Ochratoxin A nanostructured electrochemical immunosensors based on polyclonal antibodies and gold nanoparticles coupled to the antigen
L.
Bonel
,
Juan C.
Vidal
*,
P.
Duato
and
Juan R.
Castillo
Analytical Spectroscopy and Sensors Group (GEAS), Institute of Environmental Sciences (IUCA), University of Zaragoza, c/ Pedro Cerbuna, 12, 50009, Zaragoza, Spain. E-mail: jcvidal@unizar.es; Fax: +34 976 761292; Tel: +34 976 762253
First published on 8th February 2010
Abstract
Competitive electrochemical immunosensors, based on disposable screen-printed electrodes (SPCEs), have been developed for the determination of the mycotoxin ochratoxin A (OTA). Two indirect immunoassays schemes were assessed using polyclonal antibodies and through the physical adsorption of OTA conjugated to albumin from bovine serum (OTA-BSA) and newly synthesized OTA-BSA bound to gold nanoparticles (OTA-BSA-AuNPs) onto the working electrode surface. After the competition step, detection was facilitated by a secondary aIgG antibody labelled with alkaline phosphatase and differential-pulse voltammetry using α-naphtyl-phosphate as substrate. The performance of the optimized immunosensors in terms of sensitivity, reproducibility and selectivity was studied. The linear working range of the described biosensors ranged between 0.9 and 9.0 ng mL−1 for the OTA-BSA based immunosensor and between 0.3 and 8.5 ng mL−1 for the gold nanostructured immunosensor, with a limit of detection (LOD) equal to 0.86 ng mL−1 (RSD = 10.6%) and 0.20 ng mL−1 (RSD = 8.0%) of OTA, respectively. The nanostructured immunosensor was applied to a certified wheat standard and a non-contaminated wheat material spiked with OTA, obtaining recoveries from about 104 ± 0.07% to 107 ± 0.08%. The influence of the wheat matrix on the analytical performance of the immunoassay was also studied.
Introduction
Mycotoxins are secondary metabolites of a range of filamentous fungi with deleterious effects for humans and animals, which can be found in agricultural commodities and animal foodstuffs.1 They are present in trace amounts, ranging from below to few nanograms per gram of sample, and therefore, there is a need for highly sensitive and selective analytical methods for these organic toxins.2 Of among all the groups of mycotoxins (aflatoxins, ochratoxins, fumonisins, zearalenone, citrinin, ergot alkaloids, etc.), ochratoxin A (OTA) is one of the most frequent contaminant of several foods and feeds, it is nephrotoxic and carcinogenic and poses a serious threat to the health of both humans and animals. For these reasons, the regulatory legislation of the European Community enforcing food safety is more restrictive every time.3Instrumental methods for the analysis of OTA use mainly chromatographic based techniques,4 mostly based on high pressure liquid chromatography (HPLC) assisted with molecular fluorescence detection (FLD).5,6 A number of methods have been validated by inter-laboratory collaborative studies, many of them under the auspices of international organisms such as AOAC International.7 Chromatographic methods generally require multiple sample preparation steps prior to detection, including extraction, sample clean up, preconcentration and sometimes derivatization of the analyte to improve the detection sensitivity.8 Particularly, sample preconcentration using solid phase extraction (SPE) and immunoaffinity columns (IAC) are very common in official methods of mycotoxin analysis using HPLC-FLD.9
To overcome the above limitations, a variety of immunoassay methods have been developed with isolated monoclonal and polyclonal antibodies raised against the specific mycotoxins,10–12 particularly for OTA.13–15 The very selective immunological basis of such methods makes them highly specific and, therefore, less dependent on sample cleanup. So far, enzyme-linked immunosorbent assay (ELISA) is the most common immunoassay technique used in OTA analysis due to simplicity and capability for parallel analysis of multiple samples.13,16,17
A number of ELISA test kits have also been commercially developed for many important mycotoxins, including OTA, which are useful tools for screening and quantification at low ppb levels18 and may be used for in situ measurements of mycotoxin occurrences. They have benefits in speed and sensitivity, but might have some drawbacks such as cross reactivity and matrix dependence which would result in over-estimation of the analyte concentration (possibility of false-positive results).16,19
More recently, the use of biosensors allows a major portability, in situ analysis of OTA with similar selectivity and sensitivity to ELISAs, simultaneously with minor consumption of reagents and much shorter times of analyses.20–23 They are mostly based on immunological principles (immunosensors), and in many recent years have also appeared devices based on specific aptamers against OTA (aptasensors).24
Amperometric immunosensors that use antibodies (or antigens) attached to the surface of electrodes are especially suitable in this sense,25 particularly using disposable screen-printed electrodes (SPCEs).26 Direct or indirect immobilizations onto the SPCEs are based on distinct functional reagents such as diazonium organic salts25 or biotin/avidin chemistries,27 or by simple physical adsorption of the proteins. Limits of detection of immunosensors on SPCEs lie in the order of 0.5–20 ng mL−1 and dynamic ranges up to concentrations of about 1–100 ng mL−1 of the mycotoxins. For instance, a surface-adsorbed antibody against aflatoxin B1 (AFB 1) gives a detection limit of around 0.15 ng mL−1 in buffer solution.28 Nitrocellulose membrane-based ELISA immunoassays have also been described for the simultaneous screening of aflatoxin B1 and OTA in chilli samples, with limits of quantitation of 2 and 10 ng g−1 respectively.29 Palleschi et al. have described a monoclonal antibody based electrochemical immunosensor for the determination of OTA, with limits of detection in the range 60 to 100 μg L−1 in direct and indirect assay formats, although the synthesis of an OTA-alkaline phosphatase conjugate is necessary for revealing the signals of the biosensors.15
On the other hand, metallic nanoparticles, in particular gold nanoparticles (AuNPs), are nowadays of great interest in new generation of bioelectronic devices with increased sensitivity, high biocompatibility and novel functions.30 Particularly, some different nanostructured surfaces on SPCEs immunosensors have been proposed, like electrochemical generated AuNPs from AuCl4−,31 adsorbed AuNPs stabilized by the synthetic membrane-like compound didodecyl-dimethyl-ammonium bromide,32 labelling of secondary antibodies,33,34 localization of colloidal gold-labeled protein A on the antigen-antibody immune complex,35 stabilizing the gold colloid with BSA and glutaraldehyde,36 PAH (poly allylamine hydrochloride)-modified, nanosized-Au particles assemblies,37 synthesis of the nanoparticles on conducting polymers38 or drop-coating AuNPs onto polythionine modified electrode surface for the determination of aflatoxin B1.39
In a previous paper, we have characterized kinetics and affinity of polyclonal antibodies against OTA by measuring small mass changes with a quartz-crystal microbalance (QCM),40 which was applied to QCM immunosensors for the mycotoxin. Covalent attachment of the OTA-BSA conjugates through gold nanoparticles produced mass-amplified signals, but this enhancement did not reach to detect some of the required very small concentrations of OTA necessary for certain kind of samples.
In this paper, indirect competitive immunosensors for OTA are developed with the same polyclonal antibodies, but using disposable screen-printed electrodes and differential-pulse voltammetry. Additionally, improvements of nanostructured immunosensors with newly synthesized molecules of the antigen bound to gold nanoparticles are also studied. Sensitivity was greatly improved with regard to our previously developed QCM immunosensors for OTA.40
Experimental
Materials and equipment
Screen-printed electrodes (SPCEs) were made in-house in a multistage deposition process onto 1 mm thick substrate of flexible fr-4 epoxy resin sheets. Graphite based ink (Electra ED7100) was used for the working electrode. A carbon based counter electrode and a silver pseudo-reference electrode were also printed onto the same substrate. In the final step, a layer of insulating ink was screen printed over the electrodes to give the disk shaped exposed working electrode (Ø = 4 mm, 0.126 cm2). The intra batch variation of ten lots of ten electrodes, calculated via the chronoamperometric determination of the bare working area of each electrode in 80 μL of a solution of α-naftol 10 mg L−1, was observed to be less than 10% (n = 10), indicating good reproducibility. The printed foils were stored dry, at room temperature, in the dark. Before using, the screen printed electrodes surface were electrochemically pretreated in 70 μL of 0.2 M H2SO4 with three cyclic voltammetry potential scans carried out between −1.2 V and +1.5 V at a scan rate of 100 mV s−1.All electrochemical measurements were performed on an Eco Chemie potentiostat model PGSTAT 1212 (Utrecht, Netherlands). The immunosensor measurements were carried out at room temperature by using differential-pulse voltammetry (DPV) with the following parameters: range potential from 0 to +600 mV (1-naphthol), or from 0 to −800 mV (for p-benzoquinone formed by HRP), step potential 2 mV, scan rate 10 mV s−1, modulation amplitude 30 mV and modulation time 70 ms.
Chemicals
Ochratoxin A (OTA) from Aspergillus Ochraceus (Aspergillus Oryzae) (≥98%) was obtained from Scharlab (Barcelona, Spain). Aliquots from a stock solution of OTA (100 μg mL−1, 10 mL) were stored at −20 °C, and diluted at room temperature with PBST buffer to the desired concentrations when required. Bovine serum albumin (>98%) (BSA), casein from bovine milk, gold colloidal nanoparticles (20 nm diameter, ∼0.01% HAuCl4, ref. G1652), ochratoxin A conjugated to BSA (OTA-BSA, about 3.8 molecules of OTA in one molecule of BSA), anti-rabbit IgG-Peroxidase (aIgG-HRP, 1.0 mg mL−1), anti-rabbit IgG-Alkaline Phosphatase (aIgG-AP, 1.1 mg mL−1), α-naphthyl phosphate disodium salt, hydroquinone (≥99%), and hydrogen peroxide (30%) were purchased from Sigma-Aldrich (Madrid, Spain). Polyclonal rabbit antibody against Aspergillus Ochraceus OTA (pAbOTA, 1 mg mL−1) was from AbD Serotec (Madrid, Spain). Other analytical reagents were from Merck (Darmstadt, Germany). Wheat certified reference material BCR-471 (OTA <0.6 μg Kg−1 at 95% level) was from IRMM (European Comission) and certified by BCR. OTA in wheat flour material (ref. B-MYC0880, lot#M07453Z), with a certified mass concentration of 2.7 ± 1 μg Kg−1 at a 95% level, was from Biopure (Tulin, Austria).Immunosensor assays with pAbOTA
All steps of the immunoassays and electrochemical measurements were carried out onto the surface of the SPCEs. In between the sequential steps (coating, binding, competition and revealing) the surface of the electrodes were carefully washed by duplicate with PBST buffer (0.1 M phosphate buffer + 0.138 M NaCl + 0.05% Tween-20, pH 7.4). First, 10 μL of 3 μg mL−1 of the OTA-BSA conjugate in CB buffer (0.05 M carbonate, pH = 9.6) was deposited onto the working electrode of the SPCEs and left to adsorb for 20 h at 4 °C. Then 10 μL of a blocking solution of 1% (w/v) casein in PBST buffer was added and left at 37 °C for 30 min. Competitive tests were made with addition of 10 μL of pre-mixed solutions (they were vortexed for about 10 s in eppendorf vials) made with 10 μL of OTA standards in PBS buffer (0–1000 μg L−1) or sample, and 10 μL of pAbOTA antibody (3 mg L−1), following incubation during 30 min at 37 °C. After washing, 10 μL of the secondary antibody aIgG-AP (1.1 mg L−1, dilution 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000) in PBS containing 1% casein was left to react for 30 min at 37 °C. At last, 60 μL of a solution α-naphthyl phosphate 1 mg mL−1 in buffer DEA (0.1 M diethanolamine buffer + 1 mM MgCl2 + 100 mM KCl + 1% casein, pH 9.6) was deposited onto the screen-printed and after 5 min the product of the enzymatic reaction (α-naphtol) was determined by DPV.
:1000) in PBS containing 1% casein was left to react for 30 min at 37 °C. At last, 60 μL of a solution α-naphthyl phosphate 1 mg mL−1 in buffer DEA (0.1 M diethanolamine buffer + 1 mM MgCl2 + 100 mM KCl + 1% casein, pH 9.6) was deposited onto the screen-printed and after 5 min the product of the enzymatic reaction (α-naphtol) was determined by DPV.
Synthesis of OTA-BSA-AuNPs conjugate
The OTA-BSA-colloidal gold conjugate (OTA-BSA-AuNPs) was prepared by adding 48.5 μL of 1 mg mL−1 of OTA-BSA conjugate to 1 mL colloid gold solution (∼0.01% HAuCl4) while gently stirring and adjusting to pH = 6.0 with 0.1 M NaOH. The mixture was incubated at room temperature for 1 h, then 200 μL of PBS with 5% BSA was added into the mixture to stabilize the colloidal gold solution and the resultant was additionally incubated for 30 min. Upon centrifugation at 13400 rpm for 10 min, two phases were obtained: a clear to pink supernatant of unbound OTA-BSA and a dark red sediment of conjugate. The supernatant was discarded and the sediment was rinsed by re-suspending in 1 mL of PBS with 0.1% BSA and collected after a second centrifugation (13400 rpm) for 10 min. At last, the conjugate was re-suspended in 100 μL of tris-buffered saline buffer (20 mM tris + 150 mM NaCl, pH 8.0 adjusted with HCl), containing 0.1% BSA added to minimize nonspecific adsorption and increase the stability of the conjugate during the immunoassays. The nanoparticle conjugates were stored at 4 °C for more than 1 month without significant decrease in activity.Immunosensor assays with polyclonal antibodies and gold nanoparticles
The immunosensor assay procedure is similar to that explained before but with slight changes, and it was also based on an indirect competitive format. The immobilization of the antigen was performed in this case with 10 μL of AuNPs-BSA-OTA conjugate (2.5 mg L−1) in 0.5 M. CB buffer (0.05 M carbonate, pH = 9.6) and kept overnight at 4 °C. An incubation temperature of 4 °C was applied to diminish the effect of possible nonspecific interactions that is more likely to arise with higher temperatures. The binding or competition studies were carried out by adding 10 μL of pAbOTA solutions or 10 μL of pre-mixed solutions of OTA + pAbOTA respectively, following incubation at 37 °C during 30 min. The competitive solutions were prepared in eppendorf vials by mixing (vortexing for about 10 s) 10 μL of OTA standard solutions (0–1000 μg L−1 in PBS buffer) or sample, with 10 μL of pAbOTA antibody (5 μg mL−1), and they were dispensed onto the SPCEs in about 10 s. All the other blocking, washing and revealing steps were as explained before.Analysis of wheat extracts
Calibration curves for OTA were produced by spiking the non-contaminated wheat extracts with OTA standard solutions. For this purpose, aliquots of the non-contaminated wheat powder (1 g) were extracted with 10 mL of acetonitrile:water 6:4 (v/v) by shaking on an orbital shaker for 20 min at room temperature. Extracts were cleared by centrifugation (10 min, 3000 g), and 1 mL of the supernatants was mixed with 4 mL of OTA standards in PBST buffer for detection by the immunosensor.
To determine the OTA in the certified material, 1 g of the powder sample (or non-contaminated wheat spiked with OTA standards) was extracted with 10 mL of the extraction solvent (acetonitrile:water 6:4 (v/v)), mixed in a rotating stirrer for 5 min and centrifuged at 3000 rpm for 10 min, 1 mL of the supernatant was then diluted in 4 mL of PBST buffer and used for the detection of OTA. Spiked samples of OTA (5 and 10 ng g−1), used recovery studies, were prepared 1 day prior to extraction by adding the appropriate volume of a methanolic OTA standard solution to 5 g of the non-contaminated wheat powder and were shaken for 20 min before extraction. The concentrations of OTA in wheat extracts were determined from the calibration curve and it was used to calculate the concentration of the original samples.
Results and discussion
Immunosensor assays using polyclonal antibodies: optimization of the experimental factors
The main objectives of this work are the development of optimized pAbOTA immunosensors for detecting OTA and the improvement of the analytical sensitivity due to nanostructuration using gold nanoparticles (AuNPs). Sandwich immunoassay schemes are not possible with OTA due to its small molecular weight, and only indirect competitive immunoassays were considered.The immunoassay schemes used are depicted in Fig. 1 and they were based on immobilization of OTA-BSA (Fig. 1A) or OTA-BSA-AuNPs (Fig. 1B) conjugates onto the SPCEs. Calibration and binding curves were obtained entirely onto the modified SPCE surface by adding a fixed quantity of antibody (pAbOTA) which competes with the immobilized antigen and OTA in solution, and the amperometric currents were revealed through secondary antibodies labelled with alkaline-phosphatase (aIgG-AP).
 | ||
| Fig. 1 Scheme of the two immunosensor designs: a) coating with OTA-BSA onto the working surface electrode; b) nanostructuration by coating with OTA-BSA-AuNPs. | ||
Checkboard titrations of OTA-BSA-AuNPs, pAbOTA and secondary aIgG-AP concentrations were optimized, as in a conventional ELISA spectrophotometric assay, to obtain the maximum electrochemical sensitivity. This optimisation was of paramount importance since the behaviour of antibodies fixed onto the SPCEs solid supports (in heterogeneous phase) can vary considerably with respect to their performances in solution. After characterization of the affinity through these binding curves, competitive conditions between immobilised OTA, pAbOTA and free OTA in solution were also optimized.
The unspecific adsorption of the primary and secondary antibodies on the carbon surface of the SPCEs can seriously affect the performance of the immunosensor protocols, for what a blocking agent is necessary. For this purpose, additions of 10 μL of BSA and casein proteins 0.01%, 0.1% and 1% (w/v) in PBS were compared as blocking agents. Measurements with bare and blocked SPCEs after incubating solutions of pAbOTA (1:1000)+aIgG-AP (1:1000), without antigen, following the electrochemical procedure, showed a significant decreasing of the currents with the protein modified electrodes. Better results were obtained for casein 1% (w/v) (blocking percentage about 92%) than with BSA 1% (w/v) (about 78%).
The sensitivities of two secondary aIgG-AP and aIgG-HRP antibodies, necessary for revealing the extent of reaction of the pAbOTA, were compared. The working electrode surfaces were modified with a fixed addition of 10 μL of 2.5 μg mL−1 of OTA-BSA, saturated with 10 μL of pAbOTA 5 μg mL−1 and blocked with 10 μL of casein 1% (w/v). Then, 10 μL of the secondary antibody solutions (in a range of concentrations 0–100 mg L−1) were added and the final currents measured after following the immunoassay protocol. The currents obtained are plotted in Fig. 2. The substrate solution used for HRP was 5 μL of 18 mM hydroquinone and 45 μL of the co-substrate 1.1 mM hydrogen peroxide in PBS buffer (0.1 M PBS + 1% BSA, pH = 7.4). The background currents measured were similar in both cases (indicating similar non-specific adsorption), whereas the maximum currents measured with the secondary antibody are higher with aIgG-AP (about 755 ± 5.1 nA) than with aIgG-HRP (about 455 ± 9.2 nA). Higher amounts of 1 mg L−1 of the secondary antibodies did not increase the currents in both cases (Fig. 2). We decided to use in further studies the aIgG labelled with AP for the voltammetric detection due to their best sensitivity with respect to the HRP labelled antibody. From a number of substrates that can be used as electrochemical mediator for alkaline-phosphatase (AP), we decided to use α-naphthyl phosphate as substrate because of the good redox properties of the α-naphthol, which is oxidised under irreversible diffusion-controlled conditions at the SPCEs.
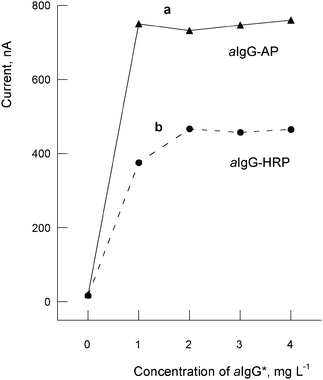 | ||
| Fig. 2 Influence of the enzyme label and concentration of the secondary antibody on the measured currents: a) aIgG-AP (10 μL); b) aIgG-HRP (10 μL). Data in x-axis are not in scale. | ||
The currents obtained did not increased with incubation times between OTA-BSA and pAbOTA greater than 30 min at 37 °C (times from 15 to 60 min were studied). Neither the signals increased with reaction times between pAbOTA and aIgG-AP longer than approximately 25 min at the same temperature. The highest currents are measured with incubation times between the substrate (α-naphthyl phosphate) and the enzyme (AP) of 6–9 min. Nevertheless, the current increases for more than 5 min are very small (about 50.6 ± 2.3 nA), so we used this time. In this way, the assay is quicker and contributes to obtain a less time consuming device, which is very important in the development of commercial immunosensors.
For the best competitive conditions, binding curves of dilutions of pAbOTA (1/10 to 1/1000) with different previously coated OTA-BSA antigen concentrations (1 to 5 μg mL−1) were obtained following the indicated procedure and with aIgG-AP 1.1 mg L−1 (1/1000 dilution). The pAbOTA quantity gives the excess with respect to analyte that competes with the sites of the immobilized antigen. The optimum dilutions of the pAbOTA (at different coated antibody concentrations) are a compromise between the maximum difference of the currents (bound-unbound) and the inflection points of the titration curves (EC50). The current asymptote differences (at zero and maximum analyte concentrations) at the binding curve increases were very small when increasing the antibody concentrations (about 15–25 ± 0.36 nA, possibly due to saturation of all accessible sites of the immobilized antigen reacted), and only a much higher antibody concentration produced a slight decrease of EC50. A pAbOTA concentration equal to 3 mg L−1 was chosen as optimum.
Competitive calibration curves using standard solutions of OTA (from 0 to 1000 ng mL−1) under the optimized conditions are shown in Fig. 3a. The currents obtained by DPV measurements increase with a decrease of OTA concentrations, a typical behaviour of an indirect competitive immunoassay. Three replicates were made of each point of the calibration. The standard curves were fitted by nonlinear regression using the 4-parameters logistic (4-PL) equation:41

 | ||
| Fig. 3 Standard calibration plots obtained for OTA standard solutions (0–1000 ng mL−1) in PBS using the SPCEs immunosensors: a) Coating with OTA-BSA (10 μL, 2.5 μg mL−1); b) Coating with conjugated antigen bound to nanoparticles (OTA-BSA-AuNPs) (10 μL, 3 μg mL−1); c) Same as in b) but spiking with OTA non-contaminated wheat-extracts. Error bars represents the relative deviation of the three measurements of each point (n = 3). | ||
| LOD a/ng mL−1 | EC50b/ng mL−1 | Linear Range/ng mL−1 | CV/(%) | |
|---|---|---|---|---|
| a LOD, limit of detection = (blank − 3 sd) of the zero point. b Concentration of OTA at 50% inhibition. c Calibration curves for OTA in PBST buffer. d Calibration curve in “blank” wheat extracts. | ||||
| OTA-BSAc | 0.86 | 2.17 | 0.9–9.0 | 10.6% |
| OTA-BSA-AuNPsc | 0.20 | 0.68 | 0.3–8.5 | 8.0% |
| OTA-BSA-AuNPsd | 0.21 | 0.72 | 0.4–7.2 | 7.8% |
Sensitivity of the OTA-BSA immunosensor was high (EC50 = 2.17 ng mL−1 OTA). The linear concentrations ranged from 0.9 to 9.0 ng mL−1, with a detection limit of 0.86 ng mL−1. The intra-electrode reproducibility was 10.6% rsd (n = 3 replicates) and the inter-assay repeatability about 15% rsd (n = 3). Once the antigen is immobilized on the SPCEs, the time consumed for the immunosensor measurements is about 2 h.
Immunosensor assays using antigen-gold nanoparticles nanostructuration
The use of AuNPs was considered with the purpose of increasing the quantity and accessibility of the immobilized OTA, besides an improvement of the amperometric currents due to dispersed metallic particles at the nanoscale. For this purpose, the OTA conjugated to BSA was covalently bound to the gold nanoparticles before being immobilized onto the working electrode surface. The competitive immunoassay scheme is similar to the previous one (Fig. 1B), but the presence of the NPs produced slight changes in the optimum concentrations of the immunoreagents. It was expected that nanostructuration with 20 nm gold particles would increase the accessibility of the conjugated OTA with higher number of effective antigen sites on the same surface of the working electrode, besides an increase of the charge-transfer kinetics in voltammetric measurements.As before, binding curves of pAbOTA (from 0 to 10 μg mL−1) at different previously coated (incubated overnight at 4 °C) OTA-BSA-AuNPs (from 0 to 4 μg mL−1), using a fixed concentration of aIgG-AP (1.1 μg mL−1, dilution 1:1000) were compared to asses optimal concentrations of the immunoassays. The saturation of the working electrode with OTA-BSA-AuNPs was obtained by coating with 10 μL of 2.5 μg mL−1 concentration, which were reacted using an excess of the antibody (pAbOTA, 10 μL, 8 μg mL−1). Maximum currents were about 1160 ± 72.8 nA (CV = 6%) at these conditions (higher than with immunosensor without nanoparticles), which signifies an increased sensitivity (due to nanoparticles) and also the maximum accessible sites of the antigen (more concentration of immobilized OTA-BSA-AuNPs did not increase signals). The optimal concentration of pAbOTA determined at about EC50 of the binding curves (defined as 50% of the inhibition concentration) for making a competitive assay was 5 μg mL−1.
Without pAbOTA in the competition step, the currents measured were similar with and without the antigen. The low measured currents (80 ± 6.6 nA) in such conditions are correlated with a low non-specific binding of the secondary antibody.
The competitive binding curve for OTA standard solutions with pAbOTA (5 μg mL−1) is plotted in Fig. 3, the corresponding mathematical data (using the four-parameter logistic equation) are in Table 1, and the analytical performance of this immunosensor is summarized in Table 2. As the reproducibility of the AuNPs immunosensor (OTA-BSA-AuNPs) was better than with the first one (OTA-BSA), especially with the highest currents at zero OTA concentrations, LOD was lower due to nanostructuration (1.5 ng mL−1, Table 2). The sensitivity was also better with the OTA-BSA-AuNPs as the smaller value of EC50 (0.68 g mL−1 OTA), and the slope of the linear part is also higher, as it can be seen in Tables 1 and 2. The linear range is slightly broader (0.3 to 8.5 ng mL−1 of OTA) with this nanostructured biosensor.
Determination of OTA in wheat samples
The more sensitive nanostructured immunosensor was applied to wheat extracts. The principal intention of the immunosensors is to minimize the matrix effects of real samples, owing to the selectivity of the appropriately selected primary antibody. The sample preparation procedures should also minimize matrix interferences and inhibition effects of the immunoassays.At first, evaluation of the matrix effect in immunoassays was carried out, and secondly the study of the extraction efficiency of OTA from contaminated (or spiked) wheat samples to verify the accuracy of the biosensors. For this purpose, we used two sample materials: a non-contaminated wheat powder with a certified content of OTA <0.6 μg Kg−1 (95% level) and a contaminated wheat flour powder with an OTA content of 2.7 ± 1 μg Kg−1 (certified at a 95% level).
As said, a variety of food commodities are subjected of OTA contamination, the most important cereals and cereal products. Conventional analysis of solid samples usually begins with extraction in organic (e.g. chloroform, hexane-acetonitrile, dichloromethane-ethanol, or ethyl acetate) or in aqueous-organic solvents (e.g. methanol-water, acetonitrile-water), containing additives like sodium hydrogen carbonate or magnesium chloride to enhance solubility and extraction efficiency of OTA. We used an extraction procedure to liberate OTA from the samples which was based on the AOAC official method 2000.3 for ochratoxin in barley,8 with minor changes, extracting the analyte by blending with acetonitrile-water (6:4 v/v) as explained.
To evaluate the matrix effect on the OTA-pAbOTA interaction, binding curves were carried out by preparing different dilutions of OTA in extracts of non-contaminated wheat. The phosphatase alkaline activity was not affected by small amounts of acetonitrile used for extraction, in accordance with other authors,15 but sensitivity changes were produced by the wheat extracts matrix (Table 1). The binding curve of the spiked wheat extracts (Fig. 3 and data of Table 2) showed minor differences between the currents at the top and the maximum (i.e. the currents of the ends of the 0 and maximum competition) with respect to that obtained without the matrix of the wheat-extracts (Fig. 3), as well as a decreased slope of the linear part of the graph, which would signify the partial inhibition of the OTA with pAbOTA reaction. Nevertheless, because the value of EC50 is only slightly higher due to wheat matrix (0.72 against 0.68, Table 1), the immunosensor allows the determination of concentrations of OTA with similar sensibility. The detection limit for wheat sample extracts was 0.72 ng mL−1 of OTA (Table 2), based on a signal to noise ratio of 3:1.
To verify the accuracy of the biosensors, the certified and spiked with OTA wheat samples were extracted and analysed for recovery calculations using the calibration curves of the matrix extracts. Recovery was estimated by analysing spiked control samples against non extracted standards. The recovery experiments were made by duplicate, and the OTA concentration was calculated by interpolation in the 4-PL calibrating equation of the “blank” extracts. The results are summarized in Table 3. The average recoveries of OTA ranged from about 104 ± 7% to about 107 ± 7.9%, with RSD lower than about 8%, which show the agreement between measured and nominal concentrations of the spiked and the certified samples.
Conclusions
There is an important need for fast, easy-to-use devices for detection of mycotoxins in complex matrixes, such as cereals, wine, or beer. In this context, disposable biosensors (SPCEs) capable of measuring in situ very few ppbs of OTA, with rapidity and small instrumentation, suppose a big advance in mycotoxin analysis. Voltammetric immunosensors combine the high selectivity of the immunoanalysis with the ease and sensitivity of the electrochemical detection. We developed and compared analytical properties of two OTA-BSA and OTA-BSA-AuNPs indirect competitive immunosensors onto disposable SPCEs, of which the second nanostructured biosensor reaches more sensitivity (EC50 = 0.68 ng mL−1) and lower LOD (1.5 ng mL−1 of OTA) due to more accessible sites of the immobilized antigen on the surface of the electrode and increased electrocatalyzed currents due to gold nanoparticles. The parameters that can influence the immunoassay procedure coupled to the affinity reaction of OTA with pAbOTA were previously optimised using the SPCEs by electrochemical measurements using alkalino-phosphatase labelling (more sensitive than DPV measurements with HRP enzyme). These immunosensors are capable of measuring OTA in wheat samples with a linear range of 0.4 to 7.2 ng g−1 of OTA, with a limit of detection of 0.21 ng g−1 OTA, and with recoveries of OTA from extracted wheat samples ranged from about 104 ± 7% to 107 ± 7.9%. The extraction procedure and the nanostructured immunosensor allow the determination of OTA below EU fixed regulatory limits for cereals.Acknowledgements
This work has been financed by the Aragon Government (Science, Technology and University Department) with Project PM 027/2007. We also should like to thank the ACP Company (Aragonesa de Componentes Pasivos S.A., Tarazona, Spain) for the supported grant of Ms Patricia Duato.References
- J. W. Bennett and M. Klich, Clin. Microbiolog. Rev., 2003, 16, 497 Search PubMed.
- I. Kralj Cigic and H. Prosen, Int. J. Mol. Sci., 2009, 10, 62 Search PubMed.
- H. P. van Egmond, R. C. Schothorst and M. A. Jonker, Anal. Bioanal. Chem., 2007, 389, 147 CrossRef.
- N. W. Turner, S. Subrahmanyam and S. A. Piletsky, Anal. Chim. Acta, 2009, 632, 168 CrossRef CAS.
- E. Chiavaro, A. Lepiani, F. Colla, P. Bettoni, E. Pari and E. Spotti, Food Addit. Contam., 2002, 19, 575 CrossRef CAS.
- G. S. Shephard, A. Fabiani, S. Stockenstrom, N. Mshicileli and V. Sewram, J. Agric. Food. Chem., 2003, 51, 1102 CrossRef CAS.
- M. W. Trucksess, C. M. Weaver, C. J. Oles, F. S. Fry, G. O. Noonan, J. M. Betz and J. I. Pader, J. AOAC Int., 2008, 91, 511 CAS.
- W. C. Stephen and K. P. Kwong, J. AOAC Int., 2007, 90, 773.
- N. H. S. Ammida, L. Micheli, S. Piermarini, D. Moscone and G. Palleschi, Anal. Lett., 2006, 39, 1559 CrossRef CAS.
- A. Logrieco, D. W. M. Arrigan, K. Brengel-Pesce, P. Siciliano and I. Tothill, Food Addit. Contam., 2005, 22, 335 CrossRef CAS.
- A. L. Sun, Q. A. Qi, Z. L. Dong and K. Z. Liang, Sens. and Instrum. Food Qual., 2008, 2, 43 Search PubMed.
- I. Y. Goryacheva, S. De Saeger, S. A. Eremin and C. Van Peteghem, Food Addit. Contam., 2007, 24, 1169 CrossRef CAS.
- A. Visconti and A. De Girolamo, Food Addit. Contam., 2005, Suplement 1, 37 CrossRef.
- F. Y. Yu, T. F. Chi, B. H. Liu and C. C. Su, J. Agric. Food Chem., 2005, 53, 6947 CrossRef CAS.
- S. H. Alarcon, G. Palleschi, D. Compagnone, M. Pascale, A. Visconti and I. Barna-Vetró, Talanta, 2006, 4, 1031 CrossRef.
- S. Fujii, E. Y. S. Ono, R. M. R. Ribeiro, F. Garcia Algarte, C. R. Takabayashi, T. C. R. M. Oliveira, E. N. Itano, Y. Ueno, O. Kawamura and E. Y. Hirooka, Braz. Arch. Biol. Techn., 2007, 50, 349 Search PubMed.
- L. Monaci and F. Palmisano, Anal. Bioanal. Chem., 2004, 378, 96 CrossRef CAS.
- European Mycotoxins Awareness Network (http://www.mycotoxins.org/).
- I. Y. Goryacheva, S. De Saeger, I. S. Nesterenko, S. A. Eremin and C. Van Peteghem, Talanta, 2007, 72, 1230 CrossRef CAS.
- C. O. Parker and I. E. Tothill, Biosens. Bioelectron., 2009, 24, 2452 CrossRef CAS.
- I. Yu. Goryacheva, S. De Saeger, I. S. Nesterenko, S. A. Eremin and C. Van Peteghem, Talanta, 2007, 72, 1230 CrossRef CAS.
- B. Prieto-Simón, T. Noguer and M. Campás, TRAC, Trends Anal. Chem., 2007, 26, 689 CrossRef CAS.
- M. Pohanka, D. Jun and K. Kuca, Drug Chem. Toxicol., 2007, 30, 253 CrossRef CAS.
- J. A. Cruz-Aguado and G. Penner, J. Agric. Food Chem., 2008, 56, 10456 CrossRef CAS.
- A. E. Radi, X. Munoz-Berbel, M. Cortina-Puig and J. L. Marty, Electrochim. Acta, 2009, 54, 2180 CrossRef CAS.
- O. D. Renedo, M. A. Alonso-Lomillo and M. J. A. Martinez, Talanta, 2007, 73, 202 CrossRef.
- B. Prieto-Simón, M. Campàs, J. L. Marty and T. Noguer, Biosens. Bioelectron., 2008, 23, 995 CrossRef CAS.
- R. M. Pemberton, R. Pittson, N. Biddle, G. A. Drago and J. P. Hart, Anal. Lett., 2006, 39, 1573 CrossRef CAS.
- D. Saha, D. Acharya, D. Roy, D. Shrestha and T. K. Dhar, A nal. Chim. Acta, 2007, 584, 1573.
- J. Wang, Small, 2005, 1, 1036 CrossRef CAS.
- V. Escamilla, D. Hernandez, M. B. Gonzalez, J. M. Pingarron and A. Costa, Biosens. Bioelectron., 2009, 24, 2678 CrossRef CAS.
- V. Shumyantseva, E. Suprun, T. Bulko and A. Archakov, Electroanalysis, 2009, 21, 530 CrossRef CAS.
- K. Idegami, M. Chikae, K. Kerman, N. Nagatani, T. Yuhi, T. Endo and D. Tamiya, Electroanalysis, 2008, 20, 14 CrossRef.
- A. Vig, A. Radoi, X. Muñoz, G. Gyemant and J. L. Marty, Sens. Actuators, B, 2009, 138, 214 CrossRef.
- K. Brainina, A. Kozitsina and J. Beikin, Anal. Bioanal. Chem., 2003, 376, 481 CAS.
- K. Z. Liang and W. J. Mu, Anal. Chim. Acta, 2006, 580, 128 CrossRef CAS.
- J. A. A. Ho, J. K. Chiu, J. C. Hong, C. C. Lin, K. C. Hwang and J. R. R. Hwu, J. Nanosci. Nanotechnol., 2009, 9, 2324 CrossRef CAS.
- K. Singh, M. A. Rahman, J. I. Son, K. C. Kim and Y. B. Shim, Biosens. Bioelectron., 2008, 23, 1595 CrossRef CAS.
- M. A. Rahman, P. Kumar, D. S. Park and Y. B. Shim, Sensors, 2008, 8, 1595 CrossRef.
- J. C. Vidal, P. Duato, L. Bonel and J. R. Castillo, Anal. Bioanal. Chem., 2009, 394, 575 CrossRef CAS.
- J. W. A. Findlay and R. F. Dillard, AAPS J., 2007, 9, E260 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |