An in vitro crystallization setup to assess the efficiency of different phosphate binders in nephrology: quantitative analytical considerations
Thorsten
Peitsch
a,
Matthias
Matthes
a,
Vincent
Brandenburg
b and
Matthias
Epple
*a
aInorganic Chemistry and Center for Nanointegration Duisburg-Essen (CeNIDE), University of Duisburg-Essen, Universitaetsstr. 5-7, 45117, Essen, Germany. E-mail: matthias.epple@uni-due.de; Fax: +49 201 1832621; Tel: +49 201 1832413
bIZKF BioMAT and Department of Nephrology, University Hospital Aachen, Pauwelsstraße 30, 52057, Aachen, Germany
First published on 19th May 2010
Abstract
An increased phosphate level in the blood (hyperphosphatemia) is a severe problem for dialysis patients. Different phosphate binders which are used to prevent hyperphosphatemia were studied in a custom-made in vitro crystallization apparatus which simulated the stomach (pH 2) and the gut (pH 7). The effective phosphate binding capacity was measured and the resulting products were identified. This apparatus permits the quantitative analysis of the phosphate binding effect under the given in vitro conditions. In particular, phosphate binders on the basis of calcium acetate (Calciumacetat Nefro®), calcium carbonate (Calciumcarbonat® Fresenius), aluminium hydroxide (Antiphosphat®), lanthanum carbonate (Fosrenol®), and poly(allylamine hydrochloride) (Renagel®) were studied, and also compared with pure calcium acetate, calcium carbonate, and aluminium sulfate.
Introduction
An increased level of phosphate in the blood, i.e. hyperphosphatemia, is one of the crucial causative factors in the development of hyperparathyroidism (HPT) in chronic kidney disease (CKD): hyperphosphatemia is a critical component of CKD–mineral and bone disorder (CKD–MBD).1,2 The usage of phosphate binders in dialysis patients to reduce the amount of phosphorus absorbed from the gut has been one of the cornerstones of hyperparathyroidism prevention and treatment over the last 40 years.3,4 Typically, a patient swallows a daily dose of tablets containing chemical compounds which bind phosphate from the food and form insoluble compounds which are then excreted. The number of available phosphate binder types has grown over time. Nephrology has started with aluminium (in the 1970s to early 1980s) and moved towards calcium-based binders (in the early 1980s to late 1990s), and finally non-calcium-, non-aluminium-containing phosphate binders came into play.5The decision for a particular binder is based on issues regarding differences in binding capacity, tolerability, and potential side effects. For patients with end-stage renal disease (ESRD), six agents have gained official approval for phosphate binding: aluminium compounds (to be used only for a limited period of time), calcium acetate, calcium carbonate, lanthanum carbonate, Sevelamer® hydrochloride, and Sevelamer® carbonate. Sevelamer is an insoluble, polycationic cross-linked poly(allyamine) polymer which can bind anions like phosphate.
Patients with progressive chronic kidney disease and especially those on dialysis often die prematurely.6,7 This increased mortality risk is due at least in part to features of CKD–MBD, including accelerated vascular calcification8 and hyperphosphatemia.9 Both epidemiological and basic research revealed that phosphate lowering is not just the correction of an abnormal biochemical parameter, but may be strongly related to clinical outcomes. As the efficiency of phosphate binders is typically simulated either with static experiments or assessed by epidemiological studies where the phosphate level of many patients is monitored, questions remain about the nature of the crystallization process under changing pH conditions (stomach: pH 2; gut: pH 7) and the final products of the crystallization process.
We have constructed an apparatus which is able to simulate the physiological system of the stomach (acidic conditions, pH ≈ 2), followed by the gut (neutral conditions, pH ≈ 7). The system does not involve biomolecules but only ionic phosphate at physiological concentration, and also HCl in the “stomach” and NaOH in the “gut” for pH regulation. Therefore, the chemical effects of the phosphate binders can be separated from other (biological) influences, and the solid products can be analyzed in pure form. The whole system is operated at 37 °C, and volumes and residence time in “stomach” and “gut” are similar to the human situation (1 h each).
Experimental
Chemicals
(NH4)2HPO4 (Merck), HCl (Sigma-Aldrich), NaOH (VWR Prolabo), Al2(SO4)3·16H2O (Raab Karcher Chemie), CaCO3 (calcite; Riedel-de Haen) and Ca(OOCCH3)2 (Merck) were obtained in p.a. quality. For all preparations, ultrapure water was used from a Purelab Ultra instrument (ELGA).Phosphate binders
We used the following samples (tablets taken from freshly opened packages; content per tablet and kind of active component according to the manufacturers):• Antiphosphat® from GRY-Pharma, Kirchzarten, Germany; active component 600 mg Al(OH)3 gel (exact composition not specified). We determined the content of aluminium by AAS to 212 mg per tablet.
• Calciumacetat-Nefro® from MEDICE, Iserlohn, Germany; active component 700 mg Ca(OOCCH3)2.
• Calciumcarbonat® from Fresenius Medical Care, Bad Homburg v.d.H., Germany; active component 500 mg CaCO3.
• Fosrenol® from Shire Pharmaceutical Contracts Ltd., Basingstoke, UK; active component 750 mg La2(CO3)3.
• Renagel® from Genzyme Europe B.V., The Netherlands; active component 800 mg Sevelamer®, a cross-linked poly(allylamine) hydrochloride. We determined the content of chloride to be 148.4 ± 1.5 mg per tablet.
