Development of a RP-HPLC method for the simultaneous analysis of diltiazem and statin: Application in pharmaceuticals and human serum
Najma
Sultana
a,
M. Saeed
Arayne
b,
Nighat
Shafi
a,
Farhan Ahmed
Siddiqui
*b and
Azhar
Hussain
c
aResearch Institute of Pharmaceutical Sciences, Faculty of Pharmacy, University of Karachi, 75270, Pakistan
bDepartment of Chemistry, University of Karachi, Pakistan. E-mail: farhanchemist@gmail.com
cDepartment of Pharmaceutics, University of Karachi, Pakistan
First published on 10th August 2010
Abstract
High-performance liquid chromatographic (HPLC) method has been developed and validated for the simultaneous determination of diltiazem and statins in raw materials, their pharmaceutical formulations and human serum. In HPLC, diltiazem and statins are chromatographed using acetonitrile–water (85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 v/v, pH 2.6 ± 0.02) as the mobile phase at a flow rate of 1.0 mL min−1 at ambient temperature. The separation is carried out on a Hiber®, 250-4.6 RP-18 column, equipped with a UV/visible detector at 230 nm. All the statins eluted at a different retention time and each showed good resolution from diltiazem. The method has been successfully applied to pharmaceutical formulations because no chromatographic interferences from the tablet excipients are found. The linearity is found to be in the range 0.625–20 μg mL−1. The suitability of the method for the quantitative determination of the drugs is proven by validation in accordance with the requirements laid down by the International Conference on Harmonization (ICH) guidelines. The validation results, together with statistical treatment of the data, demonstrated the reliability of this method.
:15 v/v, pH 2.6 ± 0.02) as the mobile phase at a flow rate of 1.0 mL min−1 at ambient temperature. The separation is carried out on a Hiber®, 250-4.6 RP-18 column, equipped with a UV/visible detector at 230 nm. All the statins eluted at a different retention time and each showed good resolution from diltiazem. The method has been successfully applied to pharmaceutical formulations because no chromatographic interferences from the tablet excipients are found. The linearity is found to be in the range 0.625–20 μg mL−1. The suitability of the method for the quantitative determination of the drugs is proven by validation in accordance with the requirements laid down by the International Conference on Harmonization (ICH) guidelines. The validation results, together with statistical treatment of the data, demonstrated the reliability of this method.
Introduction
Hypertension and hyperlipidemia are correlated to each other and have an additional effect on coronary heart disease and the associated mortality rate, since cardiovascular disease is closely related to factors such as hypertension, high cholesterol levels or diabetes. These factors are related to family history, sex, age, obesity and diabetes.1–10 Coadministration of antihypertensive, hypolipemiant and antidiabetic drugs is frequently used in treatment.11–14 One of the most used combinations consists of a synergic association of a diuretic (chlorthalidone, hydrochlorothiazide, etc.) and an angiotensin II receptor antagonist (valsartan, telmisartan, etc.) to control the hypertension, with a statin (fluvastatin, simvastatin, etc.) to reduce the cholesterol levels.15 Therefore, it has been hypothesized that coadministration of an antihypertensive agent and statin might be an effective therapeutic option for multiple cardiovascular risk factors and concomitant management of elevations in blood pressure and LDL cholesterol.16–21Diltiazem is a peripheral and coronary vasodilator with limited negative inotropic activity inhibiting cardiac conduction, particularly at the sino-atrial and atrioventricular nodes. It is available for intravenous administration in the treatment of various cardiac arrhythmias (atrial fibrillation or flutter and paroxysmal supraventicular tacchycardia) and Raynaud's syndrome.22,23 The statins, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors are the most effective among all hypolipidemic agents.24 These agents are highly effectual in enhancing the HDL levels while, reducing LDL cholesterol, overall cholesterol, apolipoprotein B and triglyceride levels.25 In addition, statins may improve the vasodilatation capacity of large arteries and may thus contribute to BP lowering in patients treated with both an anti-hypertensive and a statin.26 Literature survey revealed that number of assay methods have been used for analysis of diltiazem in bulk drug, pharmaceutical preparations and serum using different techniques including colorimetry,27 stripping voltammetry and flow amperometry,28 Raman spectroscopy,29 capillary electrophoresis 30, polarimetry,31 spectrophotometry32 and RP-HPLC.33–35 However, several analytical procedures have also been described for simultaneous separation and enantio-separation of diltiazem, its analogs, possible degradation products and metabolites.33,36,37 A number of assays have been reported for the quantitative determination of statins in pharmaceutical dosage forms, bulk material and human plasma or serum. Nearly all these assay methods are based on either HPLC or GC techniques38 like assay scheme for rosuvastatin,39 simvastatin40,41 and atorvastatin.42 An HPLC method for simultaneous determination of HMG-CoA reductase inhibitors in pharmaceutical formulations and plasma or serum has been reported.39,43,44
There are very few analytical methods developed for the simultaneous determination of a combination of different kind of drugs used in treatment (antihypertensive, hypolipemiant, antidiabetic, antithrombotic). These methods have been applied to pharmaceuticals and plasma samples.45–47 The present study was conducted to analyze the diltiazem and statins in pharmaceutical formulations and human serum. We present the development and validation of a HPLC assay using UV detector for measurement and allowed determination of each agent without the need for development of separate and distinct methods for each analyte.
Experimental
Instrumentation
A Shimadzu HPLC system equipped with LC-10 AT VP pump and SPD-10 A VP UV-VIS detector was utilized. The chromatographic system was integrated via Shimadzu model CBM-102 to P-IV computer loaded with Shimadzu CLASS-VP software (Version 5.03) for data acquisition and mathematical calculations. Rheodyne manual injector fitted with a 20 μL loop, a Hiber®, RT 250-4.6 Purospher® STAR RP-18 column and DGU-14 AM on-line degasser and Mettler Toledo electronic balance, microlitre syringe and micropore filtration assembly were used in this study.Materials and reagents
Diltiazem, was gifted from Hilton Pharma (Private) Limited. Rosuvastatin (X-plended 20 mg), atorvastatin (Atopitar 10 mg) and simvastatin (Atcol 10 mg) Pharm Evo (Pvt.) Ltd., Atco Pharma(Pvt.) Ltd., and Geofman Pharma (Pvt.) Ltd., respectively, were purchased locally. Reference standards of all these statins were supplied by lab-9 of the Department of Chemistry at the University of Karachi. Each product was labelled and had an expiry date of not less than 365 days at the time of study. All reagents used were of HPLC grade. Methanol and phosphoric acid 85% (Merck, Germany) and HPLC grade deionized filtered water were used to prepare the mobile phase. Stock solutions of diltiazem and statins were prepared in the mobile phase. Fresh working solutions were prepared daily. All solutions were filtered (0.45 μm) and degassed using a sonicator.Preparation of solutions
Standard solutions of diltiazem and each statin were prepared by dissolving appropriate amounts of drug in mobile phase acetonitrile–water (85:15, v/v) to yield final concentrations of 100 μg mL−1. Seven concentrations of each drug were prepared by making serial dilutions from stock solutions. For the assay preparation, the content of 20 tablets were powdered, a weighed portion of the powder equivalent to a suitable amount of drug (according to the labeled claims) was transferred into a 50 mL volumetric flask. The drug was fully dissolved in mobile phase and then diluted with this solvent up to the mark, seven dilutions of each drug were prepared, a portion of this solution was filtered through a disposable 0.45 μm filter and then injected.
Serum drug solution
Blood samples were collected from healthy volunteers and then centrifuged at 3000 rpm for 10 min. The supernatant obtained was stored at −20 °C. After thawing, serum was deprotonated by acetonitrile and spiked daily with working solutions to produce desired concentrations of diltiazem and statins. A 20 μL volume of each sample was injected and chromatographed under the above conditions.Chromatographic conditions
The chromatographic analysis was performed at ambient temperature with isocratic elution. The mobile phase consisted of acetonitrile–water (85:15 v/v) with pH adjusted to 2.6 ± 0.02 with phosphoric acid (85%) and pump at a flow rate of 1 mL min−1, sample volume of 20 μL was injected in triplicate onto the HPLC column and effluents were screened over the dative UV region at 230 nm.
Results and discussion
The primary target in developing this LC method was to achieve simultaneous determination of these drugs in pharmaceutical formulations and also in human serum under common conditions that are applicable for routine quality control, research and development of these drugs in ordinary laboratories. To the best of our knowledge no method is available in literature which can analyze these drugs simultaneously, so there was a need to develop a method which could simultaneously determine all these drugs.The development of HPLC methods for the determination of drugs has received considerable attention in recent years because of their importance in the quality control of drugs and drug products.
This work was intended to develop a precise, reliable, and least time consuming method, based on reverse-phase HPLC separation combined with UV detection, for simultaneous drug assay in raw materials, bulk drug samples, dosage formulations and, especially, in human serum.
Method development
Method development is very important for drug quality control, stability, metabolism, pharmacokinetics and toxicity studies. Simple, efficient and economical analytical methods are very critical requirements for all these types of investigation.48:10, 80:20, 70:30, 60:40, 50:50, 40:60, 30:70, 20:80 and 10:90) as well as methanol and water were tried.
An HPLC-UV method was developed and successfully validated for the simultaneous quantitation of diltiazem and statins. Chromatographic parameters were isocratically optimized on a set of different C18 stationary phases with different lengths and particle sizes. Isocratic scouting does not involve re-equilibration of the column.49 First, a Hypersil, ODS, C18 (150 × 4.6 mm, 5 μm) column was experimented with a variety of mobile phases but the consequences still were not adequate. Analyte peaks were not efficient, reproducible with short analyses time (<10 min) and acceptable resolution, because of peak tailing and placebo obstruction in the formulations. Best choice of the stationary phase that provided satisfactory peak shape, resolution and run time was based on a Hiber®, RT 250-4.6 Purospher® STAR RP-18 endcapped (5 μm). A variety of mobile phases were investigated for optimization of chromatographic conditions with respect to the quantity and different rates of organic modifier. In order to select an appropriate organic modifier, different compositions of acetonitrile and methanol were experienced. Beneficial partition was established with acetonitrile. Various mobile phase compositions were attempted to separate diltiazem and statins and the amount of acetonitrile varied, when it was less than 65% of the total volume of mobile phase diltiazem was retained on the column. Mobile phase between 65–82% of acetonitrile showed that statins could not be separated but each showed a good resolution from diltiazem. The optimum results (capacity factor (k′), specificity, good resolution and short omega peak) were obtained when the copmposition of acetonitrile in the mobile phase was 85%. The fitness of the mobile phase was achieved on the foundation of the sensitivity, suitability for stability studies, run time required for the investigation, simplicity of preparation, and utilization of readily available cost-effective solvents. Variation in pH of the mobile phase had a great influence on the retention time, chromatographic peak and efficiency of the chromatographic system. pH of the mobile phase varied between 2.5 to 3.5 and best results were attained at pH 2.6 ± 0.02, if the pH is less than 2.6 peak tailing was observed while at a higher pH the diltiazem and statins peak distorted and retention times were higher. For detection wavelength, UV spectra of both diltiazem and statins overlapped. It was found that both the drugs exhibited the highest response (maximum UV absorption) at 230 nm and offered a greatest intensity with smallest interference. A typical chromatogram of the separation of all the drugs is shown in Fig. 1.
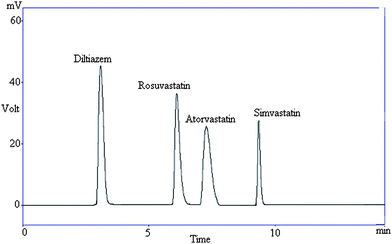 | ||
| Fig. 1 Representative chromatogram showing resolution between diltiazem and statins in a reference standard. | ||
Method validation
Method validation has received considerable attention in the literature, by industrial committees and regulatory agencies.50 The developed method was validated according to the ICH guidelines51 under such parameters as system suitability, selectivity, specificity, linearity, accuracy, precision and sensitivity (detection and quantification limit).| Parameters | % RSD | |||
|---|---|---|---|---|
| DLZ | ROS | ATR | SIM | |
| a Diltiazem; DLZ, Rosuvastatin; ROS, Atorvastatin; ATR, Simvastatin; SIM. | ||||
| Retention time (Rt in minutes) | 0.1846 | 0.3497 | 0.4859 | 0.3783 |
| Capacity factors (K′) | 0.1851 | 0.3492 | 0.4856 | 0.3786 |
| Theoretical plates/N | 1.3241 | 0.0440 | 0.5192 | 0.0385 |
| Tailing factor (T) | 1.0247 | 1.5655 | 1.5890 | 1.9241 |
| Resolution (R) | 0.6251 | 0.4417 | 2.5923 | 2.7442 |
Tailing (symmetry) factor (T) and number of theoretical plates (N) was calculated by formulas: T = W/(2Wa) and N = 5.545(tR/W1/2)2, where W is the peak width at 5% height from baseline, Wa the peak front edge width at the same height and W1/2 is the peak width at half height. According to data of a robustness test study we proposed criteria for system suitability test (tailing factors <2, numbers of theoretical plates >4000 and repeatability (RSD) of five replicate samples to be not more than 1.5, (peak area for replicate analysis). The robustness test is used to verify that the resolution and repeatability of the system are adequate for the analysis intended.
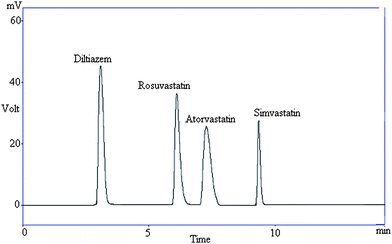 | ||
| Fig. 2 Representative chromatogram showing resolution between diltiazem and statins in pharmaceutical formulations. | ||
Specificity was also determined by screening different samples of controlled human serum, which were free from interfering endogenous plasma components. This is evidenced by the lack of interfering peaks in the chromatograms of serum samples. Fig. 3 represents a typical chromatogram of human serum containing all drugs analysed. A typical chromatogram of blank serum is shown in Fig. 4. The method demonstrated good resolution between the peaks and was found to be free of interferences.
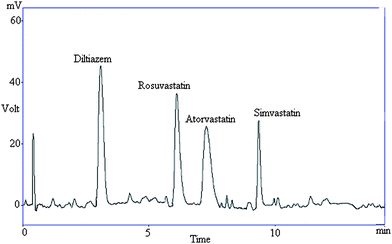 | ||
| Fig. 3 Representative chromatogram showing resolution between diltiazem and statins in human serum. | ||
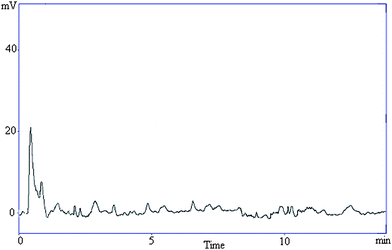 | ||
| Fig. 4 A representative chromatogram of blank serum. | ||
| Analytes | Goodness of fit (R2) | Standard error | Standard error of estimate | Intercept | Slope | LOD | LOQ |
|---|---|---|---|---|---|---|---|
| a Diltiazem; DLZ, Rosuvastatin; ROS, Atorvastatin; ATR, Simvastatin; SIM. | |||||||
| Bulk material (0.625–20 μg mL−1) | |||||||
| DLZ | 0.9997 | 2417 | 4256.05 | 148112 | 25454 | 0.0029 | 0.0089 |
| ROS | 0.9997 | 3002 | 5287.49 | 83864 | 27214 | 0.0094 | 0.0286 |
| ATR | 0.9997 | 2641 | 4651.52 | 71340 | 23788 | 0.0017 | 0.0053 |
| SIM | 0.9995 | 1366 | 2405.85 | 15723 | 9934 | 0.0042 | 0.0127 |
| Serum (0.625–20 μg mL−1) | |||||||
| DLZ | 0.9996 | 2424 | 4264 | 148105 | 25467 | 0.0031 | 0.0093 |
| ROS | 0.9994 | 3018 | 5281 | 83859 | 27211 | 0.0098 | 0.0345 |
| ATR | 0.9995 | 2657 | 4624 | 71389 | 23752 | 0.0025 | 0.0064 |
| SIM | 0.9995 | 1359 | 2412 | 15716 | 9945 | 0.0051 | 0.0138 |
| % Recovery | Recovered conc. μg mL−1 | |||||||
|---|---|---|---|---|---|---|---|---|
| Conc. μg mL−1 | DLZ | ROS | ATR | SIM | DLZ | ROS | ATR | SIM |
| a 80% = 12 μgmL−1; 100% = 15 μgmL−1; 120% = 18 μgmL−1, n = No of sample injected. Diltiazem; DLZ, Rosuvastatin; ROS, Atorvastatin; ATR, Simvastatin; SIM. | ||||||||
| 0.625 | 99.99 | 100.0 | 99.98 | 100.09 | 0.62 | 0.62 | 0.62 | 0.62 |
| 1.25 | 100.0 | 99.65 | 99.99 | 99.87 | 1.25 | 1.24 | 1.24 | 1.24 |
| 2.5 | 100.0 | 100.59 | 100.0 | 100.15 | 2.50 | 2.51 | 2.500 | 2.50 |
| 5 | 99.9 | 99.75 | 99.92 | 99.97 | 4.99 | 4.98 | 4.99 | 4.99 |
| 10 | 100.07 | 100.07 | 99.79 | 99.98 | 10.00 | 10.00 | 9.97 | 9.99 |
| 15 | 99.99 | 100.07 | 99.71 | 99.99 | 14.99 | 15.01 | 14.95 | 14.99 |
| 20 | 99.94 | 99.95 | 100.87 | 100.03 | 19.98 | 19.99 | 20.17 | 20.00 |
| Spiking (bulk material % level) | ||||||||
| 80 | 99.80 | 101.5 | 99.96 | 100.21 | 11.97 | 12.18 | 11.99 | 12.02 |
| 100 | 100.19 | 100.11 | 100.18 | 102.15 | 15.02 | 15.01 | 15.02 | 15.32 |
| 120 | 100.17 | 100.14 | 99.97 | 100.53 | 18.03 | 18.02 | 17.99 | 18.09 |
| Spiking (serum % level) | ||||||||
| 80 | 98.95 | 101.78 | 97.99 | 99.97 | 11.87 | 12.21 | 11.75 | 11.99 |
| 100 | 98.12 | 100.01 | 99.45 | 101.54 | 14.71 | 15.00 | 14.91 | 15.23 |
| 120 | 99.01 | 100.75 | 98.65 | 101.04 | 17.82 | 18.13 | 17.75 | 18.18 |
Recoveries of different concentrations of all the drugs from pharmaceutical formulations and serum were calculated by dividing the integrated peak area by the respective nominal drug concentration for calibration curve and serum samples, and expressed as the recovery (%).
| Day 1 (CV) | Day 2 (CV) | |||||||
|---|---|---|---|---|---|---|---|---|
| Conc. μg mL−1 | DLZ | ROS | ATR | SIM | DLZ | ROS | ATR | SIM |
| a Diltiazem; DLZ, Rosuvastatin; ROS, Atorvastatin; ATR, Simvastatin; SIM, Cofficient of variation; CV. | ||||||||
| 0.625 | 0.13 | 0.10 | 0.58 | 0.24 | 0.65 | 0.85 | 0.93 | 0.95 |
| 1.25 | 0.22 | 0.63 | 0.12 | 0.183 | 0.46 | 0.79 | 0.13 | 0.32 |
| 2.5 | 0.67 | 0.78 | 0.48 | 0.159 | 0.67 | 0.84 | 0.12 | 0.35 |
| 5 | 0.88 | 0.55 | 0.29 | 0.96 | 0.13 | 0.97 | 0.56 | 0.65 |
| 10 | 0.69 | 0.92 | 0.65 | 0.176 | 0.59 | 0.86 | 0.16 | 0.59 |
| 15 | 0.27 | 0.65 | 0.64 | 0.89 | 0.33 | 0.14 | 0.28 | 0.22 |
| 20 | 0.83 | 0.57 | 0.48 | 0.28 | 0.61 | 0.19 | 0.48 | 0.69 |
| Serum/μg mL−1 | ||||||||
| 0.625 | 0.18 | 0.18 | 0.49 | 0.96 | 0.98 | 0.65 | 0.76 | 0.86 |
| 1.25 | 0.35 | 0.59 | 0.16 | 0.112 | 0.99 | 0.78 | 0.69 | 0.93 |
| 2.5 | 0.68 | 0.68 | 0.65 | 0.161 | 0.97 | 0.58 | 0.98 | 0.89 |
| 5 | 0.73 | 0.64 | 0.38 | 0.98 | 0.67 | 0.97 | 0.101 | 0.97 |
| 10 | 0.37 | 0.39 | 0.28 | 0.13 | 0.58 | 0.68 | 0.113 | 0.94 |
| 15 | 0.74 | 0.94 | 0.45 | 0.143 | 0.98 | 0.84 | 0.98 | 0.92 |
| 20 | 0.75 | 0.67 | 0.39 | 0.99 | 0.97 | 0.98 | 0.89 | 0.99 |
:1 and 10:1 summarized in Table 2. The minimum limits at which the analytes could be readily detected and quantified for diltiazem, rosuvastatin, simvastatin and atorvastatin were 0.0029, 0.0094, 0.0017, 0.0042 μg mL−1 and 0.0089, 0.0286, 0.0053, 0.0127 μg mL−1 respectively, which suggested that a nanogram quantity of each drug can be estimated accurately.
