A new method for rapid degree of substitution and purity determination of chloroform-soluble cellulose esters, using 31P NMR†
Alistair W. T.
King
*a,
Jarno
Jalomäki
a,
Mari
Granström
b,
Dimitris S.
Argyropoulos
c,
Sami
Heikkinen
a and
Ilkka
Kilpeläinen
*a
aLaboratory of Organic Chemistry, Department of Chemistry, University of Helsinki, P.O. Box 55 (A. I. Virtasen Aukio 1), FIN-00014, Finland. E-mail: alistair.king@helsinki.fi; ilkka.kilpeläinen@helsinki.fi; Tel: +358 50527 9446
bBASF SE, GCI/R, M311 67056, Ludwigshafen, Germany
cDepartment of Forest Biomaterials, North Carolina State University, Raleigh, NC 27695-8005, USA
First published on 31st August 2010
Abstract
Chloroform-soluble palmitic and decanoic acid esters of cellulose were synthesized from the reaction of MCC with acid chlorides in LiCl/DMA and the ionic liquid [amim]Cl, as novel cellulose solvents. A process of derivatization of the remaining hydroxyl groups, as phosphite esters and subsequent 31P NMR analysis, allowed for simultaneous degree of substitution (DS) determination and quantification of the aqueous-quench acid by-product impurity, after the appropriate calculation. The full mathematical treatment for DS determination is presented, including scripts for the Python and Java programming languages for rapid interpretation of results. This method for DS determination was validated against traditional analyses of the palmitoyl cellulose and the fully substituted p-nitrobenzoyl palmitoyl diester product, from additional reaction with p-nitrobenzoyl chloride. DOSY NMR and SCORE analyses were also employed to demonstrate the utility of this rapid 31P derivatization and analytical process over traditional 1D NMR analyses.
Introduction
Cellulose, derived from natural sources (commonly bacteria and plants), has been long promoted as an abundant renewable resource for advanced materials. Cellulose is chemically modified at its C-2, C-3 and C-6 hydroxyl groups to form esters, ethers, carbonates or carbamates.1,2 However, determination of accurate values for degree of substitution (DS) and regioselectivity in the functionalization of cellulose can often be a complex task. This becomes more troublesome the more complex the reaction pathway or the substituent is, but is absolutely crucial for the characterisation of novel and technically advanced materials. Optimisation of these reactions from novel polar media such as lithium chloride/N,N-dimethylacetamide (LiCl/DMA) or imidazolium-based ionic liquids (ILs)2 such as 1-allyl-3-methylimidazolium chloride ([amim]Cl) is significantly more challenging than analysis of a purified product. A typical case in point is the esterification of cellulose with long-chain ‘fatty’ acid chlorides (Fig. 1). Several problems exist for the efficient optimization of these reactions using traditional DS determination methods such as gravimetric analysis, 1H NMR, elemental analysis (EA), IR or GPC. The main problem occurs during reaction workup. At this stage, addition of a quenching reagent, typically water, destroys any remaining reactive electrophile (e.g. acid chloride or anhydride) converting it to carboxylic acid. The main role of the water is to precipitate the product and thus separate it from the liquid reaction media as many solvents for cellulose chemistry are comprised of compounds with low vapour pressures (e.g. ionic liquids, LiCl, and DMA). Co-precipitation of product and by-products undoubtedly occurs, often requiring tedious purification procedures. Furthermore, this is compounded by the fact that 1H NMR,1 as the best method for DS determination at present, can suffer from poor resolution of by-products from the anhydroglucose (AGU) unit and main substituent resonances, frequently causing inaccuracies in DS determination. This is particularly a problem for long-chain acyl substituents such as for fatty acid esters. Building upon traditional methods, recent articles have appeared based upon saponification of weighed samples of cellulose esters under standardised conditions, followed by GC analysis of the liberated acids.3 These techniques, however, also suffer from the fact that the samples must be acid free to give accurate results. One method, recently published, that attempts to alleviate these problems, is an improved IR analysis procedure. This involves resolution of the free acid and ester carbonyl stretches using monoethanolamine to form a conjugate base–acid pair, with the free acid in the analytical sample and leaving the esters untouched.4 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch of the conjugate base–acid pair is shifted out of the region of the ester allowing increased accuracy in acid impurity determination. The IL 1-ethyl-3-methylimidazolium acetate ([emim][OAc]) was used for dissolution of their samples, as one of a range of accessible ILs capable of dissolving wood and cellulose,5 allowing for analysis of esters of cellulose in the low to medium range (0–1.5) in addition to those which are fully organically soluble.
O stretch of the conjugate base–acid pair is shifted out of the region of the ester allowing increased accuracy in acid impurity determination. The IL 1-ethyl-3-methylimidazolium acetate ([emim][OAc]) was used for dissolution of their samples, as one of a range of accessible ILs capable of dissolving wood and cellulose,5 allowing for analysis of esters of cellulose in the low to medium range (0–1.5) in addition to those which are fully organically soluble.
![Typical fatty acid esterification of cellulose to give the ester and acid by-product, from [amim]Cl 1, as reaction media.](/image/article/2010/AY/c0ay00336k/c0ay00336k-f1.gif) | ||
| Fig. 1 Typical fatty acid esterification of cellulose to give the ester and acid by-product, from [amim]Cl 1, as reaction media. | ||
Despite the high novelty and utility of this method, IR analysis is still limited in its accuracy and in the information that it provides. However, the use of cellulose-dissolving media, such as ILs, in the analysis procedure is an important concept as it may allow for the analysis of normally insoluble products such as low DS cellulose esters.
In the present publication, it is our objective to validate and demonstrate the utility of 31P derivatization and NMR analyses for the optimisation of cellulose functionalization reactions, in comparison to traditional methods. For this, we have initially chosen a short set of long-chain cellulose esters in the organically soluble DS range of ∼1.5 to 3 and of variable purity. Based upon our existing and growing knowledge of wood and wood biopolymer dissolution and functionalization in ILs, aided by 31P labelling and NMR analysis,6,7 it is our goal ultimately to develop this technique for the study of a wider range of wood bio-polymer chemical modification reactions.
Experimental
Materials
Allyl chloride, 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (2-Cl-TMDP), anhydrous N,N-dimethylacetamide (DMA), endo-N-hydroxy-5-norbornene-2,3-dicarboximide (e-HNDI), anhydrous lithium chloride (LiCl), microcrystalline cellulose (MCC), N-methylimidazole and p-nitrobenzoyl chloride were purchased from Aldrich (Finland), and were used without further purification. Palmitoyl and decanoyl chloride were purchased from TCI Europe, and were used without further purification. [amim]Cl 1 was prepared according to a previous report.6Measurements
Quantitative 31P and 13C NMR spectra were recorded at 27 °C using inverse gated proton decoupling sequences on a Varian Unity Inova 600 spectrometer (600 MHz proton frequency) equipped with a 5 mm direct detection broadband probe-head. Quantitative 31P spectra were collected with 512 transients using 90° pulse flip angle, 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz spectral width, 1 s acquisition time, and 10 s relaxation delay. The quantitative 13C spectrum was collected with 20000 transients utilizing a 90° pulse flip angle, 36000 Hz spectral width, 1 s acquisition time and 60 s relaxation delay. Quantitative 1H spectra and DOSY (diffusion-ordered spectroscopy) data were recorded at 27 °C on a Varian Unity Inova 500 spectrometer (500 MHz proton frequency) equipped with 5 mm triple-resonance (1H, 13C, and 15N) gradient probe-head. For 1H measurements, 60 transients were collected with 45° pulse flip angle, 1.9 s acquisition time, and 13 s relaxation delay. Spectral width for the 1H spectra was 8000 Hz. DOSY datasets were recorded using Bipolar Pulse Pair Stimulated Echo pulse sequence (BPPSTE)8 using 1 ms gradient duration, 0.2 ms gradient recovery delay, 150 ms diffusion time, 224 transients, 1 s acquisition time, 2 s relaxation delay, and spectral width of 8000 Hz. In DOSY datasets, 60 diffusion gradient amplitudes were used (ranging from 0.5 to 20 G cm−1). DOSY data were analyzed with the Speedy COmponent REsolution (SCORE)-algorithm9 incorporated into the DosyToolBox 0.53-software10 running on Matlab 7.5.0 (MathWorks, Natick, MA, USA). SCORE analysis was carried out with standard parameters over the region of 0–6 ppm for a 2-component system.
000 Hz spectral width, 1 s acquisition time, and 10 s relaxation delay. The quantitative 13C spectrum was collected with 20000 transients utilizing a 90° pulse flip angle, 36000 Hz spectral width, 1 s acquisition time and 60 s relaxation delay. Quantitative 1H spectra and DOSY (diffusion-ordered spectroscopy) data were recorded at 27 °C on a Varian Unity Inova 500 spectrometer (500 MHz proton frequency) equipped with 5 mm triple-resonance (1H, 13C, and 15N) gradient probe-head. For 1H measurements, 60 transients were collected with 45° pulse flip angle, 1.9 s acquisition time, and 13 s relaxation delay. Spectral width for the 1H spectra was 8000 Hz. DOSY datasets were recorded using Bipolar Pulse Pair Stimulated Echo pulse sequence (BPPSTE)8 using 1 ms gradient duration, 0.2 ms gradient recovery delay, 150 ms diffusion time, 224 transients, 1 s acquisition time, 2 s relaxation delay, and spectral width of 8000 Hz. In DOSY datasets, 60 diffusion gradient amplitudes were used (ranging from 0.5 to 20 G cm−1). DOSY data were analyzed with the Speedy COmponent REsolution (SCORE)-algorithm9 incorporated into the DosyToolBox 0.53-software10 running on Matlab 7.5.0 (MathWorks, Natick, MA, USA). SCORE analysis was carried out with standard parameters over the region of 0–6 ppm for a 2-component system.
Methods
High purity palmitoyl cellulose 2. Microcrystalline cellulose (MCC, 0.617 g, 3.81 mmol) was heated in DMA (anhydrous, 30 ml) at 130 °C for 3 h under inert atmosphere. The sample (2% w/w in solution) was allowed to cool to 90 °C upon which LiCl (anhydrous, 2.080 g, 6.73% w/w in solution) was added in one portion and the sample allowed to cool to room temperature with stirring. The sample was allowed to stir for a further 18 h under inert atmosphere before addition of palmitoyl chloride (2.5 ml, 8.24 mmol) and pyridine (anhydrous, 1.2 ml, 14.90 mmol). The resulting mixture was allowed to stir for 72 h at room temperature and under inert atmosphere. The reaction mixture was poured into methanol (200 ml) and allowed to stir for 30 min. The mixture was filtered. The filtrand was dissolved in chloroform (100 ml) and precipitated by pouring into methanol (300 ml). The solution was filtered and the filtrand washed with methanol (100 ml) and deionised water (100 ml). The resulting solid was dried under vacuum at 55 °C for 18 h to give high purity palmitoyl cellulose 2 (1.97 g, 219% WPG (weight % gain), 92% yield at a DS of 1.67), as a white powder (found: C, 69.87, H, 11.0. Calc. for C32.72H60.10O6.67 (DS 1.67): C, 70.1, H, 10.8%); νmax (ATR)/cm−1 3481, 2922, 2851 and 1742; δH (500 MHz, CDCl3, Me4Si) 0.68–1.09 (3H, br t, J 6.8, CH3), 1.09–1.98 (26H, br m, COCH2(CH2)13CH3), 1.98–2.70 (2H, br m, COCH2(CH2)13CH3), 2.70–5.62 (7H, br m, AGU); DS31P: 1.67 ± 0.012 (calculated from 31P NMR analysis of the phosphitylated product, performed in triplicate); DS1H: 1.65 (calculated from 1H NMR analysis); DSEA: 1.62 (calculated from C-content determined from the EA); decanoic acid impurity: 0.94 ± 0.078% w/w (calculated from 31P NMR analysis of the phosphitylated product, performed in triplicate).
High purity p-nitrobenzoyl-palmitoyl cellulose 3. Palmitoyl cellulose (2, 100 mg, 0.179 mmol) was dissolved in a mixture of chloroform
:pyridine (1:1, 2 ml). p-Nitrobenzoylchloride (150 mg, 0.809 mmol) was added in one portion and the mixture was allowed to stir at room temperature for 18 h under inert atmosphere. Methanol was added until the product precipitated and the solution became clear. The solution was removed with a pipette and this precipitation process repeated 2 times. The solid was then finally precipitated from chloroform:hexane (1:1) with methanol addition. The solution was decanted and the product dried under vacuum at 55 °C for 18 h to give high purity p-nitrobenzoyl-palmitoyl cellulose 3 (128 mg, 28% WPG, 97% yield at a combined DS of 3), as a white solid (found: C, 65.64, H, 8.7, N, 2.29. Calc. for C42.03H64.09N1.33O10.66 (DS 1.67): C, 72.6, H, 9.3, N, 2.7%); νmax (ATR)/cm−1 2922, 2853 and 1741; δH (500 MHz, CDCl3, Me4Si) 0.68–1.09 (3H, br t, J 6.8, CH3), 1.09–1.98 (26H, br m, COCH2(CH2)13CH3), 1.98–2.70 (2H, br m, COCH2(CH2)13CH3), 2.70–5.62 (7H, br m, AGU). δC (125 MHz, CDCl3) 14.46, 23.05, 25.01, 29.28–30.39, 32.29, 34.04, 61.84, 70.75–75.81 (br m), 100.83, 123.91, 130.62, 134.25, 151.18, 162.65–164.37 (br m), 171.96–173.46 (br m); DS1H: 1.67 (calculated from 1H NMR analysis); DS13C: 1.67 (calculated from 13C NMR analysis); DSEA: 1.53 (calculated from C-content determined from the EA); DSEA: 1.75 (calculated from N-content determined from the EA).
Crude decanoyl cellulose 5. MCC (100 mg, 0.617 mmol) was dissolved in [amim]Cl (1.90 g) by heating at 100 °C for 18 h in a sealed vessel (5% w/w in solution). Pyridine (500 µl, 1.87 mmol) was added at high temperature and the mixture agitated for 10 s at 2500 rpm, using an Janke & Kunkel Vibrofix VF1 Electronic orbital shaker. Decanoyl chloride (640 µl, 3.09 mmol) was added and the mixture agitated for 1 min. The sample was heated at 100 °C for 2 h before deionised water (4 ml) was added and the solution agitated for 30 s. This mixture was heated at 100 °C for 1 h before being filtered and the filtrand washed with further deionised water (20 ml). The filtrand was dried in a vacuum oven for 18 h at 55 °C to give crude decanoyl cellulose 5 (181 mg, 81% WPG of impure material), as a white solid; νmax (ATR)/cm−1 3474, 2956, 2922, 2853, 1741 and 1709; δH (500 MHz, CDCl3, Me4Si) 0.74–1.00 (3H, br m, CH3), 1.00–1.95 (14H, br m, COCH2(CH2)7CH3), 1.95–2.60 (2H, br m, COCH2(CH2)7CH3), 3.12–5.47 (7H, br m, AGU); DS31P: 2.29 (calculated from 31P NMR analysis of the phosphitylated product); decanoic acid impurity: 25.2 wt% (calculated from 31P NMR analysis of the phosphitylated product).
:CDCl3/3:2, 0.0152 mmol) was added in one portion and the solution was vortexed (∼10 s). 31P NMR spectra (243 MHz for 31P) were recorded with 700 µl samples, in a 5 mm o.d. NMR tube.
A description for the calculation of DS values and impurities from cellulose functionalization reactions follows; the experimental variables required for DS determination from phosphite ester derivatization and 31P NMR analysis are: (1) MWS (substituent molecular weight). This is the molecular weight (g mol−1) of the substituent, not including the linking oxygen atom (between the cellulose backbone and the substituent); (2) ISvol (internal standard volume, µl); (3) ISmol (internal standard molarity, mmol); (4) IR (integration ratio of remaining functionalized cellulose hydroxyls against internal standard); and (5) WS (sample weight, mg). In addition, the constants DSmax (maximum achievable DS value of 3 for unsubstituted cellulose) and OHC (the free hydroxyls per weight unit of cellulose, 3 × 162 = 0.01852 mol g−1) are also required. The eqn (1) for calculation of the free hydroxyls per weight unit of substrate (OHS, mol g−1) is as follows:
 | (1) |
The final eqn (2) required to calculate the DS from 31P labelling and NMR analysis (DS31P), is as follows:
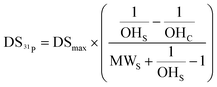 | (2) |
In addition to calculating the DS of pure products, the integration ratio (against internal standard) for any impurity (II), e.g. carboxylic acids, in combination with the molecular weight of the impurity (MWI, g mol−1) can be used to determine the weight of the impurity (WI, mg) in the original sample. This is determined using eqn (3):
 | (3) |
If impurity exists in a sample a corrected DS value can be eventually attained by simply subtracting WI from the original sample weight (WS) and recalculating OHS, as shown in eqn (4):
 | (4) |
The above equations may be adapted to starting polymers other than unsubstituted cellulose, for any given reaction. A good example of this is if an additional substituent is added to an existing substituted cellulose product. In this case, if you have predetermined the DS value of the initial substituent, by 31P labelling and NMR analysis, one can analyse the product of the second substitution reaction in a similar manner. To determine the DS of the second substituent from these results you can replace OHC in the second calculation of eqn (2) with OHS from the first calculation of eqn (1), and for the second calculation of eqn (2), DSmax is replaced with the DS of the first substituent. The result will be DS31P values for both substituents from analysis of both products. Other non-cellulosic polymers or polymer mixtures, such as xylan, lignin, starch or wood, may also be analysed by replacing OHC and DSmax values from their experimental or theoretically calculated values. As some polymers may have a non-integer number of hydroxyls per monomer unit, or non-regular monomer units, DS31P may be represented as a percentage substitution of the total sample by replacing DSmax with 100.
Eqn (1) and (2) have been incorporated in to scripts for the Python and Java programming languages13 in order to allow for rapid processing of data (see ESI†).
Results and discussion
As the objective of this article is to assess the 31P labelling and NMR analysis procedure, against traditional NMR analyses, suitable materials or reactions were required to demonstrate the advantages of using the 31P procedure over the traditional analyses, while still allowing for validation of the DS values against each other. To achieve this, a common acylation reaction in cellulose chemistry was chosen. That is esterification of cellulose with long-chain fatty acids.DS31P determination and method validation using high purity palmitoyl cellulose 2
To initially determine the accuracy of the 31P method on a CDCl3 soluble sample, MCC was reacted to give palmitoyl cellulose 2, employing the standard method of dissolution of MCC into LiCl/DMA followed by reaction with palmitoyl chloride, in the presence of excess pyridine (Fig. 2). Care was taken to prepare the best quality 1H NMR spectra possible for DS determination (see ESI†). | ||
| Fig. 2 Synthesis of palmitoyl cellulose 2 and p-nitrobenzoyl-palmitoyl cellulose 3. | ||
As a secondary method, outside 31P labelling and analysis, the remaining hydroxyls in the palmitoyl cellulose product 2 were further esterified as p-nitrobenzoyl esters. This was achieved by quantitatively reacting the chloroform soluble starting material with the p-nitrobenzoyl chloride (Fig. 2) to give, after purification, fully substituted p-nitrobenzoyl-palmitoyl cellulose 3. The reaction was determined to be complete (almost no remaining free hydroxyl groups) by the absence of any OH-stretch in the IR spectra (see ESI†) or absence of any aliphatic phosphite ester resonances, after 31P derivatization and NMR analysis. In addition to providing an alternative DS1H value, this functionality also provided the opportunity to analyse the product by EA, observing both carbon and nitrogen percentages, and quantitative 13C NMR, with the carbonyl resonances providing a suitable region for integration and DS13C determination (see ESI†). The inclusion of the chromophore also would allow for GPC with UV detection, although this is not a suitable method for determining DS values. Although it was possible to determine DS13C by straightforward integration of the carbonyl resonances, with sufficient exponential line broadening, the signal to noise ratios and resolution, after 20000 transients (∼66 h collection time), were not adequate enough to allow for accurate determination of regioselectivity, as has been previously observed for cellulose acetates.14
31P derivatization and NMR analysis of palmitoyl cellulose 2, shown in Fig. 3, were performed by dissolution of the substrate 2 into CDCl3 and reaction with 2-Cl-TMDP under basic (pyridine) conditions. The internal standard e-HNDI was also included in the phosphitylating mixture, which also reacted with the phosphite acid chloride (2-Cl-TMDP) or anhydride (TMDP-anhydride), in solution, to form its corresponding phosphite ester (e-HNDI-TMDP). The spectrum was calibrated with TMDP-anhydride at 132.2 ppm. e-HNDI-TMDP resonates at 152.0 ppm under these solvent conditions. Alkoxy phosphite esters (alkoxy-TMDP) are located in the region between 151.5 and 143.5 ppm and both decanoic acid and palmitic acid–phosphite mixed anhydrides (decanoate or palmitate–TMDP) resonate at 134.9 ppm. The reaction was performed in triplicate and the original sample weights were corrected, based upon the presence of 0.94% w/w palmitic acid impurity (as determined by the 31P NMR integrations of palmitate–TMDP). DS31P for this compound 2 was determined to be 1.67 with a standard deviation for the full procedure of 0.012. It is expected that as you approach DS values close to 0 or have very large substituents, the standard deviation will increase although for the chloroform soluble regime (DS ca. 1–3) the error will remain approximately the same.
 | ||
| Fig. 3 Derivatization mixture and 31P NMR analysis of TMDP-phosphitylated palmitoyl cellulose 4. | ||
From the initial palmitoyl cellulose substrate 2 and its subsequent characterization, the complete set of determined DS values are listed in Table 1, including short self-explanatory descriptions of the methods used to obtain them. From a comparison of these methods, we can see that EA predictably shows the most deviation from the mean value. This is very much dependent on both purity of materials and skill of the analyst. Due to the simplicity of integration, and the ability to resolve low molecular weight hydroxylated impurities from the polymeric material in solution, 31P derivatization and NMR analysis offer a more accurate and thorough characterization method, in combination with the traditional methods.
| Analyte | Method | Integral regions/element | Integral regions (ppm) | Calculation | DS |
|---|---|---|---|---|---|
| 2 | DS1H | COCH2(CH2)13CH3vs. AGU region | 0.5–1.9 vs. 2.5–5.5 | ((CH2)/29)/(AGU/7) | 1.65 |
| 2 | DSEA | Calculated from C-content | — | — | 1.62 |
| 3 | DS1H | ArH vs. AGU regions | 8.5–7.5 vs. 2.7–6.0 | 3-((ArH)/4)/(AGU/7) | 1.67 |
| 3 | DS13C | COAr region vs. COAlk | 162.7–164.4 vs. 172.0–173.5 | COAlk × 3/(COAr + COAlk) | 1.67 |
| 3 | DSEA | Calculated from C-content | — | — | 1.53 |
| 3 | DSEA | Calculated from N-content | — | — | 1.75 |
| 4 | DS31P | e-HNDI-TMDP vs. alkoxy-TMDP | 151.5–152.5 vs. 151.0–142.0 | See eqn (1) and (2) | 1.67 |
| 2 | DSmean | Mean analysis value | — | — | 1.65 |
DS31P determination and DOSY analysis of crude decanoyl cellulose 5
In order to demonstrate the true value of 31P derivatization and NMR analysis, e.g., in the optimization of cellulose acylation reactions, crude decanoyl cellulose 5 was synthesized, by carrying out a similar procedure to that used for the synthesis of palmitoyl cellulose 2, but avoiding any purification steps, beyond quenching the reaction by heating with water.As ILs, such as [amim]Cl, are predicted to be potential environmentally benign media for cellulose chemistry, based upon their recyclability and low vapour pressure, [amim]Cl was chosen as media for this reaction. A typical workup procedure for cellulose reactions, from ILs, involves stirring with a solvent such as water to quench the reactants and precipitate the product. Additionally, heating with the ‘quenching’ solvent is often required to remove traces of IL from the product. This procedure will typically preserve the yield and polydispersity of the product by precipitating most of the functionalised polymer provided the functionality is sufficiently hydrophobic, as is the case with most acyl species. This will also co-precipitate a large portion of the quenched reactants, with the desired product. These co-precipitated species are often impossible to resolve using traditional 1H NMR analysis, preventing rapid, consistent and reliable optimisation of these reactions.
The crude decanoylated cellulose product 5 was subjected to DOSY NMR to attempt to resolve any small organic species, including the decanoic acid quenching by-product, from the functionalized biopolymer. Inspection of the SCORE analysis results (Fig. 4), for a 2-component system, clearly shows resolution of the decanoic acid quenching by-product from the desired product 5. The SCORE analysis estimates this impurity at 20.2% mol/mol (of hydrogen) which, when converted into % w/w, gives a value of 18.6% w/w. From the extracted product 5 spectra (Fig. 4), no accurate DS determination is possible.
 | ||
| Fig. 4 SCORE analysis of the crude decanoyl cellulose 5 DOSY dataset, for a 2-component system. The separate spectra are from the two major components with widely differing diffusion rates, indicating low and high molecular weight species. | ||
When the same mixture was phosphitylated with 2-Cl-TMDP and analysed by 31P NMR, according to the above method, accurate values of 25.2% w/w for decanoic acid impurity and 2.29 for DS31P of the impure product were rapidly determined (see ESI†). A proper comparison between the % w/w impurity values from the 31P and DOSY experiments is not meaningful as the DOSY procedure is unable to produce quantitative data. When the DOSY spectrum for the palmitoylated cellulose sample 2 was collected, it was not possible to process the data, using the SCORE algorithm, with the purpose of obtaining any meaningful results that would allow for quantification of the palmitic acid impurity. This was due to the very low quantity of acid by-product impurity present in the sample. On the other hand, however, the 31P NMR analysis method allowed for determination of this impurity to be 0.94 ± 0.078% w/w. The significant advantage of DOSY over 1H NMR or 31P NMR seems to be only in a qualitative discrimination between two abundant components in a mixture, where one component is a high molecular weight polymer. This lack of quantitivity with DOSY is mostly dictated by differences in T1 and T2 relaxation that occur during the DOSY pulse sequence. T1 in particular can be greatly affected by the molecular weight of the resonating species.
This 31P-based analysis method limits the optimisation of these reactions to a aqueous quench, drying of the crude product and one in situ derivatization and NMR analysis procedure, to simultaneously determine DS and purity of the product. This is unrivalled by any other NMR procedure, such as DOSY or 1H NMR, which must serve as complementary qualitative techniques, due to long collection times and poor resolution, in the analysis of these crude reaction mixtures.
Conclusions
A new method for DS determination of functionalized cellulose products has been developed, based upon derivatization of the remaining hydroxyl groups, as phosphite esters. Under standardized solvent and 31P NMR acquisition conditions, these phosphite esters can be quantitatively integrated against an internal standard, allowing for calculation of the DS of the functionalized cellulose starting material. This method is at the moment applicable to the analysis of chloroform soluble products and was validated against a palmitoylated cellulose sample, which was analysed in detail by standard DS determination methods. A crude decanoylated cellulose reaction product was also analysed by this 31P derivatization and NMR procedure, allowing for rapid determination of both decanoic acid impurity and DS of the unpurified product. Within a potential comprehensive list of polymeric substrates and chemical modifications, this study initially highlights the potential utility of this method at least for optimising cellulose acylation reactions, with a view to more widespread application and process optimization.Acknowledgements
The authors wish to acknowledge the Academy of Finland (project number 122534), the Finnish Funding Agency for Technology and Innovation (TEKES) and Finlands Distinguished Professor Program (FiDiPro).References
- D. Klemm, B. Philipp, T. Heinze, U. Heinze and W. Wagenknecht, Comprehensive Cellulose Chemistry: Fundamentals and Analytical Methods, Wiley-VCH, Weinheim, 1998 Search PubMed.
- J. Wu, J. Zhang, H. Zhang, J. He, Q. Ren and M. Guo, Biomacromolecules, 2004, 5, 266–268 CrossRef CAS; S. Barthel and T. Heinze, Green Chem., 2006, 8, 301–306 RSC; M. Granström, J. Kavakka, A. W. T. King, J. Majoinen, V. Mäkelä, J. Helaja, S. Hietala, T. Virtanen, S. Maunu, D. S. Argyropoulos and I. Kilpeläinen, Cellulose, 2008, 15, 481–488 CrossRef.
- C. S. Freire, A. J. D. Silvestre, C. P. Neto and R. M. A. Rocha, Cellulose, 2005, 12, 449–458 CrossRef CAS; J. Peydecastaing, C. Vaca-Garcia and E. Borredon, Cellulose, 2009, 16, 289–297 CrossRef CAS.
- P. Dominguez de Maria and A. Martinsson, Analyst, 2009, 134, 493–496 RSC.
- I. Kilpeläinen, H. Xie, A. W. T. King, M. Granström, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142–9148 CrossRef; R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS; S. Köhler, T. Liebert, M. Schöbitz, J. Schaller, F. Meister, W. Günther and T. Heinze, Macromol. Rapid Commun., 2007, 28, 2311–2317 CrossRef; Y. Fukaya, K. Hayashia, M. Wada and H. Ohno, Green Chem., 2008, 10, 44–46 RSC.
- A. W. T. King, I. Kilpeläinen, S. Heikkinen, P. Järvi and D. S. Argyropoulos, Biomacromolecules, 2009, 10, 458–463 CrossRef CAS.
- A. W. T. King, L. Zoia, I. Filpponen, A. Olszewska, H. Xie, I. Kilpeläinen and D. S. Argyropoulos, J. Agric. Food Chem., 2009, 57, 8236–8243 CrossRef CAS.
- M. D. Pelta, H. Barjat, G. A. Morris, A. L. Davis and S. J. Hammond, Magn. Reson. Chem., 1999, 37, 706–714.
- M. Nilsson and G. A. Morris, Anal. Chem., 2008, 80, 3777–3782 CrossRef CAS.
- The Mathias Nilsson Research Group, The DOSY Toolbox (Matlab plugin): Processing PFG-NMR Diffusion Data, http://personalpages.manchester.ac.uk/staff/mathias.nilsson/software.htm.
- D. S. Argyropoulos, J. Wood Chem. Technol., 1994, 14, 45–63 CrossRef CAS; A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- H. Xie, A. W. T. King, I. Kilpeläinen, M. Granström and D. S. Argyropoulos, Biomacromolecules, 2007, 8, 3740–3748 CrossRef CAS.
- Python Software Foundation, Homepage for the Python Programming Language, http://www.python.org/; Sun Microsystems Inc., Homepage for the Java Programming Language, http://www.sun.com/java/.
- C. M. Buchanan, K. J. Edgar, J. A. Hyatt and A. K. Wilson, Macromolecules, 1991, 24, 3050–3059 CrossRef CAS; T. Miyamoto, Y. Sato, T. Shibata, H. Inagaki and M. Tanahashi, J. Polym. Sci., Polym. Chem. Ed., 1984, 22, 2363–2379 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Python and Java scripts for determination of DS values, including spectroscopic data for the cellulose ester products, for a visual measure of sample quality. See DOI: 10.1039/c0ay00336k |
| This journal is © The Royal Society of Chemistry 2010 |