Methods
X-Ray powder diffraction (XRD) was carried out with a Siemens D500 diffractometer (Cu Kα radiation, λ = 1.54 Å) in Bragg-Brentano mode. UV-visible absorption spectra were recorded with a Varian Cary WinUV spectrophotometer in 1 cm quartz cuvettes. Orthophosphate was determined by UV spectrometry using the molybdenum blue method (λ = 725 nm). Aluminium and calcium were determined by atomic absorption spectroscopy (Thermo Electron Corporation, M-Series AA spectrometer). Chloride was determined potentiometrically with a Metrohm device model 716 DMS Titrino. The samples were solubilized according to the Schoeninger method and titrated with 0.01 M AgNO3 solution.The physiological situation of the intestinal tract was modeled as shown in Fig. 1.
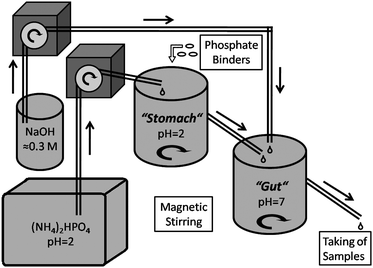 | ||
| Fig. 1 Schematic picture of the physiological simulation apparatus. | ||
The whole setup was constructed from glassware. The first vessel (the “stomach”; 1000 mL) was filled with a solution of (NH4)2HPO4 (c = 3 mM and 6 mM, respectively) that was brought to pH 2 ± 0.05 with HCl. The same solution was continuously added with a peristaltic pump with a flow rate of 886 mL h−1. As the water level in the “stomach” was increased by the stirring, the effective volume was about 890 mL, and the average hydrodynamic residence time was 60 min. From this vessel, an overflow led into the second vessel (the “gut”; 950 mL). Into this “gut”, a solution of NaOH (0.34 M; pH 13–14) was continuously pumped with a flow rate of 49 mL h−1. Therefore, the total flow into the “gut” was 935 mL h−1, and the average hydrodynamic residence time was 61 min. The pH in the “gut” was 7 ± 0.05. The whole system was thermostated at 37 ± 0.5 °C. An overflow led from the “gut” to a Buchner funnel with a filter and a suction flask which permitted the isolation of the solid products. Except for this outlet, the access of air into the system was limited as far as possible.
The pH and the temperature in both vessels were continuously monitored with WTW pH340 instruments, and the data were transferred to a computer every 12 s. The system was allowed to equilibrate for 2 h (with all liquid flows turned on) before the addition of the phosphate binders. Then, the dose of phosphate binder for one meal was added into the “stomach” by simply dropping the tablets into the vessel. Only in the case of Fosrenol®, the tablet was ground before the addition to follow the manufacturers' advice (it is a chewing tablet); in all other cases, the tablets were added intact. Every phosphate binder was studied twice at 3 mM and 6 mM phosphate concentration, respectively. The model compounds calcium carbonate (as powder and as tablet), calcium acetate, and aluminium sulfate were studied only once.
The amount of bound phosphate was determined by measuring the phosphate concentration at the outlet of the “gut”. Typically, samples of 10 mL were taken at defined time intervals (10 min at the beginning, then 20 and then 30 min). The phosphate concentration was determined photometrically as described above. The resulting curves were numerically integrated as follows: the average phosphate concentration value of every two data points was computed. The difference between this value and the baseline for the phosphate concentration was taken as y-value and then multiplied with the time difference between the two data points (x-value). This corresponds to the trapezoidal rule for numerical integration. As baseline for the phosphate concentration, we used the average of the starting points (t = 0) of all experiments because a small error in the starting pH value would strongly change the whole integral. This baseline value was 5.79 mM with a standard deviation of 0.08 mM (N = 15) for 6 mM and 2.89 mM with a standard deviation of 0.06 mM (N = 12) for 3 mM.
Results and discussion
The properties of the different phosphate binders and control compounds are summarized in Table 1. We used two different phosphate concentrations: 3 mM and 6 mM to elucidate the effect of the total phosphate concentration on the phosphate-binding efficiency of the different phosphate binders. All compounds led to an increase of the pH in the “stomach” because they are all more or less basic in nature. The corresponding pH data are shown in Fig. 2 and 3. In extreme cases, the pH increased up to 5.5 after administration of the phosphate binder. After an induction period which was due to the slow transfer of the solution from the “stomach” into the “gut” including mixing effects, the pH in the “gut” increased as well. Exceptions were phosphate binders with calcium acetate where the buffering effect of the acetate caused a decrease of the pH in the “gut” to about 6. These effects were confirmed by repeating the experiments with the chemically pure compounds calcium carbonate and calcium acetate.| Sample | Kind and amount of active pharmaceutical ingredient per tablet | Active chemical ingredient | Amount of active chemical ingredient per tablet | Expected chemical product after phosphate binding | Amount of expected chemical product after phosphate binding per tablet | Amount of bound phosphate per tablet (theoretical) | Number of tablets per experiment (as recommended) | Amount of bound phosphate per experiment (theoretical) |
|---|---|---|---|---|---|---|---|---|
| a The content of aluminium was determined by AAS. Phosphate can be absorbed either by reaction with dissolved Al3+ ions to form solid AlPO4 or by adsorption on solid Al(OH)3 or AlO(OH) (see text). b Depending on the pH during precipitation, a mixture of hydroxyapatite, Ca5(PO4)3OH (HAP), and octacalcium phosphate, Ca8(PO4)4(HPO4)2·5H2O (OCP), will precipitate. The stoichiometry of the precipitate is therefore variable to some extent. The given computations refer to the extreme values of a Ca : P molar ratio of 1.33 (for OCP, binds less phosphate) and 1.67 (for HAP, binds more phosphate). c Sevelamer® is a crosslinked poly(allylamine) hydrochloride (PAH). d The content of chloride was determined beforehand by dissolution of one tablet and subsequent titration. | ||||||||
| Phosphate binders | ||||||||
| Antiphosphat® | 600 mg Al(OH)3 gel | Al3+ | 212 mg (7.86 mmol)a | Aluminium phosphate, AlPO4 | 959 mg (7.86 mmol) as AlPO4 | 747 mg (7.86 mmol) as AlPO4 | 2 | 1494 mg as AlPO4 |
| Calciumacetat-Nefro® | 700 mg Ca(OOCCH3)2 | Ca2+ | 177 mg (4.42 mmol) | HAP and/or OCPb | 444 mg (0.884 mmol, HAP) to 542 mg (0.552 mmol, OCP) | 252 mg (2.66 mmol, as HAP) to 314 mg (3.32 mmol, as OCP) | 3 | 757 to 946 mg |
| Calciumcarbonat® Fresenius. | 500 mg CaCO3 | Ca2+ | 200 mg (4.99 mmol) | HAP and/or OCPb | 501 mg (0.998 mmol, HAP) to 612 mg (0.623 mmol, OCP) | 285 mg (3.00 mmol, as HAP) to 356 mg (3.75 mmol, as OCP) | 3 | 854 to 1068 mg |
| Fosrenol® | 1240 mg La2(CO3)3 | La3+ | 750 mg (5.40 mmol) | Lanthanum phosphate, LaPO4 | 1263 mg (5.40 mmol) | 513 mg (5.40 mmol) | 1 (ground) | 513 mg |
| Renagel® | 800 mg Sevelamer®c | Cationic R-NH3+ groups | 148.4 ± 1.5 mg Cl− (4.19 mmol)4 | H2PO4− ions bound to PAH | 800 + 398 = 1198 mg | 398 mg (4.19 mmol) | 3 | 1193 mg |
| Model compounds | ||||||||
| Aluminium sulfate (p.a., powder) | 2695 mg Al2(SO4)3·16H2O | Al3+ | 230 mg (8.52 mmol)1 | Aluminium phosphate, AlPO4 | 1039 mg (8.52 mmol) as AlPO4 | 809 mg (8.52 mmol) as AlPO4 | — | 809 mg as AlPO4 |
| Calcium acetate (p.a., powder) | 210 mg Ca(OOCCH3)2 | Ca2+ | 532 mg (13.28 mmol) | HAP and/or OCP2 | 1336 mg (2.66 mmol, HAP) to 1631 mg (1.66 mmol, OCP) | 757 mg (7.97 mmol, as HAP) to 946 mg (9.96 mmol, as OCP) | — | 757 to 946 mg |
| Calcium carbonate (p.a., powder) | 1500 mg CaCO3 | Ca2+ | 600 mg (14.99 mmol) | HAP and/or OCP2 | 1502 mg (2.99 mmol, HAP) to 1837 mg (1.87 mmol, OCP) | 854 mg (8.99 mmol, as HAP) to 1068 mg (11.24 mmol, as OCP) | — | 854 to 1068 mg |
| Calcium carbonate (p.a., pellet) | 1500 mg CaCO3 | Ca2+ | 600 mg (14.99 mmol) | HAP and/or OCP2 | 1502 mg (2.99 mmol, HAP) to 1837 mg (1.87 mmol, OCP) | 854 mg (8.99 mmol, as HAP) to 1068 mg (11.24 mmol, as OCP) | — | 854 to 1068 mg |
 | ||
| Fig. 2 Measured pH values in the “stomach” (starting at pH 2) and in the “gut” (starting at pH 7) for a phosphate concentration of 6 mM. (A) Antiphosphat®; (B) Calciumacetat-Nefro®; (C) Calciumcarbonat® Fresenius; (D) Fosrenol®; (E) Renagel®; (F) Calcium carbonate powder; (G) Calcium acetate powder; (H) Calcium carbonate pellet. | ||
 | ||
| Fig. 3 Measured pH values in the “stomach” (starting at pH 2) and in the “gut” (starting at pH 7) for a phosphate concentration of 3 mM. (A) Antiphosphat®; (B) Calciumacetat-Nefro®; (C) Calciumcarbonat® Fresenius; (D) Fosrenol®; (E) Renagel®; (F) Calcium carbonate powder. | ||
The phosphate binding capacity was determined at the exit of the “gut” by monitoring the outcoming phosphate concentration. These data are shown in Fig. 4 and 5. After induction times of less than one hour, the phosphate concentration at the exit decreased strongly, in some cases down to almost zero. Interestingly, all phosphate binders showed a different behavior. In some cases, a distinct minimum of the phosphate concentration was reached after one hour, in other cases, the minimum of the phosphate concentration was broad. After 6 to 7 hours, the phosphate concentration almost regained its original value in all cases. This corresponds to the human digestion system and more or less to the time between two meals. By integration of the area over the phosphate concentration curves, the total amount of absorbed phosphate was computed. For the experiments with a phosphate concentration of 3 mM less phosphate was bound which can be explained by the lower supersaturation (ion product of the insoluble compounds).
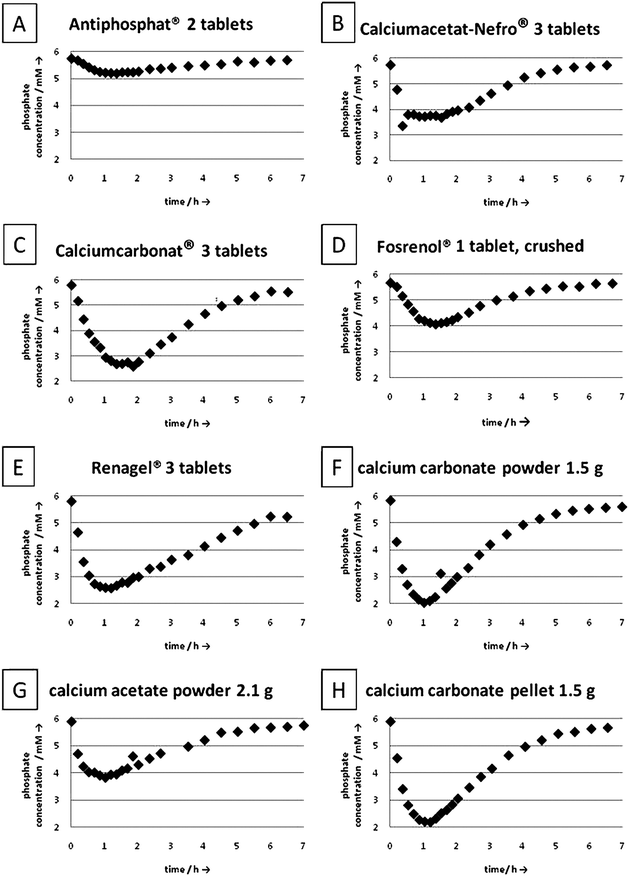 | ||
| Fig. 4 Measured phosphate concentration at the exit of the apparatus for a phosphate concentration of 6 mM. (A) Antiphosphat®; (B) Calciumacetat-Nefro®; (C) Calciumcarbonat® Fresenius; (D) Fosrenol®, (E) Renagel®; (F) Calcium carbonate powder; (G) Calcium acetate powder; (H) Calcium carbonate pellet. | ||
 | ||
| Fig. 5 Measured phosphate concentration at the exit of the apparatus for a phosphate concentration of 3 mM. (A) Antiphosphat®; (B) Calciumacetat-Nefro®; (C) Calciumcarbonat® Fresenius; (D) Fosrenol®, (E) Renagel®; (F) Calcium carbonate powder. | ||
The results for phosphate binding are summarized in Table 2. The degree of phosphate binding was different for all samples. It can be compared to the theoretical value by making assumptions on the nature of the solid product that was precipitated (Table 1). The solid products were collected at the exit of the “gut” and analyzed by X-ray powder diffraction (Fig. 6). Fig. 7 summarizes all phosphate binding capacities in graphical form.
| Sample | 6 mM phosphate concentration | 3 mM phosphate concentration | Solid product according to X-ray diffraction | ||
|---|---|---|---|---|---|
| Bound phosphate/mg | % of theory | Bound phosphate/mg | % of theory | ||
| Phosphate binders | |||||
| Antiphosphat® (2 tablets) | 222 ± 19 | 15 ± 1. | 239 ± 54 | 16 ± 4% | Aluminium phosphate (poorly crystalline) |
| Calciumacetat-Nefro® (3 tablets) | 732 ± 21 | 79 to 99% (±5%) | 427 ± 8 | 46 to 58% (±2%) | Octacalcium phosphate |
| Calciumcarbonat® Fresenius (3 tablets) | 1089 ± 9 | 102 to 128% (±1%) | 969 ± 35 | 91 to 113% (±4%) | Hydroxyapatite |
| Fosrenol® (1 tablet) | 564 ± 18 | 110 ± 3% | 568 ± 17 | 111 ± 3% | Lanthanum phosphate |
| Renagel® (3 tablets) | 1286 ± 34 | 108 ± 3% | 1001 ± 24 | 84 ± 2% | Amorphous |
| Model compounds | |||||
| Aluminium sulfate hydrate (p.a., powder) | 762 | 94% | — | — | Amorphous |
| Calcium acetate (p.a., powder) | 594 | 63 to 79% | — | — | Octacalcium phosphate |
| Calcium carbonate (p.a., powder) | 1117 | 105 to 131% | 913 | 86 to 107% | Hydroxyapatite |
| Calcium carbonate (p.a., pellet) | 1074 | 101 to 126% | — | — | Hydroxyapatite |
 | ||
| Fig. 6 Selected X-ray powder diffractograms of the solid products collected at the exit of the “gut”. The crystalline products were hydroxyapatite (all calcium carbonate samples), octacalcium phosphate (calcium acetate samples), lanthanum phosphate (Fosrenol®), aluminium phosphate and aluminium oxide hydroxide (AlO(OH); diaspor and boehmite) (Antiphosphat®). The product from Renagel® was fully X-ray amorphous. | ||
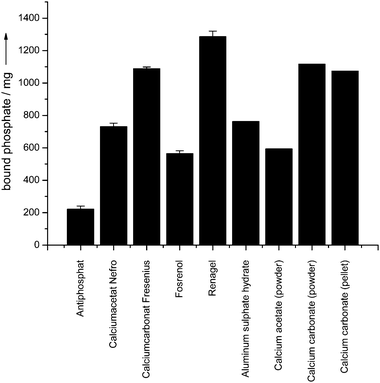 | ||
| Fig. 7 Experimental phosphate binding capacities at a phosphate concentration of 6 mM with respect to the number of tablets used per giving (Table 1). | ||
The phosphate binders showed a different performance in the binding studies. Fig. 7 shows the experimental phosphate binding capacities of all investigated drugs and compounds. In the following, the results are discussed for each phosphate binder and the corresponding model compounds.
Antiphosphat®
For Antiphosphat®, we observed a comparatively low degree of phosphate binding under these experimental conditions, i.e. 15 to 16% of the theoretical value if a formation of aluminium phosphate was assumed. This is due to the insufficient dissolution of the tablet in the “stomach” as it could be visually observed, i.e. not the whole amount of aluminium hydroxide was dissolved. This was confirmed by parallel experiments where we tried to dissolve the ground tablets in diluted and concentrated acids (HCl or H2SO4) and observed a slow and incomplete dissolution. The X-ray diffraction analysis of ground samples of Antiphosphat® showed aluminium oxide hydroxide, AlO(OH), either as diaspor or as boehmite (JCPDS 01-084-0185; 01-076-1871) with very broad peaks, indicating small and disordered crystallites. Only a part of the aluminium oxide hydroxide was dissolved in the “stomach” at low pH and then reprecipitated in the “gut” at neutral pH as aluminium phosphate, AlPO4 (eqn (1)). The analysis of the solid product by X-ray diffraction confirmed the presence of aluminium phosphate, although in a very poorly crystalline state, together with unreacted AlO(OH).| Al3+ (aq) + HPO42− (aq) + H2O (l) → AlPO4 (s) + H3O+ (aq) | (1) |
As control, we used an equivalent amount of aluminium sulfate hydrate (2.695 g, containing 0.23 g aluminium). This salt readily dissolves in water. In this case, the pH in the “stomach” practically did not change, but after a few minutes a drop in the pH in the “gut” was observed, down to about 3 after 30 min. After 3 h, the pH in the “gut” increased again to the original value (pH 6.96 after 7 h). The solid product was fully X-ray amorphous in this case, as proven by X-ray powder diffraction. The phosphate binding capacity was 762 mg, corresponding to 94% of the theory. Aluminium itself is well suited to bind phosphate, but it must be present in a dissolved state, i.e. as ion, Al3+ (aq). Undissolved AlO(OH) cannot convert into aluminium phosphate without prior dissolution; it only adsorbs phosphate ions.10 The slow dissolution of AlO(OH) is underscored by the small change of the pH during the experiments with Antiphosphat®.
A static experiment was performed to verify the phosphate binding capacity of Antiphosphat® (Fig. 8). 2 L of a 6 mM phosphate solution with pH 7 were stirred in a beaker at 37 °C, and the phosphate concentration in solution was monitored. Two tablets of Antiphosphat® were put into the phosphate solution at pH = 7. The tablets disintegrated into small particles but did not dissolve. After one hour, the pH was lowered to 2 by adding 4 mL of concentrated hydrochloric acid, simulating the “stomach”. During the following hour the particles dissolved only partially (the pH rose only very slowly). Then the pH was increased to 7 again with NaOH, simulating the “gut”. The experiment was continued for 4 more hours but no change was observed. After 6 h the experiment was finished and the precipitate was filtered off and dried. The yield was 1.19 g. X-Ray powder diffraction showed a poorly crystalline mixture of AlPO4 and AlO(OH). The phosphate concentration showed a drop at exactly the moment when the pH was increased from 2 to 7. This underscores that only dissolved Al3+ ions are capable to bind significant amounts of phosphate at pH 2 and 7. Only minor amounts of phosphate were bound onto the aluminium oxide/hydroxide surface (eqn (2)):
| AlO(OH) (s) + x PO43− (aq) → AlO(OH)·(PO43−)x (s) (x ≪ 1) | (2) |
 | ||
| Fig. 8 pH and phosphate concentration during a static experiment with Antiphosphat®. Efficient phosphate binding occurs only at pH 7 due to the precipitation of AlPO4. | ||
The two tablets of Antiphosphat® will have a maximum phosphate binding capacity of 1.5 g (15.75 mmol) if the whole aluminium content (425 mg; 15.75 mmol) is used to bind one PO43− ion per Al3+ ion to form AlPO4. This is the upper chemical limit for phosphate binding. The adsorption of phosphate onto the surface of AlO(OH)10 always binds less phosphate. The 2 L of the prepared solution contained 1.11 g (11.72 mmol) phosphate, therefore all phosphate should have precipitated, but only 391 mg (4.12 mmol) phosphate were removed from the solution according to Fig. 7. Antiphosphat® bound only 35% of the phosphate that was present in solution, and reached only 26% of its theoretical phosphate binding capacity with respect to aluminium phosphate formation. These numbers corroborate the low phosphate binding capacities in the continuous experiments. If 4.12 mmol phosphate were bound as aluminium phosphate and the remaining aluminium (11.63 mmol) was still present as AlO(OH), a total mass for the precipitate of 503 mg AlPO4 + 698 mg AlO(OH) = 1201 mg can be computed. This is very close to the mass of the solid precipitate (1190 mg).
Calciumacetat-Nefro®
This material rapidly increased the pH in the “stomach” and considerably decreased the pH in the “gut” due to buffering by the administered acetate (acetic acid has a pKs of 4.76). The precipitation occurred at a pH between 6 and 7 in the “gut” which led to octacalcium phosphate (OCP), Ca8(PO4)4(HPO4)2·5H2O, and not to hydroxyapatite (HAP), Ca5(PO4)3OH, which would preferentially precipitate at a higher pH.11 OCP was identified by X-ray powder diffraction (eqn (3)).| 8Ca2+ (aq) + 6HPO42− (aq) + 9H2O (l) → Ca8(PO4)4(HPO4)2·5H2O (s) + 4H3O+ (aq) | (3) |
However, it cannot be excluded that a mixture with hydroxyapatite was present because these two compounds are difficult to distinguish by XRD and also because OCP can transform into hydroxyapatite.11 Furthermore, precipitated hydroxyapatite is usually calcium-deficient, i.e. it has the formula Ca10−x(PO4)6−x(HPO4)x(OH)2. Because the calcium to phosphate ratio in the final product is determining the phosphate binding capacity for a given amount of calcium, it is difficult to predict the exact phosphate binding capacity. For OCP, the molar Ca : P ratio is 1.33 and for hydroxyapatite it is 1.67. For a calcium-deficient hydroxyapatite (CDHA), the molar Ca : P ratio can range between about 1.5 and 1.67.11 Therefore, the composition of the product can range between OCP and HAP which influences the phosphate binding capacity (see Table 1). The measured calcium binding capacity for Calciumacetat-Nefro® ranged between 46 and 97%, depending on the assumed calcium phosphate phase. This was confirmed by an experiment with pure calcium acetate.
Calciumcarbonat® Fresenius
In this case, the pH in the “stomach” was considerably increased but there was no decrease of the pH in the “gut” as in the case of calcium acetate because carbonate left the “stomach” as gaseous CO2. Therefore, the calcium phosphate was precipitated at a higher pH (between 7 and 8.2), and the more basic product hydroxyapatite was formed as confirmed by X-ray powder diffraction (eqn (4)).| 5Ca2+ (aq) + 3HPO42− (aq) + 5H2O (l) → Ca5(PO4)3OH (s) + 4H3O+ (aq) | (4) |
91 to 128% of the theoretically expected amount of phosphate were bound, again depending on the formed calcium phosphate phase. This was confirmed by control experiments with synthetic calcium carbonate, as powder as well as tablet. All gave the same results, indicating a rapid dissolution of calcium carbonate in the “stomach”, followed by efficient binding in the stomach. Interestingly, the increase of pH in the “stomach” was much faster with the chemically pure calcium carbonate, highlighting the slower dissolution of the pharmaceutically formulated phosphate binder.
Fosrenol®
Fosrenol contains acid-soluble lanthanum carbonate as active ingredient that reacts with acids under release of carbon dioxide (eqn (5)).| La2(CO3)3 (s) + 2H2PO4− (aq) + 2H3O+ (aq) → 2LaPO4 (s) + 3CO2 (g) + 5H2O (l) | (5) |
As solid product, lanthanum phosphate is expected which is practically insoluble under acidic, neutral and basic conditions.12 Therefore, the precipitation occurred already in the “stomach”. Lanthanum phosphate was observed as solid product by X-ray diffraction. The binding capacity was about 110% of the theoretical value. This indicates that the reaction was quantitative, and also that some hydrogen phosphate was bound, e.g. as La1−x/3(PO4)1−x(HPO4)x. An adsorption of phosphate on the surface of lanthanum phosphate also cannot be excluded.
Renagel®
Renagel is binding phosphate by ion exchange of chloride by phosphate from a solid polymer. Therefore, the absolute binding capacity is difficult to predict because it depends on the degree of ion exchange. We have analytically determined the amount of chloride per tablet beforehand. We then assumed an exchange by dihydrogen phosphate H2PO4− which is reasonable at about neutral pH and also if we assume that each ammonium group binds one anion (eqn (6)).| R–NH3+Cl− (s) + H2PO4− (aq) → R–NH3+H2PO4− (s) + Cl− (aq) | (6) |
The nature of the bound phosphate species depends on the pH because phosphoric acid has three dissociation steps. We observed a binding capacity of 84–108%. The mismatch is probably due to a non-quantitative saturation of ammonium groups by chloride in the starting materials and a possible binding of hydrogen phosphate instead of dihydrogen phosphate.
Renagel increased the pH both in the “stomach” and in the “gut”, probably due to protonation of previously unsaturated amine groups (eqn (7)):
| R–NH2 + H3O+ ← → R–NH3+ + H2O | (7) |
X-Ray diffraction showed that the product was amorphous as expected for a non-crystalline, cross-linked polymer. A static experiment (as that shown in Fig. 1 for Antiphosphat® showed that about 1/3 of the phosphate was already bound at pH 2 and the remaining 2/3 after increase of the pH to 7.
Comparison of the phosphate binders
It is instructive to compare the place of phosphate binding (stomach or gut) and also the amount of bound phosphate per tablet weight (Table 3). Calcium carbonate and Renagel showed the highest phosphate-binding capacity in this respect.| Phosphate binder | Place of phosphate binding | Weight of one tablet | Tablets per giving | Amount of phosphate bound (at 6 mM) per served tablet mass |
|---|---|---|---|---|
| Antiphosphat® | Gut | 734 mg | 2 | 151 mg g−1 |
| Calciumacetat-Nefro® | Gut | 906 mg | 3 | 269 mg g−1 |
| Calciumcarbonat® Fresenius | Gut | 698 mg | 3 | 520 mg g−1 |
| Fosrenol® | Stomach | 3139 mg | 1 | 180 mg g−1 |
| Renagel® | Stomach (1/3) and gut (2/3) | 927 mg | 3 | 462 mg g−1 |
All investigated phosphate binders were active and in some cases, the observed phosphate binding capacity was very close to the chemically expected value (Calciumacetat-Nefro®, Calciumcarbonat® Fresenius, and Renagel®). One compound consistently exceeded the expected potential for phosphate binding (Fosrenol®). The largest discrepancy we found was the low phosphate binding capacity of the aluminium containing drug Antiphosphat®. It is noteworthy that aluminium-based phosphate binders are used successfully to reduce hyperphosphatemia since almost forty years.13 As mentioned before, an incomplete dissolution of the active compound aluminium hydroxide is the likely reason. Larson et al.10 reported that different phosphate binders on the basis of aluminium hydroxide vary strongly in their phosphate binding capacity, depending on the crystalline phases in the drug. They showed that boehmite, AlO(OH), and gibbsite, Al(OH)3, were not sufficiently soluble under conditions similar to our apparatus to release Al3+ ions in the large quantities which are required for the precipitation of AlPO4. Nevertheless, phosphate was removed from the system by adsorption to the aluminium hydroxide particles in comparable amounts as in our experiments, with a strong influence of the specific particle surface area.14,15 It is also important to note that dissolved aluminium in the stomach may cause aluminium accumulation in brain or bone,16 therefore it is reasonable to make use of the adsorption of phosphate on the surface of aluminium hydroxide although this reduces the theoretical capacity compared to aluminium phosphate.
The two calcium-containing drugs Calciumacetat-Nefro® and Calciumcarbonat® showed good phosphate binding, especially at high phosphate concentrations, but did not act in the same way. Calcium ions are the active compound in both drugs, and their amount is similar when the drugs are administered in the recommended doses of three tablets per meal. The buffering effect of the acetate ion led to different pH-values in the “gut” during the occurring crystallization processes and therefore to different crystalline products. With Calciumacetat-Nefro® at pH 6, mostly octacalcium phosphate (OCP) precipitated, whereas Calciumcarbonat® at a pH above 8 generated hydroxyapatite (HAP). Therefore, Calciumcarbonat® bound almost 50% more phosphate than Calciumacetat-Nefro® although it contains only 13% more calcium. These results can be explained by the findings of Sheikh et al.17 whose experiments over a wide pH-range showed that phosphate binding capacities of calcium-containing drugs rise with increasing pH value. This indicates that pH-changing drugs (e.g. against reflux) may interfere with phosphate binders. However, in the physiological situation where the residence time in the gut is longer, this pH-dependency may be smaller or even absent.
The other two examined drugs contained neither aluminium nor calcium. Fosrenol® consists of lanthanum carbonate and precipitates LaPO4 in the presence of phosphate18 so that it is comparatively easy to predict how much phosphate should be bound per tablet. Lanthanum phosphate is insoluble under acidic, neutral and basic conditions, contrary to calcium phosphate which is acid-soluble. The theoretical binding capacity was in all cases exceeded by about 10% which leads to the assumption that the precipitate is not totally stoichiometric. Despite this high efficiency, Fosrenol® bound only the second-smallest amount of phosphate per giving. Two tablets of Fosrenol® per giving would easily increase the amount of bound phosphate to that of the other phosphate binders.
The active compound of Renagel®, i.e. Sevelamer® hydrochloride, is a water-insoluble polymer with chloride groups that can be exchanged by other anions, in this case hydrogen phosphate ions. It gave the highest absolute phosphate binding capacity of all drugs and chemicals in our experiments, consistent with other studies.19 Other investigators found a superiority of Fosrenol® compared to Sevelamer®20 because lanthanum carbonate binds phosphate over a wide pH-range and also in the presence of bile-acids (unlike Sevelamer®).
An important limitation of our setup is the flow rate of acid introduction into the “stomach” and of base introduction into the “gut”. This determines the pH change in both compartments after the introduction of the phosphate binders. It is likely that the pH in the gut in the physiological situation is more stable than in our setup. This will influence the nature of the precipitated compounds if their solubility is pH-dependent, notably, for calcium phosphate. The solubilities of lanthanum phosphate, aluminium phosphate, aluminium oxide/hydroxide, and Sevelamer® are almost independent of pH. However, the fact that the phosphate binding capacity was lower for calcium acetate than for calcium carbonate which contradicts earlier results5,21–25 may be explained by this fact.
The phosphate-binding action in relation to the tablet weight showed considerable differences for the five phosphate binders. This may represent a clinical advantage in some cases with respect to patient compliance. We deliberately did not relate the phosphate binding capacity to the price of the daily tablet giving, but this would also give interesting relationships like “mg of phosphate bound per Euro spent”.
Conclusion
A custom-made analytical apparatus is presented which allows the quantitative assessment of the binding capacity of different phosphate binders. This is possible by a thorough control over the experimental conditions and also by the application of quantitative analytical methods. The investigated phosphate binders are based on different chemical mechanisms which bind phosphate. The phosphate binding capacity not only depends on the chemical reaction that binds the phosphate, but also on the dissolution kinetics under acidic (stomach) and neutral conditions (gut). The phosphate binders may change the pH in both organs, thereby in turn influencing the phosphate binding capacity. At higher phosphate concentrations, more phosphate is bound, except for the lanthanum phosphate-based drug Fosrenol® which shows the same activity under both conditions. However, the absolute amount of phosphate bound by a given drug depends on its chemical “efficiency” and also on the amount of chemicals per giving. It can of course be increased by giving more tablets per serving.Acknowledgements
We thank Mrs K. Genesius, Mrs L. Dreyer, Mrs K. Brauner, Mrs V. Hiltenkamp and Mrs C. Fischer for experimental assistance.References
- M. Denda, J. Finch and E. Slatopolsky, Am. J. Kidney Dis., 1996, 28, 596 Search PubMed.
- M. Rodriguez, S. Canadillas, I. Lopez, E. Aguilera-Tejero and Y. Almaden, J. Bone Miner. Metab., 2006, 24, 164 CrossRef CAS.
- J. A. Coladonato, J. Am. Soc. Nephrol., 2005, 16, 107.
- E. Slatopolsky, J. Am. Soc. Nephrol., 2003, 14, S297 CrossRef.
- A. J. Hutchison, Kidney Int., 2009, 75, 906 CrossRef CAS.
- G. A. Block, T. E. Hulbert-Shearon, N. W. Levin and F. K. Port, Am. J. Kidney Dis., 1998, 31, 607 Search PubMed.
- A. S. Go, G. M. Chertow, D. J. Fan, C. E. McCulloch and C. Y. Hsu, N. Engl. J. Med., 2004, 351, 1296 CrossRef CAS.
- G. A. Block, P. Raggi, A. Bellasi, L. Kooienga and D. M. Spiegel, Kidney Int., 2007, 71, 438 CrossRef CAS.
- R. N. Foley, A. J. Collins, C. A. Herzog, A. Ishani and P. A. Kalra, J. Am. Soc. Nephrol., 2009, 20, 397 CrossRef CAS.
- E. A. Larson, S. R. Ash, J. L. White and S. L. Hem, Kidney Int., 1986, 29, 1131 CrossRef CAS.
- S. V. Dorozhkin and M. Epple, Angew. Chem., Int. Ed., 2002, 41, 3130 CrossRef CAS.
- F. H. Firsching and S. N. Brune, J. Chem. Eng. Data, 1991, 36, 93 CrossRef.
- A. C. Alfrey, G. R. Legendre and W. D. Kaehny, New Engl. J. Med., 1976, 294, 184 CAS.
- W. H. Vanriemsdijk and J. Lyklema, J. Colloid Interface Sci., 1980, 76, 55 CrossRef CAS.
- W. H. Vanriemsdijk and J. Lyklema, Colloids Surf., 1980, 1, 33 CrossRef CAS.
- A. B. Hodsman, D. J. Sherrard, A. C. Alfrey, S. Ott, A. S. Brickman, N. L. Miller, N. A. Maloney and J. W. Coburn, J. Clin. Endocrinol. Metab., 1982, 54, 539 CAS.
- M. S. Sheikh, J. A. Maguire, M. Emmett, C. A. Santa Ana, M. J. Nicar and L. R. Schiller, J. Clin. Invest., 1989, 83, 66 CrossRef CAS.
- K. Breitenborn, Dtsch. Arztebl., 2006, 103, 3359 Search PubMed.
- D. P. Rosenbaum, S. R. Holmes-Farley, W. H. Mandeville, M. Pitruzzello and D. I. Goldberg, Nephrol., Dial., Transplant., 1997, 12, 961 CrossRef CAS.
- V. Autissier, S. J. P. Damment and R. A. Henderson, J. Pharm. Sci., 2007, 96, 2818 CrossRef CAS.
- F. Caravaca, I. Santos, J. J. Cubero, J. F. Esparrago, M. Arrobas, J. L. Pizarro, R. Robles and E. Sanchez-Casado, Nephron, 1992, 60, 423 CrossRef CAS.
- P. Morinière, M. Djerad, B. Boudailliez, N. El Esper, F. Boitte, P. F. Westeel, M. Compagnon, M. Brazier, J. M. Achard and A. Fournier, Nephron, 1992, 60, 6 CrossRef CAS.
- F. Ben Hamida, I. el Esper, M. Companon, P. Moriniere and A. Fournier, Nephron, 1993, 63, 258 CrossRef CAS.
- L. Jegadeesan, B. Ramakrishna, G. T. John and J. C. M. Shastry, Nephrology, 1996, 2, 53 CrossRef.
- M. Wallot, K. E. Bonzel, A. Winter, B. Geörger, B. Lettgen and M. Bald, Pediatr. Nephrol., 1996, 10, 625 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |