Gas chromatography-mass spectrometric analysis of trimethylsilyl derivatives of toxic hydrolyzed products of nerve agent VX and its analogues for verification of Chemical Weapons Convention†
Deepak
Pardasani
,
Ajay
Purohit
,
Avik
Mazumder
and
D. K.
Dubey
*
Vertox Laboratory, Defence Research and Development Establishment, Jhansi Road, Gwalior, 474002, India. E-mail: dkdubey@rediffmail.com; Fax: +91-751-2341148; Tel: +91-751-2233488
First published on 20th April 2010
Abstract
The formation of trimethylsilyl (TMS) derivative of hydrolyzed product of nerve agent VX, namely S-2-(N,N-diisopropylaminoethyl) methylphosphonothiolate (EA-2192), was reported to be hampered due to its zwiterionic character. Contrary to this assumption, we have synthesized and analyzed the TMS derivative of EA-2192 and its analogues. Initially, the problem of non-detectability in GC-MS analysis was found to be due to their condensation followed by decomposition on GC column. An optimized temperature program, starting from 175 °C column temperature, favours partitioning of TMS derivatives of EA-2192 and its analogues into mobile phase, which facilitates their detection. It reduces their on-column condensation and decomposition. Excellent reproducibility (<1% RSD) of TMS derivatives of S-2-(N,N-dialkylaminoethyl) alkylphosphonothiolates was achieved by GC-MS analysis with temperature program of 175 °C (2 min)–10 °C min−1– 300 °C (5 min). Under the optimized conditions LODs ranged from 8–10 μg mL−1 for different analytes at a split ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10.
:10.
1. Introduction
Retrospective detection and identification of chemical warfare agents (CWAs), such as nerve agents, and their degradation products is of utmost importance for the verification program of the Chemical Weapons Convention (CWC).1–3 The verification regime of CWC relies on inspections, on-site analysis, or sampling and off-site analysis in a network of laboratories designated by the Organization for Prohibition of Chemical Weapons (OPCW).4–7 For verification analysis of the CWC, the most reliable and widely accepted analytical technique is gas chromatography coupled to mass spectrometry (GC-MS).5–12 Therefore, methods allowing improved GC-MS analysis of CWAs and their degradation products become important for the success of verification analysis of the CWC.The chemical warfare agent O-ethyl S-2-(N,N-diisopropylaminoethyl) methylphosphonothiolate (VX) and its analogues (Fig. 1) are the most toxic nerve agents that are listed in the schedule 1 category of CWC.2
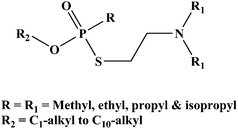 | ||
| Fig. 1 O-ethyl S-2-(N,N-diisopropylaminoethyl) methylphosphonothiolate (VX) and its analogues. | ||
These agents are more toxic than other nerve agents such as sarin or tabun, and have been used for terrorist purposes in Japan.13 Therefore, retrospective identification of these agents that relies on identification of live agents themselves and their characteristic degradation products (so called markers) is crucial, both for civilian and military protection, and obtaining judicial proof of illegal use, preparation or stockpiling.
Analysis of VX and its analogues is not a problem by itself; straightforward methods are documented for their detection and identification using GC-MS as the analytical technique.8 What apparently remains as a problem is GC-MS analysis of the trimethylsilyl (TMS) derivative of S-2-(N,N-diisopropylaminoethyl) methylphosphonothiolate (EA-2192), the hydrolytic product of, and equitoxic to, nerve agent VX.14–16
The hydrolysis of VX and analogues can take place via P–S, P–O or S–C bond cleavages (Scheme 1). Predominance of a particular route depends on pH and concentration. In dilute aqueous solutions (≤0.01 M), VX gives rise to non-toxic ethyl methylphosphonic acid (EMPA) through P–S bond cleavage and highly toxic EA-2192 through P–O bond cleavage in an approximate ratio of 87:13.17–19 Ethyl methylthiophosphonic acid (EMPTA) is formed in minute quantities via S–C bond cleavage.
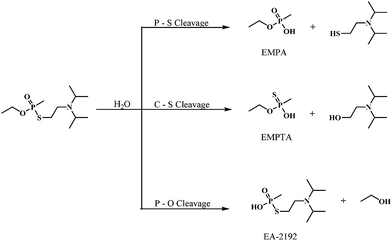 | ||
| Scheme 1 Hydrolytic pathways of VX. | ||
Verification analysis of CWC requires the identification of live agents and their degradation products; hence identification of VX and all its possible degradation products becomes important. Identification of EMPA, EMPTA, N,N-diisopropylaminoethane thiol and N,N-diisopropylaminoethanol by GC-MS can be easily carried out after silylation, which is routine and well documented in open literature.13,15,16 However, the identification of the TMS derivative of EA-2192 is reported to be hampered due to its zwiterionic character.14,15 EA-2192 is however reported to be identified as methyl derivative by GC-MS, whereas, methylation is carried out by diazomethane, trimethylsilyl diazomethane or trimethylphenylammonium hydroxide (TMPAH).15,20 Disadvantages of derivatization of EA-2192 by methylation are, use of carcinogenic and detonable diazomethane, highly corrosive nature of TMPAH (pH ∼ 13 which leads to column deterioration) and poor peak shapes.15 However trimethylsilylation is a simple, efficient and widely used derivatization technique for GC-MS analysis of various degradation products of CWAs.13–16
Moreover, for verification analysis of CWC, it is mandatory to identify a derivatisable analyte by at least two derivatizing techniques, because if only one derivatizing technique (e.g. methylation) is used, the absence of this particular derivative has to be demonstrated in the sample,21 it necessitates the availability of another derivatizing technique. Thus, even if EA-2192 is identified as its methyl derivative, it would entail an alternative derivatization to prove that the methyl derivative of EA-2192 (O-methyl S-2-(N,N-diisopropylaminoethyl) methylphosphonothiolate, the methyl analogue of VX) was originally absent in the sample. Thus, this stringent requirement of verification analysis of CWC demands the development of a GC-MS method of analysis of TMS derivative of EA-2192 (EA-2192-TMS) and its analogues.
In this paper, we report the optimization of GC-MS analysis conditions for EA-2192-TMS and its analogues. Contrary to the reported difficulty of synthesis and analysis, we found that these derivatives could be prepared by a straight forward derivatization technique,13–16 and were able to be detected and identified under the optimized GC-MS conditions.
2. Experimental
2.1 Materials
Chemicals required for synthesis of S-2-(N,N-dialkylaminoethyl) alkylphosphonothiolates (1a–4d, Table 1) were obtained from Sigma-Aldrich (Mumbai, India) and were of 99% purity. These included N,N-dimethyl-, N,N-diethyl, N,N-dipropyl and N,N-diisopropylaminoethyl chloride hydrochloride, diisopropylamine, 2-chloroethanol and N,O-bis(trimethylsilyl) trifluoroacetamide (BSTFA). Solvents and general chemicals were obtained from E. Merck (India) Ltd (Mumbai, India).2.2 Synthesis of S-2-(N,N-dialkylaminoethyl) alkylphosphonothiolates (1a–4d)
The acids were synthesized by heating the pyridine salt of alkylphosphonothioic acids with N,N-dialkylaminoethyl chlorides in acetonitrile at 70 °C for 2 h. Alkylphosphonothioic acids were synthesized as per the reported procedure.22 Typical synthesis of EA-2192 is described as follows:Methylphosphonothioic acid (56 mg, 0.5 mmoles) taken in a 2 mL Teflon capped reaction vial and dissolved in 250 μL of dry acetonitrile. To this solution, dry pyridine (79 mg, 1.0 mmoles) was added and the mixture was purged with dry argon. Subsequently, N,N-diisopropylaminoethyl chloride hydrochloride (100 mg, 0.5 mmoles) was added in one shot and the contents were heated to 70 °C for 2 h after replacing the Teflon cap of the vial. After completion of reaction, the reaction mixture was diluted with 500 μL of dry acetonitrile and centrifuged. The clear supernatant was transferred into another vial and used for the analysis. No attempt was made to further purify the acid (1c) as it showed ∼90% purity by 31P NMR and GC-MS analysis (after methylation with diazomethane).
Caution: These compounds are extremely toxic and must be prepared and handled by trained professionals only. The laboratory must be equipped with efficient fume hoods and persons handling them must wear a face mask and protective clothes. Decontamination solution made of bleach and alkali in water must be kept aside to decontaminate the glassware.
2.3 Derivatization of acids (1a–4d)
Solutions (200 μg mL−1) of acids were prepared in dry acetonitrile. An aliquot (100 μL) from these solutions was derivatized with 100 μL of BSTFA in a sealed vial by heating at 70 °C for 30 min.For making the methyl derivatives, aliquots (100 μL) were acidified with 10 μL of acidic methanol and after cooling to ∼0 °C in an ice/salt mixture, diazomethane was passed through these aliquots till the yellow colour persisted.
2.4 GC-MS analysis
Typical GC-MS conditions unless mentioned otherwise were as follows. The GC-MS analyses were performed in EI mode (70 eV) with an Agilent 6890 GC, equipped with model 5973 MSD (Agilent Technologies, Palo, Alta, CA, USA). SGE BPX5 capillary column (5% phenyl 95% methylpolysiloxane) with 30 m length × 0.32 mm i.d. × 0.25 μm film thickness was used at temperature program of 90 °C (2 min)–10 °C min−1–300 °C (5 min). Helium was used as carrier gas at a constant flow of 1.2 mL min−1. The samples were analyzed in split (split ratio 1:10) mode at injection temperature of 250 °C, EI source temperature 230 °C and quadrupole analyzer at 150 °C, unit mass resolution, scan range m/z 35–500, with a scan cycle of 3.2 scans/s. The injected volume was 1 μL. The injection port comprised of quartz glass liner (silanized) having 4 mm i.d and 900 μL of internal volume. It was cleaned and changed frequently to ensure that no decomposition of analytes takes place due to contamination of injection port.
Modified temperature program that was optimized for the GC-MS analysis of TMS derivatives of acids 1a–4d was as follows: 175 °C (2 min)–10 °C min−1– 300 °C (5 min). Samples were analyzed in split mode (split ratio 1:10) at an injection port temperature of 225 °C. Other conditions were similar to those described above.
For quantitative determinations, eicosane was added to the derivatized reaction mixture as an internal standard so as to give the concentration of 100 μg mL−1. Relative peak area of analyte to internal standard was taken for quantitative determinations of TMS derivatives of acids 1a–4d.
3. Results and discussion
To investigate the synthesis and stability of TMS derivatives of EA-2192 and of analogue acids, as depicted in Fig. 2, the GC-MS analysis of TMS derivatives of S-2-(N,N-dialkylaminoalkyl) alkylphosphonothiolates (1a–4d) was undertaken. | ||
| Fig. 2 S-2-Dialkylaminoethyl methylphosphonothiolates selected for the study. | ||
To optimize the GC-MS analysis conditions of TMS derivatives of acids 1a–4d, acid EA-2192 (1c) was selected as a model compound. TMS derivative of 1c was made as per the procedure described in the experimental section, and was subjected to GC-MS analysis employing temperature program starting from 90 °C with a hold time of 5 min, which was subsequently raised to 300 °C at a rate of 10 °C min−1. Other instrumental conditions remained the same as described in the experimental section. The analyte showed an irreproducible response in the total ion chromatogram (TIC). In first analysis, it was detected as a discernable signal in TIC (Fig. 3A), while in duplicate and subsequent analyses, the peak due to EA-2192-TMS was not observed at all (Fig. 3B). This observation initially indicated the instability of the selected analyte (EA-2192-TMS, this was true as discussed later). The compound was not detected due to irreversible binding onto the column. However, we extended the investigation by subjecting the derivatized analyte to 31P NMR, in which, two important observations were made; first, the chemical shift due to phosphorus (δ = 41.5 ppm) remained unchanged before and after heating the derivatization mixture at 100 °C for 2 h, and second, the multiplicity of signals due to ‘phosphorus’ was found to be doublet of sextet indicating the intact P–O–Si bond. These observations led us to think that the EA-2192-TMS was formed and stable; and its irreproducibility in GC-MS analysis could be due to its decomposition in the heated injection port (250 °C) of GC. Hence, in subsequent attempts, the GC-MS chromatograms were recorded by gradually reducing the injection port temperature to 100 °C and correspondingly reducing the initial temperature of the column also. But no traces of EA-2192-TMS were observed. These observations indicated that the derivative is stable ‘in-tube’ while decomposing on GC column due to irreversible physico-chemical interactions with the column material.
Considering these observations, we wondered whether during a chromatographic run, if the analyte is partitioned preferentially in the mobile phase (i.e. allowed to interact minimally with the column), it might survive in the column and probably be detected. To verify this hypothesis, three approaches were followed, i) analyzing the analyte by keeping the initial temperature of the column high, ii) reducing the column length and iii) changing the column stationery phase.
3.1 Analysis at elevated column temperature
Initially, the GC-MS analyses of EA-2192-TMS were performed in isothermal mode, where column temperatures were varied from 150 °C to 225 °C by successive increments of 25 °C. Quintuplicate runs were performed to check the reproducibility of the GC-MS analyses, the results are depicted in Fig. 4. | ||
| Fig. 4 TICs obtained from isothermal GC-MS analysis of TMS derivative of EA-2192. | ||
It is evident from these figures that the EA-2192-TMS was detected with good response at 175 °C and higher temperatures, at column temperatures of 150 °C and lower, the peak height of analyte was found to be reduced significantly. In the following sets of experiments, the column was programmed from 150, 175, 200 and 225 °C (hold time 2 min) and increased at the rate of 10 °C min−1 to 300 °C with a hold time of 5 min. Internal standard (IS) eicosane (100 μg mL−1) was added in the EA-2192-TMS and ratios of the peak areas of analyte to IS were used to assess the chromatographic response of the derivative. These results are shown in Fig. 5, which corroborate the assumption that TMS derivative decomposes on column if allowed to condense on it. Increasing the initial program of column from 150 °C to 175 °C drastically increased its response with subsequent gradual increment up to 225 °C. Beyond this temperature, it was not possible to analyse the derivative as it eluted along with the solvent.
 | ||
| Fig. 5 Effect of initial oven temperature on the response of TMS derivative of EA-2192. | ||
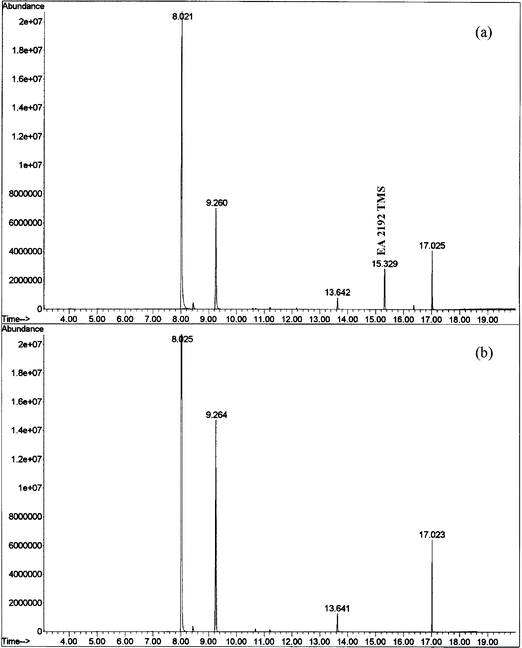
It is important to note that the threshold value of column temperature to obtain a good signal of EA-2192-TMS is 175 °C. Below this temperature, the signal will either be too weak (e.g., at 150 °C) or undetectable. The temperature program that is recommended for the retrospective detection of nerve agents,4,8 starts from 50 °C; beginning from such a low temperature causes the condensation and decomposition of EA-2192-TMS on GC column. Thus, the non-detectability of EA-2192-TMS is not due to zwiterionic character that hampers its trimethysilylation,14,15 but rather, is due to its decomposition on the GC column caused by condensation. The initial high temperature of the column shifts the partitioning of the derivative in the mobile phase thereby not allowing it to condense and decompose. A typical TIC and mass spectrum obtained from GC-MS analysis of EA-2192-TMS is depicted in Fig. 6.
 | ||
| Fig. 6 TIC (a) and mass spectrum (b) of TMS derivative of EA 2192 obtained under the optimized GC-MS conditions. | ||
To test the general applicability of this temperature program for other TMS derivatives of analogues acids of EA-2192 (Fig. 2, acids 1a–4d), all acids were derivatized individually and analyzed by the GC-MS under the optimized GC temperature program starting at 175 °C. TMS derivatives of acids 1a–4d were detected and their mass spectra were recorded (available as supporting information S1 †) along with the C-series retention indices. RI data of these analytes are given in Table 1. GC-RI and MS data are important for the retrospective identification and verification analysis of VX and its analogues.
| Series No. | Acid (TMS derivative) | RI (C-series) | Limit of detection/μg mL−1 |
|---|---|---|---|
| 1 | 1a | 1508 | 10 |
| 2 | 1b | 1644 | 8 |
| 3 | 1c | 1754 | 8 |
| 4 | 1d | 1790 | 8 |
| 5 | 2a | 1576 | 10 |
| 6 | 2b | 1709 | 8 |
| 7 | 2c | 1821 | 8 |
| 8 | 2d | 1854 | 8 |
| 9 | 3a | 1610 | 10 |
| 10 | 3b | 1738 | 10 |
| 11 | 3c | 1846 | 10 |
| 12 | 3d | 1879 | 10 |
| 13 | 4a | 1638 | 8 |
| 14 | 4b | 1776 | 8 |
| 15 | 4c | 1882 | 8 |
| 16 | 4d | 1914 | 8 |
3.2 Analysis with reduced column length
As discussed above, another method to reduce the analyte-column interaction is to shorten the column length. In these sets of experiments, column length was reduced to half (i.e. 15 meters) and analysis was performed employing the same temperature programs as were performed with full length (30 m) column. No traces of any of the TMS derivatives of 1a–4d were detected when initial column temperature was kept at 90 °C; at 150 °C, derivatives were detected inconsistently with poor signals, while at temperature programs starting from 175 °C or higher, intense signals with excellent reproducibility were observed. Thus, for efficient detection and identification of EA-2192-TMS and its analogues, the optimized temperature program is 175 °C (2 min)–10 °C min−1–300 °C (5 min).3.3 Analysis on columns of different polarity
Analysis of EA-2192-TMS on GC columns of varying polarity (BPX1-non-polar, BPX5-non-Polar and BP10-mild-polar) were also performed. On these three columns, the analyte was detected only when the column was programmed from 175 °C or above; indicating that the minimum temperature required to analyze these TMS derivatives is 175 °C.3.4 Reproducibility and limits of detection
Having optimized the GC-MS analytical conditions of TMS derivatives of acids 1a–4d, their precision and limits of detection (LOD) were determined under the optimized analytical conditions. The intra-day repeatability and inter-day reproducibility were determined for quintuplicate runs for five consecutive days. For this, the concentrations of TMS derivatives and IS were fixed at 100 μg mL−1 each. The relative standard deviations (RSDs) were observed to be within the 1.5% for both intra- and inter-day experiments indicating excellent reproducibility of the method.Since all analyses were performed in split mode (split ratio 1:10), the LODs were observed to be somewhat higher (Table 1) than those that would have been achieved had the analyses been done in splitless mode. Since the optimized temperature program starts from 175 °C, analysis with splitless mode could not be done as it adversely affected the resolution.
4. Conclusion
The development of GC-MS based identification method of TMS derivative of extremely toxic hydrolyzed product of VX, namely EA-2192 and of its various analogues, is of utmost importance to obtain the judicial proof of its illegal use and verification analysis of CWC. This study is the first disclosure of GC-MS based analytical method for identification of TMS derivatives of EA-2192 and its analogues. Contrary to the difficulty of synthesizing the EA-2192-TMS, owing to zwiterionic character of the acid EA-2192, we could synthesize and analyze it under the optimized GC-MS conditions. The hampered GC-MS analysis of TMS derivatives of this acid and its analogues is attributed to their condensation and decomposition on GC column. This limitation was overcome by systematically investigating the stability and detectability of selected analytes employing various temperature programs. When derivatives were preferentially partitioned into the mobile phase by initiating the column temperature from 175 °C or above, the analytes were successfully detected and identified with excellent reproducibility.References
- Convention on the Prohibition of the Development, Production, Stockpiling and use of Chemical Weapons and on their Destruction, Technical Secretariat of the Organization for Prohibition of Chemical Weapons: The Hague, 1997; accessible through internet http://www.opcw.org Search PubMed.
- W. Krutzsch, R. Trapp, A Commentary of CWC, ed. Martinus Nijhoff, Dordrecht, 1994 Search PubMed.
- M. Mesilaakso, Chemical Weapons Convention Chemical Analysis Sample Collection, Preparation and Analytical Methods, John Wiley & Sons, Chichester, UK, 2005 Search PubMed.
- S. Krüger, in Chemical Weapons Convention Chemical Analysis, Sample Collection, Preparation and Analytical Methods, ed. M. Mesilaakso, John Wiley & Sons, Chichester, UK, 2005, pp. 33 Search PubMed.
- M. Sokolowski, in Chemical Weapons Convention Chemical Analysis, Sample Collection, Preparation and Analytical Methods, ed. M. Mesilaakso, John Wiley & Sons, Chichester, UK, 2005, pp. 51 Search PubMed.
- J. Hendrikse, in Chemical Weapons Convention Chemical Analysis, Sample Collection, Preparation and Analytical Methods, ed. M. Mesilaakso, John Wiley & Sons, Chichester, UK, 2005, pp. 89 Search PubMed.
- M. Mesilaakso, in Chemical Weapons Convention Chemical Analysis, Sample Collection, Preparation and Analytical Methods, ed. M. Mesilaakso, John Wiley & Sons, Chichester, UK, 2005, pp. 151 Search PubMed.
- E. R. J. Wils, in Chemical Weapons Convention Chemical Analysis, Sample Collection, Preparation and Analytical Methods, ed. M. Mesilaakso, John Wiley & Sons, Chichester, UK, 2005, pp. 249 Search PubMed.
- H. D. Durst, R. J. Mays, J. L. Ruth, B. R. Williams and R. V. Duevel, Anal. Lett., 1998, 31, 1429 CAS.
- D. K. Dubey, D. Pardasani, P. K. Kanaujia, V. Tak and H. K. Gupta, Rapid Commun. Mass Spectrom., 2008, 22, 2526 CrossRef CAS.
- T. J. Reddy, S. Prabhakar, U. V. R. Vijaya Saradhi, V. Jayathirtha Rao and M. J. Vairamani, J. Am. Soc. Mass Spectrom., 2004, 15, 547 CrossRef CAS.
- Central OPCW Analytical Database, version 8, Released January 2008, Technical Secretariat of OPCW, The Hague, The Netherlands Search PubMed.
- M. Katagi, M. Nishikawa, M. Tatsuno and H. J. Tsuchihashi, J. Chromatogr. B, 1997, 689, 327 CrossRef CAS.
- W. R. Creasy, J. R. Stuff, B. Williams, K. Morrissey, J. Mays, R. Duevel and H. D. Drust, J. Chromatogr., 1997, 774, 253 CrossRef CAS.
- R. M. Black and B. Muir, J. Chromatogr. A, 2003, 1000, 253 CrossRef CAS.
- D. K. Rohrbaugh, J. Chromatogr. A, 1998, 809, 131 CrossRef CAS.
- Y. C. Yang, F. J. Berg, L. L. Szafraniec, W. T. Beaudry, C. A. Bunton and A. J. Kumar, Chem. Soc. Perk Trans. 2, 1997, 607 Search PubMed.
- Y. C. Yang, Acc. Chem. Res., 1999, 32, 109 CrossRef CAS.
- Y. C. Yang, J. A. Baker and J. R. Ward, Chem. Rev., 1992, 92, 1729 CrossRef CAS.
- J. A. Tornes and B. A. Johnsen, J. Chromatogr., 1989, 467, 129 CrossRef CAS.
- Work instructions for the evaluation of the results of OPCW proficiency tests, QDOC/LAB/WI/PT03, 27 March 2009.
- G. M. Kosolapoff, L. Maier, Organic Phosphorus Compounds, John Wiley & Sons, New York, USA, 1976, pp. 39 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimental details. See DOI: 10.1039/c0ay00089b |
| This journal is © The Royal Society of Chemistry 2010 |