, 508–511 CrossRef CAS.
- C. M. Ajila and U. J. S. Prasada Rao, Food Chem. Toxicol., 2008, 46, 303–309 CrossRef CAS.
- K. N. Prasad, B. Yang, J. Shi, C. Yu, M. Zhao, S. Xue and Y. Jiang, J. Pharm. Biomed. Anal., 2010, 51, 471–477 CrossRef CAS.
- S. Rodrigues, G. A. S. Pinto and F. A. N. Fernandes, Ultrason. Sonochem., 2008, 15, 95–100 CrossRef CAS.
- S. Y. Kim, S. M. Jeong, S. J. Kim, K. I. Jeon, E. Park, H. R. Park and S. C. Lee, Biosci., Biotechnol., Biochem., 2006, 70, 999–1002 CrossRef CAS.
- Y. Pan, K. Wang, S. Huang, H. Wang, X. Mu, C. He, X. Ji, J. Zhang and F. Huang, Food Chem., 2008, 106, 1264–1270 CrossRef CAS.
- R. Zadernowski, S. Czaplicki and M. Naczk, Food Chem., 2009, 112, 685–689 CrossRef CAS.
- S. Rodrigues and G. A. S. Pinto, J. Food Eng., 2007, 80, 869–872 CrossRef CAS.
- M. C. Nicoli, M. Anese and M. Parpinel, Trends Food Sci. Technol., 1999, 10, 94–100 CrossRef CAS.
- W. B. Setianto, S. Yoshikawa, R. L. Smith Jr., H. Inomata, L. J. Florusse and C. J. Peters, J. Supercrit. Fluids, 2009, 48, 203–210 CrossRef CAS.
- N. Ruenroengklin, B. Yang, H. Lin, F. Chen and Y. Jiang, Food Chem., 2009, 116, 995–998 CrossRef CAS.
- N. Ruenroengklin, J. Sun, J. Shi, S. J. Xue and Y. Jiang, Food Chem., 2009, 115, 1253–1256 CrossRef CAS.
- R. C. Chisté, L. J. G. de Faria, A. S. Lopes and R. D. A. Mattietto, Revista Brasileira de Fruticultura, 2009, 31, 416–422.
- K. Robards, J. Chromatogr., A, 2003, 1000, 657–691 CrossRef.
- J. Li and Y. Jiang, Molecules, 2007, 12, 745–758 Search PubMed.
- S. Zibadi, R. Farid, S. Moringuchi, Y. Lu, L. Y. Foo, P. M. Tehrani, J. B. Ulreich and R. R. Watson, Nutr. Res., 2007, 27, 408–416 CrossRef CAS.
- L. G. Chen, L. L. Yang and C. C. Wang, Food Chem. Toxicol., 2008, 46, 688–693 CrossRef CAS.
- L. Yu, M. Zhao, B. Yang, Q. Zhao and Y. Jiang, Food Chem., 2007, 104, 176–181 CrossRef CAS.
- L. Yu, M. Zhao, B. Yang and W. Bai, Food Chem., 2009, 116, 969–973 CrossRef CAS.
- E. B. Walker, J. Sep. Sci., 2007, 30, 1229–1234 CrossRef CAS.
- K. Ohguchi, C. Nakajima, M. Oyama, M. Iinuma, T. Itoh, Y. Akao, Y. Nozawa and M. Ito, Biol. Pharm. Bull., 2010, 33, 122–124 CrossRef CAS.
- L. Feltl, V. Pacáková, K. Stulík and K. Volka, Curr. Anal. Chem., 2005, 1, 93–102 CrossRef.
- A. Arora, D. Choudhary, G. Agarwal and V. P. Singh, Int. J. Food Sci. Technol., 2008, 43, 1913–1921 CrossRef CAS.
- M. Herrero, J. A. Mendiola, A. Cifuentes and E. Ibáñez, J. Chromatogr., A, 2010, 1217, 2495–2511 CrossRef CAS.
- M. Takahashi, H. Watanabe, J. Kikkawa, M. Ota, M. Watanabe, Y. Sato, H. Inomata and N. Satto, Anal. Sci., 2006, 22, 1441–1447 CrossRef CAS.
- J. Y. N. Philip, J. D. C. Francisco, E. S. Dey, J. Buchweishaija, L. L. Mkayula and L. Ye, J. Agric. Food Chem., 2008, 56, 9350–9354 CrossRef CAS.
- S. V. Shobha and B. Ravindranath, J. Agric. Food Chem., 1991, 39, 2214–2217 CrossRef CAS.
- K. Vilkhu, R. Mawson, L. Simons and D. Bates, Innovative Food Sci. Emerging Technol., 2008, 9, 161–169 CrossRef CAS.
- M. H. Entezari, S. H. Nazary and M. H. K. Khodaparast, Ultrason. Sonochem., 2004, 11, 379–384 CAS.
- E. Destandau, A. Toribio, M. Lafosse, V. Pecher, C. Lamy and P. André, J. Chromatogr., A, 2009, 1216, 1390–1394 CrossRef CAS.
- Z. Yang and W. Zhai, Innovative Food Sci. Emerging Technol., 2010, 11, 470–476 CrossRef CAS.
- T. S. Ballard, P. Mallikarjunan, K. Zhou and S. O'Keefe, Food Chem., 2010, 120, 1185–1192 CrossRef CAS.
- J. A. Pérez-Serradilla and M. D. Luque de Castro, Food Chem., 2011, 124, 1652–1659 CrossRef CAS.
- M. Pinelo, B. Zornoza and A. S. Meyer, Sep. Purif. Technol., 2008, 63, 620–627 CrossRef CAS.
- B. B. Li, A. B. Smith and M. M. Hossain, Sep. Purif. Technol., 2006, 48, 189–196 CrossRef CAS.
- R. Dhara, D. K. Bhattacharyya and M. Ghosh, J. Oleo Sci., 2010, 59, 169–176 Search PubMed.
- Y. Palapol, S. Ketsa, D. Stevenson, J. M. Cooney, A. C. Allan and I. B. Ferguson, Postharvest Biol. Technol., 2009, 51, 349–353 CrossRef CAS.
- W. Chen, C. Fu, Y. Qin and D. Huang, Food Chem., 2009, 114, 874–880 CrossRef CAS.
- K. S. Nagabhushana and B. Ravindranath, J. Agric. Food Chem., 1995, 43, 2381–2383 CrossRef CAS.
- Y. A. Lee, E. J. Chuo and T. Yokozawa, Biol. Pharm. Bull., 2008, 31, 1265–1269 CrossRef CAS.
- G. Gopalakrishnan, B. Banumathi and G. Suresh, J. Nat. Prod., 1997, 60, 519–524 CrossRef CAS.
- M. T. S. Trevisan, B. Pfundstein, R. Haubner, G. Würtele, B. Spiegelhalder, H. Bartsch and R. W. Owen, Food Chem. Toxicol., 2006, 44, 188–197 CrossRef CAS.
- L. T. Ling, A. K. Radhakrishnan, T. Subramaniam, H. M. Cheng and U. D. Palanisamy, Molecules, 2010, 15, 2139–2151 Search PubMed.
- N. Berardini, M. Knödler, A. Schieber and R. Carle, Innovative Food Sci. Emerging Technol., 2005, 6, 442–452 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |