Chromatographic analysis of diverse fruit components using HPLC and UPLC†
I.
Epriliati
*a,
G.
Kerven
b,
B.
D'Arcy
b and
M. J.
Gidley
c
aFood Technology, Widya Mandala Surabaya Catholic University, Jl. Dinoyo 42–44, Surabaya, 60265, Indonesia. E-mail: margarethaiev@gmail.com; Fax: +62 31 5610818; Tel: +62 31 5678478 ext. 146
bSchool of Land, Crop and Food Sciences, The University of Queensland, Hartley Teakle Bldg, Brisbane, 4072, Australia. E-mail: b.darcy@uq.edu.au; g.kerven@uq.edu.au; Fax: + 61 (7) 3365 1177; Tel: + 61 (7) 3346 9190
cCentre for Nutrition and Food Sciences, The University of Queensland, Hartley Teakle Bldg, St Lucia, 4072, Australia. E-mail: mike.gidley@uq.edu.au; Fax: + 61 (7) 3365 1177; Tel: + 61 (7) 3365 2145
First published on 8th September 2010
Abstract
The nutritional properties of fruits are due to the combined action of many categories of molecules. Studies on molecular nutrition, therefore, requires analytical methods that can cope with diverse molecular groups, including metabolites that may be currently unknown. Fractionation and identification protocols are crucial to validate identified compounds, and can be adapted from methodologies developed for metabolome studies. Pectin-related problems during fractionation of samples from ripe fruit products, both undigested and after digestion, as well as samples from in vitro cell culture bioassay were addressed, and chromatographic separations of target compounds (carotenoids, phenolics, sugars, and organic acids) were developed based on RP chromatography using HPLC and UPLC systems. The final method comprised of a chloroform–methanol–water (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3) extraction to simultaneously extract as many polar and nonpolar compounds as possible, followed by chromatographic analysis using (a) a HPLC-ELSD-UV system with a Prevail Carbohydrate column for sugar analysis, (b) Prevail Organic Acid column for organic acid analysis, (c) LC-MS ESI of eluants from a XTerra C-18 column for phenolics and (d) UPLC-PDA systems in a C-18-Bridged Ethane in Hybrid matrix column for carotenoids and other hydrophobic components. Acetonitrile-based mobile phases can be used to run all chromatographic systems developed.
:1:3) extraction to simultaneously extract as many polar and nonpolar compounds as possible, followed by chromatographic analysis using (a) a HPLC-ELSD-UV system with a Prevail Carbohydrate column for sugar analysis, (b) Prevail Organic Acid column for organic acid analysis, (c) LC-MS ESI of eluants from a XTerra C-18 column for phenolics and (d) UPLC-PDA systems in a C-18-Bridged Ethane in Hybrid matrix column for carotenoids and other hydrophobic components. Acetonitrile-based mobile phases can be used to run all chromatographic systems developed.
Introduction
A simultaneous analysis of diverse molecular species is a critical part of nutriomic studies. The nutriomic approach involves comprehensive studies of molecular composition and changes affected by, for example, intrinsic food characteristics and the history of food treatments. Moreover, in the human gut, ingested nutrients may be chemically converted into currently unknown compounds which may exist at minute levels. No such method is available for a complex food system.This article reports achievements and limitations of a developed method aiming at simultaneous analysis of diverse components with regard to reliability, reproducibility and robustness. One problem is that fruit samples have diverse and problematic (e.g., gelatinous) tissue matrices. A second challenge is to devise a fractionation method and chromatographic analyses of chemically diverse components such as sugars, organic acids, phenolics and carotenoids that are present at different levels from abundant (e.g., sugars, organic acids) to low (e.g., carotenoids). Methods developed should be suitable for the analysis of ripe fruits and their products, for in vitro digest solutions, and intestinal passage samples using Caco-2‡ cell monolayers (the current standard cell model for intestinal uptake). Because intermediate products of in vitro digestion, or metabolites from Caco-2 cell monolayers can be from different compound classes to the parent molecules, the method should be able to fractionate the target compounds into neutral-aqueous fractions (e.g., sugars and organic acids); aqueous-methanolic fractions (e.g., phenolics and flavonoids); and nonpolar-chloroform fractions (e.g., carotenoids) and their possible metabolites. The approach is different from conventional chemical analysis, which usually is concerned with separate extraction and detection of only one group of compounds. Moreover, an obvious advantage of the developed method is to reducing tedious chemical analysis work loads in nutriomic studies compared to conventional protocols.
The critical aspects of HPLC-based simultaneous analysis include column selection by which all compounds in each group will be separated satisfactorily, and optimizing the solvent mixture in the mobile phase to selectively elute each type of compound. In column selection, matrix particle sizes closely relate to column life time, separation performances and the pressure drop inside columns during runs.1
Detectors used include a diode array detector (DAD) that is able to identify functional groups of analytes with different wavelengths in a single run.2 Thus, this detector is compatible with a simultaneous analysis, for instance HPLC/PAD-MS analysis for flavonoids including flavonols, flavones, and flavanones in fruit and vegetables.3 Peak purification of UV-vis chromatograms is critical when DAD is used because of the difficulty in making unique assignments. Hence, there is a need to couple the detector with mass spectrometry to achieve peak identification or use standard compounds run under the same conditions.
The most common detector coupled with HPLC is the UV detector, although sensitivity and therefore peak identification decrease when concentrations of analytes are very low.4 On the other hand, electrospray ionisation-mass spectroscopy (ESI-MS) identifies analytes based on ion molecule abundance, and can overcome the sensitivity limitation of UV. MS is capable of coping with dynamic changes of molecules studied in metabolism or a chain of pathway reactions. However, it is possible that molecular weights of different molecules may be the same, thus using MS and UV detectors will be complementary. The use of a UV detector combined with retention times but without MS often fails to identify slight differences among various analytes.5
Miscibility and viscosity of solvent mixtures also need to be considered in separating extremely heterogeneous compounds. There are few miscible solvents with extreme polarity differences.6 Since HPLC is operated at relatively high pressures, a stable pressure drop along columns is important, thus viscous solvents are excluded. Although increasing the column temperature typically decreases solvent viscosity, analysing thermo-labile compounds such as several phytochemicals, is incompatible with use of this technique.2
Fig. 1 shows a general scheme of sample preparations, the sample conditions that have to be dealt with and the expected outcomes. In digest solutions and bioassay samples, additional exogenous reagents from animal extracts, enzymes, buffers such as HEPES/PIPES, and salts need to be removed. In addition, ripe fruits contain soluble pectins, which can cause viscosity problems during sample preparations. Methods should also be relatively rapid, as nutriomic study preferably involves immediate chemical analysis in order to obtain metabolites during digestions and absorptions, in particular those from bioactive compounds.
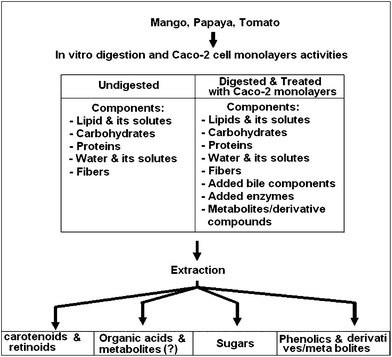 | ||
| Fig. 1 General scheme of sample preparation methods. | ||
Evaluation of fractionation method developed
The selected method is adapted from a protocol that has been used for metabolite profiling of plants by Morris et al.7 with modifications. In outline, the method involves extraction of hydrophilic and some hydrophobic compounds initially using methanol. The pellet is then re-extracted with chloroform to solubilise nonpolar components. The methanol extract is added to water and combined with the chloroform extract for further separation based on solvent polarity from methanol–water and methanol–chloroform layers. However, when this procedure was trialled in the present study on fruits, it resulted in pectin precipitation producing impermeable pellets after centrifugation. Consequently, optimal extractions could not be achieved with the method of Morris et al. Basic separations of polar and nonpolar target compounds were finally obtained by using the scheme in Fig. 2: some aspects of the chromatography method and analyses have been reported previously.8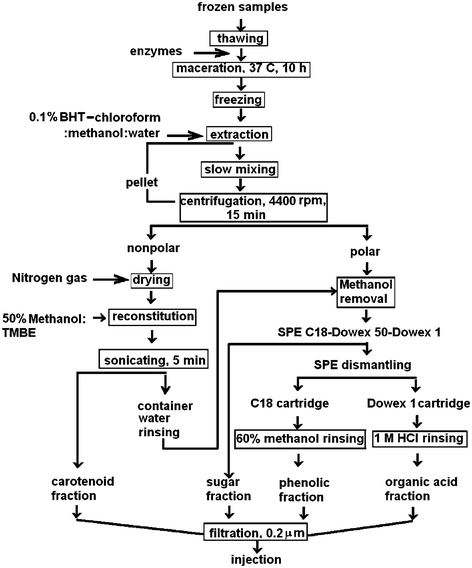 | ||
| Fig. 2 Method for separating polar and non-polar target compounds. | ||
Similar to requirements in wood metabolite profiling,7 cold extraction conditions are important because some of the target compounds are unstable. Frozen samples were used to start extractions, instead of freeze–dried samples. Attempts at using freeze dried samples encountered problems such as excessively long rehydration times in extraction solutions. In addition, digest solutions and bioassay samples that contained soluble pectins and proteins foamed significantly during freeze drying causing sample losses under vacuum.
Pectin is solubilised maximally in fully ripe fruits causing extraction problems due to its viscous or gelatinous physicochemical characteristics. Pectin-related extraction problems were found in this study for both undigested and digested samples. The main problems are firstly inefficient extraction due to pectin gelation under acid conditions which are required for phenolic extraction, and secondly pectin precipitation when in contact with alcohol. Exposure to 70–80% alcohol is known to cause cell wall material to precipitate.9 However, a higher water proportion dissolved pectin which is then difficult to remove from the aqueous extract.10 Centrifugation and filtration is ineffective for removal of soluble pectin.11 In the present study, such problems resulted in lower amounts of sugars and carotenoids released from blended mango than from larger particles of mango. This is probably due to greater exposure of pectin in the blended samples to the digest solutions. Therefore, digest solutions had to be treated with pectinase enzymes.
Both undigested and digested samples were treated with a mixture of enzymes (Optivin, The enzyme solution, Ltd., Australia) prior to the extraction step to hydrolyse plant cell wall polymers, and thereby optimize extraction, at an optimal pH of 2.5–5.5, which well fitted the pH values of fruits. This treatment was found to be effective at 2.5 < pH < 7.0. Cinar12 used commercial pectinase and cellulase for 24 h for enzyme-assisted carotenoid extraction and the use of exogenous pectin methyl esterase improved extraction yields of protoplast extraction affected by locular gel in tomato.13
Optivin contains arabinanase, pectinase, hemicellulase and protease, the combination of which is important to degrade the structural macromolecules in plant cell wall matrices. Slow maceration was found to be necessary to avoid too rapid a release of polymers which can cause cloudiness and be difficult to separate.10 Optimal use of Optivin was found to be at a ratio of 1:1 (unit activity/g equivalent fresh weight fruit products) or 1:2 (unit activity/mL digest solutions). Incubation was at 37 °C for 10 h and was carried out in closed containers wrapped with aluminium foil. To minimise degradation of labile compounds, reduced light was used throughout sample preparations. At the end of maceration, samples were stored at −20 °C overnight to prevent further enzyme action. This controlled maceration process degrades structural macromolecules (probably mostly pectins) such that residual cellulosic material can be removed by centrifugation (4–10 °C, 3000 g in an Eppendorf centrifuge). Further extractions were carried out at 4–16 °C to limit subsequent enzyme action.
After maceration, carotenoids were extracted with chloroform–methanol–water (2:1:3). The mixture was mixed in a rotational suspension mixer (Ratek, Australia) for 15–30 min. When consecutive extraction with chloroform and methanol was attempted, immiscible water and chloroform decreased recovery of the target compounds. Following methanol extraction alone, precipitated pectin trapped the target compounds, thus carotenoids were not extracted. Chloroform/methanol extracts were then dried under nitrogen gas stream. The dried extracts were stored at −20 °C to preserve labile compounds.
Chloroform containing 0.1% butylated hydroxytoluene (BHT) was used in this study to prevent oxidation of carotenoids during separation and storage prior to chromatographic analyses. Immediate evaporation of chloroform extracts containing BHT resulted in better recovery after storage (Table 1). Scott also found that the best storage form for carotenoids is the residue of a solution containing BHT.14 Recoveries of target compounds, simulated by a mixture of catechin, citric acid and sugars in various sample matrices were acceptable (Table 1).
| Sample systems | Fructose (HPLC-ELSD) | Glucose (HPLC-ELSD) | Sucrose (HPLC-ELSD) | Catechin (UPLC) | Lycopene (UPLC) | Carotene (UPLC) |
|---|---|---|---|---|---|---|
| a U: undigested, G: gastric, I: intestinal, G + I: consecutive gastric – intestinal digestions; n = 3–13 depending on batch numbers of experiments. b Y: unit area of peaks in the chromatograms; x: concentration levels of standard compounds. c Initial samples were acidified with formic acid and SPE C18. d Following the scheme in Fig. 2 phenolic fractions were rinsed with 100% methanol for Caco-2 bioassay recovery experiments (lower than that using LC-MS, i.e. 108.76 ± 7.78).7 e Data from apical sample solution compared to prepared apical solution which were not subjected to Caco-2 cell monolayers. | ||||||
| Regression and (r2)b | Y = 2E − 4x + 0.0241 (0.99) | Y = 2.6E − 3x + 0.1253 (0.99) | Y = 956.69x (0.99) | Y = 1.82E6x (0.99) | Y = 8E − 4x + 0.3823 (0.99) | Y = 1.4E − 3x + 0.53 (0.99) |
| Sugars–citric–catechin (U) | 69.78 ± 0.26 | 75.61 ± 0.56 | 96.42 ± 16.52 | |||
| Sugars–citric–catechin (G) | 86.97 ± 2.70 | 91.55 ± 0.61 | 90.02 ± 6.18 | |||
| Sugars–citric–catechin (I) | 83.53 ± 4.52 | 89.56 ± 0.58 | 96.09 ± 1.39 | |||
| Sugars–citric–catechin (G + I) | 85.52 ± 4.35 | 88.68 ± 0.91 | 102.69 ± 4.74 | |||
| Acidified catechinc | 46.37 ± 2.59 | |||||
| Catechin solutions–SPE C18d | 65.197 ± 0.031 | |||||
| Lycopene – HBSSe | 100 | |||||
| β-carotene – HBSSe | 100 |
The combination of solvents (chloroform: methanol: water) fits the requirement for simultaneous carotenoid and phenolic extraction; however, a strategy to avoid pectin precipitation was needed. Firstly, chloroform is immiscible in aqueous based fruit extracts, thus requiring an additional solvent with intermediate polarity. Methanol or ethanol is a good choice for solubilising sugars and phenolics, but not for carotenoids, and causes precipitation of pectins. Chloroform dissolves the nonpolar carotenoids well. Addition of methanol was found to retain carotenoid solubilising ability and to decrease the tendency to precipitate soluble pectin substances. The optimal mixture was found to be 2:1:3 chloroform:methanol:water, improving liquid–liquid phase separation and dissolving all components of interest within one of the two separable phases.
Usually Na2SO4 is used to remove traces of water in nonpolar extracts. However, phenolic components were found to be absorbed by solid Na2SO4, lowering the level of free phenolics. Rinsing Na2SO4 with water will improve recovery of absorbed ester phenolics.15 Adding a sufficient amount of Na2SO4 without it absorbing carotenoids is also difficult, especially for small volume samples from e.g., Caco-2 bioassays. In the present study, Na2SO4 was not used. Instead, traces of water (containing phenolics) unintentionally carried over into chloroform extracts was also dried under nitrogen gas together with carotenoid extract. Then, any traces of dried phenolic compounds were recollected by rinsing the containers with water after the dried-carotenoid extract was reconstituted with 50% tert-butyl methyl ether (TBME)-methanol because the phenolics were insoluble in 50% TBME-methanol. The washing was combined with aqueous fractions.
Aqueous methanol extracts were fractionated further using C-18–Solid phase extraction (SPE) for cleaning up and separating phenolics from sugars and organic acids. Aqueous fractions after C-18–SPE of the digest and Caco-2 sample solutions were desalted using the ion exchange resins Dowex 50W-X8 (to remove cationic salts) and Dowex 1-X8 (to bind anions including organic acids). Therefore, the Dowex 50W-X8 extraction was placed after the C-18–SPE extraction, which was then followed by Dowex 1-X8. Sugar containing fractions were the first eluates passing through these tandem cartridges, whereas organic acids were collected from Dowex 1-X8 by elution using 1 N HCl.
Detection of sugars could not be achieved from UV spectroscopy alone. The commonly preferred detector for sugar analysis is the refractive index detector, but in the present research, an ELSD (Evaporative Light Scattering Detector) was preferred, as it is more sensitive for sugar analysis. However, sugar analysis of salt-rich digests using HPLC-ELSD has a problem of interferences from salt effects on mobile phases.16 Therefore, salt removal is required. Because sodium, chloride and HEPES are abundant in the artificial digest reagents; two ion exchange resins were used, i.e., Dowex 1-X8 Chloride forms 100–200 mesh and Dowex 50W-X8 hydrogen form 100–200 mesh. Preparation of the Dowex 1-X8 and Dowex 50W-X8 followed the protocol provided by the suppliers. A controlled flow rate of one drop per 5 s with the correct amount of ion exchange resins improved subsequent chromatographic analysis.
To retain phenolic acids in the methanol–water extracts by C-18–SPE, acidification was required. Initial acidification before extraction is a common procedure for phenolic extraction in order to free bound phenolics. Acidification was inappropriate for fully ripe fruits because it enhanced gelling. Using acidified methanol-free water fractions was found to be better than acidification prior to extraction. For the simultaneous fractionations, acidification in the presence of carotenoids was considered inappropriate because of the potential for cis-trans isomerization.
Finally, C-18 SPE binds weakly ionized phenolics, making the presence of methanol in the treated samples undesirable. Therefore, methanol was evaporated in a rotary evaporator at 40 °C. This also concentrated metabolites and helped to improve the limit of detection levels of HPLC/UPLC.
Evaluation of chromatographic method developed
The diverse and often low levels of fruit metabolites presents a challenge to the development of a single method that maximises simultaneous separations by being able to dissolve as many compound groups as possible. However, it is possible that a simultaneous analysis may not extract particular metabolites or only partially extract them.Reverse–phase (RP) liquid–chromatography has potential for simultaneous analysis with a wide variety and flexible combinations of mobile phases, stationary phases and analyte applications.2,17 Chromatographic analyses were developed for HPLC-UV/ELSD and Ultra Performance Liquid Chromatography (UPLC)-PDA.
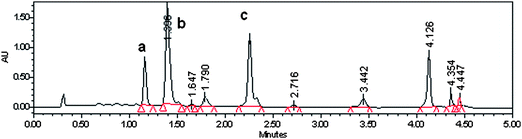
UPLC is different from HPLC in terms of the elution system. UPLC applies solvents to elute analytes that are separated through porous particles of 1.7 μm diameter, with a pore size of 80–125 Å. The pores separate the small amount of analytes through hydrophobic interactions with the C-18 stationary phase, and high pressures (max 15 000 Psi) force the analytes into narrowed bands. As a result, separation performances, peak shapes and sensitivity are improved significantly during short elution periods (Fig. 3 compared to Fig. 4). These fulfil the requirements for analysing minute concentrations of metabolites, such as those produced by Caco-2 cell monolayers in 0.33 cm2 growing areas. Caco-2 cell samples were about 100–400 μL and UPLC was suitable to detect minute concentrations of analytes due to its high sensitivity.
 | ||
| Fig. 3 LC-ESI-MS/DAD chromatograms obtained from elution using a Grace Vydac C-18 Polymeric Reverse Phase column (Grace, Illinois, USA) with MeOH:THF:water (67:27:6) for carotenoid papaya products. | ||
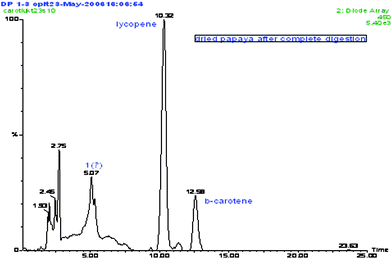
 | ||
| Fig. 4 Carotenoid UPLC/PAD chromatogram from undigested dried papaya; eluted in a C-18 Bridged Ethanes in Hybrid (BEH) column with a 0.1% formic acid in acetonitrile–TBME. a β-Cryptoxanthin, b lycopene, and c β-carotene. | ||
The shorter columns used with UPLC also give an opportunity for better resolution and short run times. Elution at increased column temperatures and very high pressures enhances mobile phase viscosity and the capacity for dissolving analytes. Therefore, the choice of mobile phase is critical for optimising the powerful separation forces that determine analyte partitioning. Overall, UPLC was found to generate sharp peaks at a very low concentration of analytes. However, UPLC has disadvantages due to limited solvent combinations for the mobile phases as a result of their behaviours and viscosities under ultra high pressures.
UPLC uses an injection system with an air gap between the sample solution in the loop and the solvent which pushes the samples. In this type of injection, the air gap following the injected sample solution prevents diffusion of analytes into the mobile phase behind the air gap, thus avoiding peak tailing. There is also a potential problem with carry over of analytes contaminating the next injected sample. Different from HPLC, each injection was followed by weak and strong needle wash solutions; thus, stronger solvents capable of dissolving all injected analytes compared to the sample solvent prevent carry over. A weak needle wash solution is similar to the chromatographic mobile phase A (in this case we used 90% methanol-water containing 0.1% formic acid because lycopene dissolves poorly in acetonitrile) and a strong needle wash solution is an organic solvent (100% methanol). This method is critical for quantification of samples from e.g., Caco-2 cell monolayer as the sample volumes available are limited and contain low levels of analytes.
UPLC separation conditions were developed from previous HPLC methods or adopted from the literature, in order that both methods covered the needs for analysis of all sample classes depicted in Fig. 1. The UPLC method used in the present study was developed from the method of Sakakibara18 which successfully separated phenolics, flavonoids and theaflavin using a methanolic mobile phase. However, the mobile phase was altered to an acetonitrile base to accommodate all the target compounds. The UPLC method was developed by modifying a previously developed HPLC method in order that they had equivalent performance. This is important in order to get the same chromatographic results from undigested, digested, and Caco-2 samples.
Amongst the chromatographic systems trialled (combination of 6 columns, 5 mobile phases, 4 detectors and 3 chromatographic devices), the best separation and identification are the three columns and chromatographic sets detailed in the experimental section. The criteria for selection were minimum column numbers, the best detectors, and best elution performances including regression equations >0.99 as shown in Table 1. At this stage, we have succeeded in using one type of column for at least two different compound groups: UPLC system for phenolics and carotenoids, eluted with different mobile phases; and HPLC for sugar and organic acid analyses, running aqueous fractions on Aminex HPX-87 or Prevail Organic Acid columns eluted with a single mobile phase system coupled with a UV-DAD and ELSD, provided samples were not rich in salts.
Acetonitrile-based mobile phases have been widely used for various chromatographic analyses of analytes from a single target compound group. Thus, the simultaneous analysis adopted acetonitrile-based solvent combinations to accommodate as many target compound groups as possible. Another common solvent is methanol–water; however, it is not suitable for organic acids and tends to generate high pressures in the UPLC system. The equivalence of buffer composition between UPLC and HPLC was calculated from aqueous based solvent (buffer in water in Sakakibara method) into acetonitrile (UPLC) using Aquity calculator which is provided with the Waters Aquity UPLC instrument. The results gave an approximation of UPLC conditions having equal separation capacity between buffer elution and the new acetonitrile-based eluant. The results were trialled using similar compositions of standard compounds; if the separation was poorer than that of Sakakibara method results, the gradient elution was adjusted further. The final UPLC elution developed was acetonitrile containing formic acid 0.1% and milliQ-filtered water containing 0.1% formic acid for phenolics, consistent with the Sakakibara method. Furthermore, the same acetonitrile based mobile phase was developed for carotenoids, organic acid and sugars.
Initially, chromatographic analyses were carried out separately for each fraction obtained from a simultaneous fractionation protocol (Fig. 2) using various methods established in the Analytical Service Laboratory, The University of Queensland, Australia, or those described in various publications for all target compounds. Because the target compounds included unknown metabolites produced from the four groups of target compounds, running previously validated chromatographic systems aids peak identifications. The limitation of each chromatographic device is that there are a maximum of four mobile phase reservoirs and mobile phase tubing installations that made it impossible to run the selected chromatographic methods in one concomitant elution.
The final simultaneous analysis (examples of chromatograms for each chromatographic analysis are shown in Fig. 3–7) of four groups of nutrient compounds was achieved using the separation scheme in Fig. 2 and chromatographic systems with acetonitrile as the mobile phase.
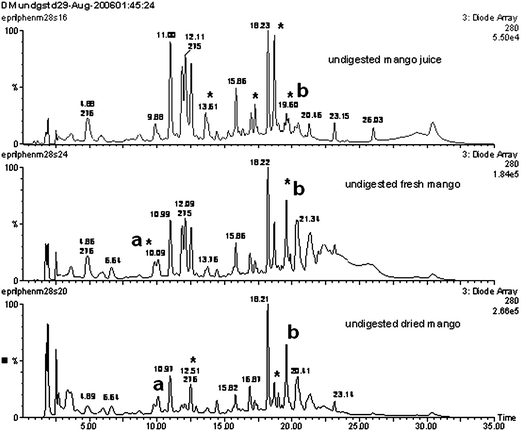 | ||
| Fig. 5 Phenolics LC-ESI-MS/DAD displayed as DAD UV-vis overlay chromatograms of local Kent mango; a (no ions, 269 nm), bm/z 131 (100%) 279 nm. Asterisk (*) indicating changes due to processing. | ||
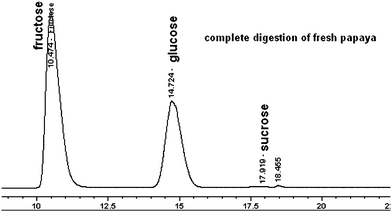 | ||
| Fig. 6 An example of sugar analysis for fresh papaya during in vitro digestions; obtained from HPLC/ELSD analysis using a Prevail Carbohydrate ES column eluted with acetonitrile–water. | ||
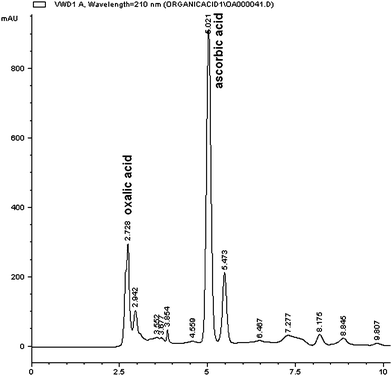 | ||
| Fig. 7 An example of HPLC/DAD chromatogram of organic acids from papaya sample after in vitro digestion run in a Prevail organic acid column eluted with acetonitrile-25 mM K2HPO4. | ||
Experimental8
Materials
Samples used in this study were fully ripe Kent mangoes, red papaya, and Roma tomatoes, all obtained from a local supermarket, which were variously chopped or blended and frozen at −20 °C prior to analyses. Their digest solutions and standard solutions, including those applied to Caco-2 cell monolayers, were analysed in triplicate.Standard compounds used were caffeic acid, p-coumaric acid, o-coumaric acid, mangiferin, quercetin, chlorogenic acid, ferulic acid, rutin, fumaric acid, naringenin, hydroxy benzeoic acid, (+) catechin, succinic acid, trans-aconitic acid, malonic acid, malic acid, citric acid, oxalic acid, L-ascorbic acid, tartaric acid, lycopene, β–carotene (Sigma, USA); lutein and β-cryptoxanthin (all were from Extrasynthese, France).
Sample preparation
Fractionation protocols to separate polar and nonpolar fruit components are summarised in Fig. 2Chromatographic methods
Chromatography of phenolics and carotenoids involved use of a C-18–based reverse phase column, whereas organic acids and sugars required ion exchange (Prevail) columns for separations. The mobile phase for phenolic UPLC analysis was: A—acetonitrile containing 0.1% formic acid and B—milliQ-filtered water containing 0.1% formic acid. It was run in a gradient of: 5% B at the start, 40% B for 1 min with a linear gradient, 73% B for the next 2 min, 10% B for 1 min and back to 5% before the next injection. UPLC carotenoid analysis applied gradient elution using acetonitrile containing 0.1% formic acid (A) and TBME (B): 2.5% B for 0.1 min, increased to 7.5% using slow increment gradient for 0.3 min, 10% for 0.1 min, 12.5% for 1 min and returned directly to 2.5% for 1.5 min to wash and equilibrate the column before the next injection. Both analyses used a C-18- Bridged Ethanes in Hybrid (BEH) matrix column; 50 × 2.1 mm using packing material with 80–125 Å pore size of 1.7 μm particles (Waters, Australia) operating at a flow rate of 0.45 mL min−1.The phenolic and carotenoid methods were also run using liquid chromatography coupled with a mass spectrometer. The phenolic fractions from the C-18 solid phase extraction cartridges were eluted with water containing 0.1% formic acid (A) and acetonitrile (B) in a 150 × 2.1 mm 3.5 μm XTerra™ MS C-18 column (Waters) connected to a 2.1 × 10 mm XTerra™ MS C-18 3.5 μm guard column (Waters). Using a flow rate of 0.4 mL min−1, the initial gradient composition was 5% B for the first 10 min and increased to 15% B at 10–15 min, 30% B at 15–20 min, 32.5% at 20–25 min, 80% at 25–28 min, and re-equilibrated to 5% B for 7 min before the next injection. The chloroform extracts were eluted through a 250 × 4.6 mm 5 μm Grace Vydac C-18 Polymeric Reverse Phase column (Grace, Illinois, USA) with MeOH–THF–water (67:27:6). The phenolic and carotenoid extracts were analyzed using a Waters Alliance 996 diode array detector (DAD) with UV spectra obtained over 200–700 nm. As there were no MS ions detected for eluted carotenoids, a MS method was not applicable for these compounds.
Sugar and organic acid analyses were run on an Agilent 1100 HPLC coupled with an Alltech 2000 evaporative light scattering detector (ELSD) and a UV detector, respectively. The elution of sugars was through a 250 × 4.6 mm 5 μm Prevail Carbohydrate ES column (Alltech, Australia) using isocratic elution with 75% acetonitrile–water at a flow rate of 1 mL min−1. N2 gas at 2 L min−1 flow rate was used for nebulization and solvent was evaporated at 65 °C. Meanwhile, organic acid analysis was conducted using a silica-based Prevail organic acids 250 × 4.6 mm, 5 μm column (Alltech, Australia). 25 mM K2HPO4 (A) and acetonitrile (B) mobile phases and a flow rate at 1 mL min−1 were used. The elution gradients were: 0–12.50 min 0% B, up to 16 min 15% B, and returned to 0% B for another 12 min. The UV detector was set at a wavelength of 210 nm.
Recovery tests were carried out using standard solutions and gave satisfactory results (Table 1). For the Caco-2 samples, the standard solutions were spiked into HBSS −25 mM HEPES (samples collected at 0 min bioassays).
Regressions for HPLC and LC-MS were determined with Excel software after running a set (3–5 concentration levels) the standard compound solutions. For UPLC regression can be determined using data analysis software provided in the Waters Aquity UPLC instrument. A linear regression line was chosen.
Furthermore, during running the chromatographic methods, every batch was initiated with a set of standard compound solutions and then every 10–20 injections of samples the standard solution was injected again (one concentration level only) to calibrate the retention time because when running many samples the retention time might drift slightly, and interfere with peak identification. At the end of elution of a batch of samples, the same level of concentration of the standard solution is injected again.
Conclusions
The extraction scheme and HPLC chromatography developed in the study can be used for the near-simultaneous analysis of sugars, organic acids, carotenoids, and phenolics. Pectin problems can be solved by using a mixture of hydrolase enzymes to macerate cell wall materials. Salt and buffer (HEPES) interference can be removed through use of ion exchange. The analysis method can be used for digest samples without bile component interferences, provided the sample does not contain polyphenols co-eluted by acetonitrile–water gradients at the end of the elution phase.In this study, analysis of diverse fruit components was achieved through multiple fractionations and HPLC/UPLC chromatography using 3 columns and 3 combinations of mobile phases that were predominantly based on acetonitrile–water. A column of porous –C18 based reverse phase packing materials was used for separation of phenolics and carotenoids, while two ion exchange columns were used for analysis of sugars and organic acids. Acetonitrile–water separated polar compounds in HPLC sugars and organic acids analyses, while acetonitrile–TBME and acetonitrile–0.1% formic acid in water separated phenolics or carotenoids in UPLC analyses, respectively.
References
- C. Cortes, M. J. Esteve, A. Frigola and F. Torregrosa, J. Agric. Food Chem., 2004, 52, 2203–2212 CrossRef.
- L. R. Snyder, J. J. Kirkland, and J. L. Glajch. Practical HPLC method development, John Wiley & Sons, Inc, New York, 1997 Search PubMed.
- U. Justensen, P. Knuthsen and T. J. Leth, J. Chromatogr., A, 1998, 799, 101–110 CrossRef CAS.
- I. E. J. Milder, I. C. W. Arts, D. P. Venema, J. J. P. Lasaroms, K. Wahala and P. C. H. Hollman, J. Agric. Food Chem., 2004, 52, 4643–4651 CrossRef CAS.
- M. Papagiannopoulos, H. R. Wollseifen, A. Mellenthin, B. Haber and R. Galensa, J. Agric. Food Chem., 2004, 52, 3784–3791 CrossRef CAS.
- P. C. Sadek The HPLC solvent guide, John Wiley & Sons, Inc, New York. 1996 Search PubMed.
- C. R. Morris, J. T. Scott, H.-M. Chang, R. R. Sederoff, D. O'Malley and J. F. Kadla, J. Agric. Food Chem., 2004, 52, 1427–1434 CrossRef CAS.
- I. Epriliati, B. D'Arcy and M. Gidley, J. Agric. Food Chem., 2009a, 57, 3363–3376 Search PubMed; I. Epriliati,, B. D'Arcy, and M. Gidley,, J. Agric. Food Chem., 2009b, 57, 3377–3388 Search PubMed.
- G. E. Hobson and J. N. Davies. in ‘The biochemistry of fruits and their products’, Vol 2 (Ed, Hulme, A. C.) Academic Press, New York, 1971, pp. 436–482 Search PubMed.
- A. Parley, L. Vanhanen and D. Heatherbell, Australian J. Grape Wine Res., 2001, 7, 146–152 CrossRef.
- A. Pollard, and C. F. Timberlake, in The biochemistry of fruits and their products. ed. Hulme, A. C.Academic press, London, 1970, pp. 573–621 Search PubMed.
- I. Cinar, Electronic Journal of Environmental Agricultural and Food Chemistry, 2003, 3, 609–616 Search PubMed.
- J. K. Burns and R. Pressey, J Am Soc Hort. Sci., 1988, 113, 624–626 Search PubMed.
- K. J. Scott, Food Chem., 1992, 45, 357–364 CrossRef CAS.
- K. Krygier, F. Sosulski and L. Hogge, J. Agric. Food Chem., 1982, 30, 330–334 CrossRef CAS.
- M. Girod, E. Beaudoin and L. Charles, Anal. Methods, 2009, 1, 128–131 RSC.
- E. Forgacs and T. CserhatiMolecular Basis of Chromatographic Separation, CRC Press, Boca Raton, 1997 Search PubMed.
- H. Sakakibara, Y. Honda, S. Nakagawa, A. Hitoshi and K. Kanzawa, J. Agric. Food Chem., 2003, 51, 571–581 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Further experimental results. See DOI: 10.1039/c0ay00244e |
| ‡ Caco-2 is abbreviation for Colon Adenocarcinoma, a cell culture obtained from cancerous cell colon commercially supplied by American Type Culture Collection (ATCC, Manassas, USA) with a reference number HTB-37 |
| This journal is © The Royal Society of Chemistry 2010 |