| Storage | 40 °C and 75% humidity | 30 °C and 65% humidity | ||||||
|---|---|---|---|---|---|---|---|---|
| DLZ | ROS | ATR | SIM | DLZ | ROS | ATR | SIM | |
| % Recovery | ||||||||
| 3 months | 100.21 | 99.07 | 100.11 | 98.6 | 99.55 | 100.08 | 100.41 | 99.38 |
| 98.44 | 98.62 | 99.28 | 99.2 | 99.18 | 98.47 | 99.03 | 100.21 | |
| 99.67 | 99.33 | 98.96 | 100.2 | 98.63 | 99.62 | 99.51 | 99.44 | |
| 99.41 | 98.49 | 99.469 | 99.5 | 99.44 | 99.53 | 99.39 | 99.62 | |
| 98.97 | 100.24 | 98.74 | 100.4 | 98.94 | 98.69 | 99.28 | 99.51 | |
| 99.51 | 100.17 | 99.48 | 98.74 | 100.37 | 100.34 | 100.27 | 98.69 | |
| 99.09 | 99.29 | 99.32 | 99.57 | 99.62 | 99.58 | 9894 | 98.94 | |
| 6 months | 99.62 | 99.58 | 100.37 | 99.44 | 100.35 | 99.69 | 98.96 | 99.17 |
| 100.37 | 98.39 | 98.74 | 98.86 | 99.26 | 99.37 | 99.49 | 99.28 | |
| 98.49 | 99.44 | 99.25 | 100.4 | 99.74 | 99.59 | 99.72 | 99.61 | |
| 98.49 | 98.85 | 99.14 | 98.66 | 99.27 | 99.22 | 100.48 | 98.79 | |
| 100.31 | 100.63 | 99.3 | 99.28 | 100.31 | 100.18 | 99.58 | 100.36 | |
| 100.24 | 99.48 | 100.38 | 99.39 | 99.57 | 99.65 | 99.12 | 99.52 | |
| 99.68 | 99.26 | 100.41 | 99.52 | 99.48 | 99.39 | 99.86 | 99.48 | |
Seven lots of commercially available drugs were retained for stability studies for up to six months duration. Two types of study were carried out, one accelerated condition and one long-term condition as described by ICH.51 Both the studies showed that all the drugs were stable under mentioned conditions, % recoveries of all the drugs obtained almost 100% (Table 5).
Conclusion
Depicts a new RP-HPLC method with UV detection for the simultaneous determination of diltiazem and statins which is found to be linear, precise, and robust. The method was applied successfully to measure serum diltiazem and statins concentrations. A short analysis time <10 min allows application in routine and quality control analyses of finished products.References
- N. Poulter, Am. J. Hypertens., 1999, 12, 92S–95S CAS.
- D. M. Lloyd-Jones, J. C. Evans and M. G. Larson, et al. , Arch. Intern. Med., 1999, 159, 2206–2212 CrossRef CAS.
- P. W. Wilson, W. B. Kannel, H. Silbershatz and R. B. D'Agostino, Arch. Intern. Med., 1999, 159, 1104–1109 CrossRef CAS.
- F. Thomas, K. Bean and L. Guize, et al. , Eur. Heart J., 2002, 23, 528–535 CrossRef CAS.
- O. Samuelsson, L. Wilhelmsen and O. K. Andersson, et al. , JAMA, J. Am. Med. Assoc., 1987, 258, 1768–1776 Search PubMed.
- D. Wood, P. Durrington and G. McInnes, et al. , Heart, 1998, 80(Suppl 2), S1–S29.
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National Cholesterol Education Program (NCEP) (Adult Treatment Panel III), JAMA, J. Am. Med. Assoc., 2001, 285, 2486–2497 Search PubMed.
- W. B. Kannel, Am. J. Hypertens., 2000, 13, 3S–10S CrossRef CAS.
- Definition, diagnosis and classification of diabetes mellitus and its complications, Department of Noncommunicable Disease Surveillance, WHO/NCD/NCS/99.2, Geneva, Switzerland, 1999 Search PubMed.
- S. M. Grundy, H. B. Brewer, J. I. Cleeman, S. C. Smith and C. Lenfant, American Heart Association and National Heart, Lung, and Blood Institute, Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition, Circulation, 2004, 109, 433–438 CrossRef.
- R. Lars-Christian, E. Baranova, O. Boguslaw, M. Weisskopf, A. Kandra and F. Philippe, Clin. Ther., 2008, 30(10), 1782–1793 CrossRef.
- W. T. Cefalu, Clin. Res. Rev., 2008, 2, 208–222 Search PubMed.
- A. O. Marcus, Diabetes Technol. Ther., 2000, 2, 101–110 CrossRef CAS.
- S. M. Grundy, J. I. Cleeman, S. R. Daniels, K. A. Donato, R. H. Eckel, B. A. Franklin, D. J. Gordon, R. M. Krauss, P. J. Savage, S. C. Smith, J. A. Spertus and F. Costa, Circulation, 2005, 112, 2735–2752 CrossRef.
- A. Markham and K. L. Goa, Drugs, 1997, 54, 299–311 CrossRef CAS.
- S. Oparil, S. Dyke and F. Harris, et al. , Clin. Ther., 1996, 18, 797–810 CrossRef CAS.
- J. L. Pool, R. Glazer, Y. T. Chiang and M. Gatlin, J. Hum. Hypertens., 1999, 13, 275–281 CrossRef CAS.
- M. Burnier and H. R. Brunner, Lancet, 2000, 355, 637–645 CrossRef CAS.
- M. R. Weir, N. Crikelair and D. Levy, et al. , J. Clin. Hypertens., 2007, 9, 103–112 Search PubMed.
- J. F. Dorval, T. Anderson and J. Buithieu, et al. , Am. J. Cardiol., 2005, 95, S249–S253.
- K. K. Koh, M. J. Quon and S. H. Han, et al. , Circulation, 2004, 110, 3687–3692 CrossRef CAS.
- D. J. Abraham Burger's Medicinal Chemistry and Drug Discovery, A John Wiley and Sons, Inc., Publication Hoboken, New Jersey,6th 3, 2003, 15–17 Search PubMed.
- A. R. Gennaro. Remington: The Science and Practice of Pharmacy., printed in the United State of America, University of the Sciences in Philadelphia 21st edn, 2005, 1364–1365 Search PubMed.
- J. L. Goldstein and M. S. Brown, Nature, 1990, 343, 425–430 CrossRef CAS.
- P. H. Jones, M. H. Davidson, E. A. Stein, H. E. Bays, J. M. McKenney, E. Miller, V. A. Cain and J. W. Blasetto, Am. J. Cardiol., 2003, 92, 152–160 CrossRef CAS.
- C. Borghi, A. Dormi and M. Veronesi, et al. , J. Clin. Hypertens., 2002, 4, 277–285 Search PubMed.
- R. C. Bindu and A. K. Sekharan, Colorimetric determination of diltiazem hydrochloride in pharmaceutical formulations, J. Indian Drugs, 1994, 31(4), 168–9 Search PubMed.
- J. Wang, Percio A. M. Farias and J. S. Mahmoud, Analyst, 1986, 111(7), 837–9 RSC.
- G. J. Vergote, C. Vervaet, J. P. Remon, T. Haemers and F. Verpoort, Eur. J. Pharm. Sci., 2002, 16, 63–67 CrossRef CAS.
- B. Chankvetadze, M. Saito, E. Yashima and Y. Okamoto, J. Chromatogr., A, 1997, 773, 331–338 CrossRef CAS.
- Y. Li and Y. Qiu, J. Zhongguo Yiyuan Yaoxue Zashi, 1998, 18(1), 29–30 Search PubMed.
- M. Akram and El-Didamony, Cent. Eur. J. Chem., 2005, 3(3), 520–536 Search PubMed.
- P. M. Lacroix, N. Beaulieu, T. D. Cyr and E. G. Lovering, J. Pharm. Sci., 1989, 78, 243–246 CrossRef CAS.
- Z. Dragica, S. Traje and S. Marina, Anal. Bioanal. Chem., 2003, 376, 843–853.
- Ke. L. X. Zhang and Z. Feilang, Biomed. Chromatogr., 2003, 17(8), 522–525 CrossRef CAS.
- R. Shimizu, K. Ishii, N. Tsumagari, M. Tanigawa and M. Matsumoto, J. Chromatogr., A, 1982, 253, 101–108 CrossRef CAS.
- R. Shimizu, T. Kakimoto, K. Ishii, Y. Fujimoto, H. Kishi and N. Tsumagari, J. Chromatogr., A, 1986, 357, 119–125 CrossRef CAS.
- Ertürk. Sıdıka, Önal Armağan and Müge Çetin Sevil, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2003, 793, 193–205 CrossRef CAS.
- B. G. Chaudhari, N. M. Patel and P. B. Shah, Indian J. Pharm. Sci., 2007, 69(1), 130–132 Search PubMed.
- L. Tan, L. L. Yang, X. Zhang, Y. S. Yuan and S. S. Ling, Sepu, 2000, 18, 232 Search PubMed.
- H. Ochiai, N. Uchiyama, K. Imagaki, S. Hata and T. Kamei, J. Chromatogr., B: Biomed. Sci. Appl., 1997, 694, 211 CrossRef CAS.
- W. W. Bullen, R. A. Miller and R. N. Hayes, J. Am. Soc. Mass Spectrom., 1999, 10, 55–66 CrossRef CAS.
- M. P. Khalid, M. Muzeeb, S. S. Jafar, B. D. Shashikumar, M. Ramesh and N. R. Srinivas, Biomed. Chromatogr., 2006, 20(3), 282–293 CrossRef CAS.
- M. J. Morris, J. D. Gilbert, J. Y. K. Hsieh, B. K. Matuszewski, H. G. Ramjit and W. F. Bayne, Biol. Mass Spectrom., 1993, 22(1), 1–8 CrossRef CAS.
- A. Mohammadi, N. Rezanour, M. A. Dogaheh, F. G. Bidkorbeh, M. Hashem and R. B. Walker, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 846, 215–221 CrossRef CAS.
- D. A. Shah, K. K. Bhatt, R. S. Mehta, M. B. Shankar, S. L. Baldania and T. R. Gandhi, Indian J. Pharm. Sci., 2007, 69, 546–549 Search PubMed.
- L. Kristoffersen, E. L. Oiestad, M. S. Opdal, M. Krogh, E. Lundanes and A. S. Christophersen, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 850, 147–160 CrossRef CAS.
- N. Zaltýn and E. Uc. Aktürk, Chromatographia, 2007, 66, S87–S91 CAS.
- S. Koichi, W. L. Lee, T. Toyohide, S. Yasuhide, S. Kiyohito, T. Yuji and K. Susumu, Anal. Bioanal. Chem., 2006, 384, 1501–1505 CrossRef CAS.
- G. S. Clarke, J. Pharm. Biomed. Anal., 1994, 12, 643 CrossRef CAS.
- ICH guideline Q2B: Validation of Analytical Procedures: Methodology, 2003 Search PubMed.
- R. L. Plackett and J. P. Burman, The Design of Optimum Multifactorial Experiments, Biometrika, 1943–1946, 33, 305–325 Search PubMed.
- E. Astm (1169–89). American Society for Testing and Materials. Standard Guide for Conducting Ruggedness Tests (Plackett-Burman Design). 100 Barr Harbor Drive, West Conshohcken PA19428–2959 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |