Glucosinolates, structures and analysis in food†
Don Brian
Clarke
*
The Food and Environment Research Agency (Fera), Sand Hutton, York, YO41 1LZ, UK. E-mail: don.clarke@fera.gsi.gov.uk; Fax: +044-1904-462133; Tel: +044-1904-462000
First published on 22nd February 2010
Abstract
Glucosinolates (GLS) are sulfur rich, anionic secondary metabolites found principally in the plant order Brassicales. This review focuses on identifying the range of GLS structures identified to date and summarises the current state of taxonomic reclassifications of GLS producing plants. Those Brassica species that are available to growers in the UK are highlighted and progress in the aspects of analytical chemistry relevant to conducting accurate determinations of GLS content of foods is reviewed. The degradation and derivatisation workflows that have been utilized for conducting “glucosinolate analysis” are summarized. A review is made of aspects of extraction, isolation, determination of purity, ultraviolet (UV) and mass spectrometry (MS) parameters, extinction coefficients, UV response factors, quantification procedures, and the availability of stable isotope labeled internal standards, and certified reference materials. An electronic database of structures, formulae and accurate masses of both the 200 known, and a further 180 predicted GLS, is provided for use in mass spectrometry.
 Don Brian Clarke | Dr Don Brian Clarke is a senior analytical chemist in the contaminants and authenticity programme of the Food and Environment Research Agency (Fera). Research interests lie within the areas of analytical chemistry and clinical trials, covering emerging environmental contaminants in food, natural toxicants and beneficial plant constituents. |
1. Introduction
Glucosinolates (GLS), β-thioglucoside-N-hydroxysulfates (cis-N-hydroximinosulfate esters) are sulfur rich, anionic secondary metabolites found almost exclusively within the plant order Brassicales (Fig. 1). Various aspects of glucosinolates research have been reviewed; nutraceutical compounds in broccoli,1 the biochemical genetics of secondary metabolites in Arabidopsis thaliana,2 the dietary role of glucosinolates,3 the role and effects of glucosinolates of Brassica species,4 the enzymatic and chemically induced decomposition of glucosinolates,5 the biology and biochemistry,6 their role in insect-plant relationships,7 their bioavailability,8,9 bio-protective effects10 and significance for human health.9,11 A large body of epidemiological evidence indicates that the chemoprotective effects of Brassica vegetables against initiation of tumours caused by chemical carcinogens may be due to glucosinolates and their metabolic products.9–13 A special issue of Phytochemistry (Issue 8, 2009, 21 papers) has reviewed the progress made in many areas of glucosinolate research. There are however obvious omissions in chemotaxonomic classifications, the recognition of new GLS structures and analytical methods for their determination.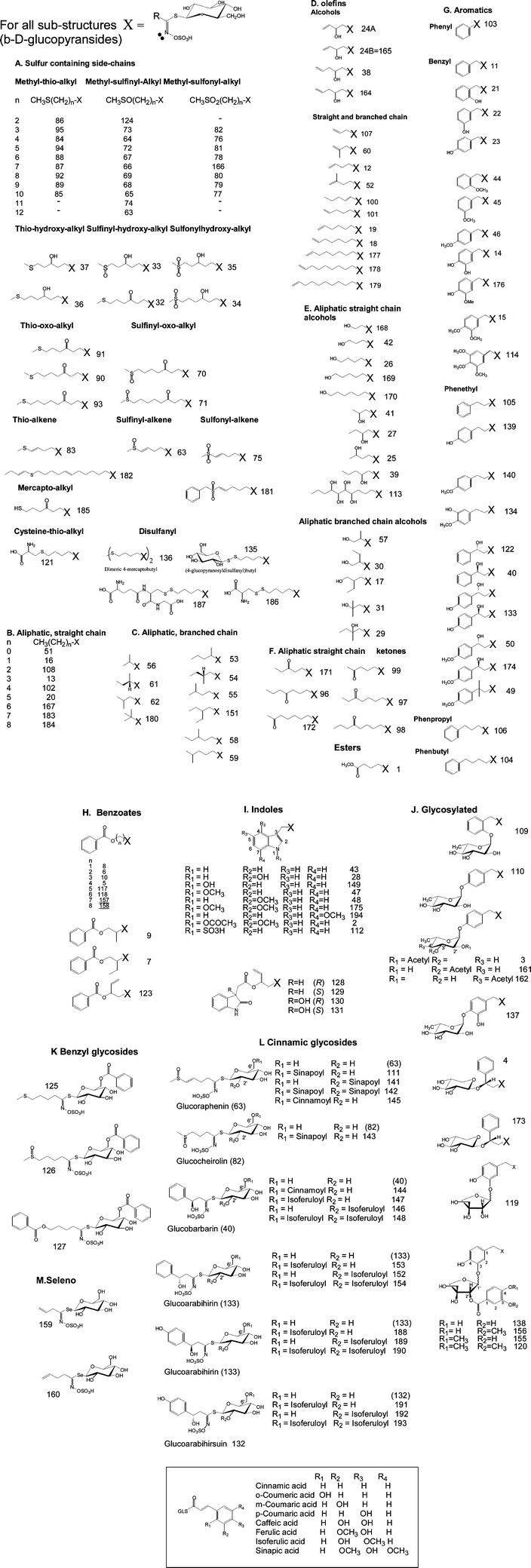 | ||
| Fig. 1 Reported glucosinolates structures by chemical class. A thorough literature review has lead to a much greater number of individual glucosinolates being characterized than previously thought. This reflects the absence of any major reviews or advances in this area since 2001. We currently have listed 200 structures, for a LC-TOF-MS screening library, with e.g. 32 of these being of relevance to the UK diet. | ||
2. Glucosinolate structures
Glucosinolates are characterized by a core sulfated isothiocyanate group, which is conjugated to thioglucose, and a further R-group. Both the glucose and the central carbon of the isothiocyanate are often further modified. This results in a diverse range of glucosinolate structures (Fig. 1). These are broadly classified as alkyl, aromatic, benzoate, indole, multiple glycosylated and sulfur containing side chains. The R chains may then contain double bonds, oxo, hydroxyl, methoxy, carbonyl or di-sulfide linkages. Since 2001 it has been generally agreed that there are 120 distinct individual glucosinolates14 and this is still almost invariably the quoted number.7 In a 2004 survey of seeds screened for 66 intact glucosinolates, four were not included in the accepted list of 120.15 Bellostas (2007) increased the number to 133, but this list has not been recognized by most subsequent researchers.16 As these three lists overlap incompletely, the Bellostas review adds a further 25 new structures raising the total to 149. Since this total has not been systematically reviewed since 2001, the number of reported glucosinolates is now approaching 200 (Fig. 1). A number of plants contain only a single glucosinolate, the majority contain 2–5, while 34 individual glucosinolates are reported in the seeds and leaves of a collection of ecotypes of Arabidopsis thaliana.17 In some respects, the number of possible structures is limited by the R-group being restricted to C1–C12 alkyl side-chains. A number of recently discovered new glucosinolates merely fill the final gaps in existing homologous sequences. Moreover, since it is difficult to verify many of the older reports, the references herein are relatively recent, but are not necessarily the first report of a new GLS, e.g. 2-ethyl-butyl-GLS (iso-hexyl) does not appear in either review,14,16 but a recent occurrence18 is referenced back to 1963.19 In this work the numbering system of Fahey is retained for structures 1–120 and simply extends as each new structure was added to our database. The database provided as supplemental information,† contains both the full, and trivial names, formulae and masses. While various abbreviated naming acronyms have been suggested, some GLS have no assigned trivial name. Moreover the three letter code used by Wathelet20 is insufficient for >100 structures, thus the only viable system is one which can combine various letter codes for the chemistry of the R-group e.g. T = thio, 3MTP = 3-methylthiopropyl, 4MOI3M = 4-methoxyindol-3-ylmethyl which is not limited in size and does not rely on the existence of trivial names.Illustrative examples of newly characterized GLS are; alkyl (hexyl),21iso-alkyl (t-butyl [1,1-dimethyl-ethyl]),22 alkene (oct-7-ene, non-8-ene, dec-9-ene),23 sulfur-chains (methylsulfonyldodecyl), benzoates (7-benzoyloxyheptyl),17 and substitution sites, 5-hydroxyindole,21 7-methoxyindole24 2-hydroxy-2-(4-hydroxylphenyl)ethyl.25 There is a range of cinnamic esters (Fig. 1) and while it is noted that these are widespread, they are not readily hydrolyzed by sulfatase and are difficult to chromatograph. These GLS derivatives have not been systematically studied to date.26 Other novel structures include benzyl-branched elongations (2-benzoyloxy-3-butene-GLS),27 5-benzylsulfonyl-4-pentenyl-GLS28 and benzyl substitutions of both the thioglucose (6′-O-benzoyloxy-glucoerucin) and apiose sugars (glucohesmatrolalin) and a variety of new indole esters (glucoisatsin).29 Furthermore, GLS structures such as 4-(cysteine-S-yl)butyl (glucorucolamine),30 dimeric 4-mercaptobutyl and 4-(gluco-disulfanyl)butyl21,31 are truly unique and indicate that additional GLS with hitherto unknown chemistries will continue to be isolated. Similar structures, but as the cysteine and glutathione disulfides, have been reported by Bennett as oxidative artifacts. These can be reverted to the parent GLS by reaction with the reducing agent TCEP (tris-2-carboxyethyl phosphine).32 The major GLS in salad rocket was then identified as 4-mercaptobutyl-GLS.33 Completing the homologous series (with e.g. C1–C10 R-group) for all these newly identified structures allows screening for a further 180 theoretical structures (Supplemental material Fig. S1†). The LC-TOF screening procedure for benzyl and cinnamoyl esters is also described (Supplemental material Fig. S2†).
3. Taxonomic classification
Knowledge of the genetic-based regulation of the accumulation of glucosinolates in plants is essential to explain the limited occurrence of individual structures in the various plant species. The mustards or cabbages are a family of flowering plants (Angiospermae) known either as the Brassicaceae or Cruciferae. Cruciferae is the older, but equally valid name, meaning “cross-bearing”, because the four petals of the flowers are reminiscent of a cross. Historically there has been confusion over the classification of this plant family, which contains about 3,700 species (Fig. 2). Glucosinolate synthesis is under enzymatic control and the structures are derived from both protein and non-protein L-amino acids.2,34 The defining feature of mustard-oil-producing plants is the system of compartmentalization of glucosinolates and the presence of myrosin cells containing the hydrolase enzyme myrosinase, which when released by e.g. herbivore damage, converts glucosinolates to biologically active isothiocyanates (mustard-oils), as part of the plants defence system (the “mustard-oil” bomb). Specific glucosinolates are often restricted within plant families, with the simplest methyl-GLS (glucocapparin) found in capers, and one of the more complex (rhamnopyranosyloxy)benzyl-GLS (glucomoringin) found in Moringa species.15,22,35 The traditional taxonomic classification placed the glucosinolate containing families in a number of different orders, implying multiple origins of the glucosinolate-myrosinase system. Since many unrelated plants were placed in the Capparaceae family, the taxonomy has been under review since 1975, with the view to placing all mustard-oil taxa within a single major clade.36–39 Naming of individual species is also ill-defined, but beyond the scope of this review. Species are simply referred to herein by the most recent name, e.g. Sinapis alba refers to the species with common name “white or yellow mustard” which is also referred to as Sinapsis alba, Brassica hirta and Brassica alba.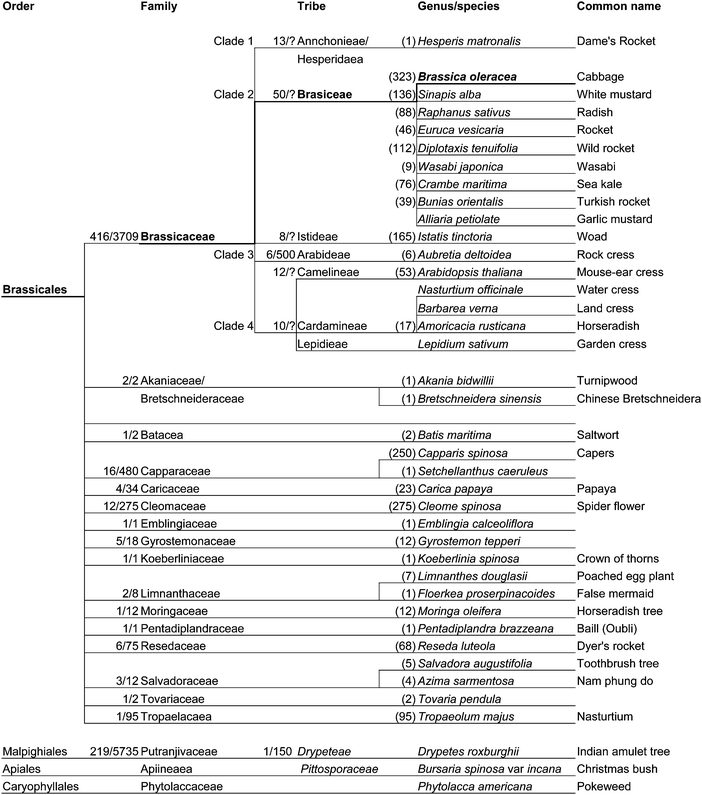 | ||
| Fig. 2 Summarized taxonomy of the order Brassicales indicating the current relationship of all the glucosinolate producing families.36–39,42 Numbers are the approximate numbers of genus and or species in each branch. These numbers are in flux and question marks denote a lack of clear data. Each branch is exemplified by a species used either in previous phylogenetics reclassifications, or as a food crop. | ||
Early work based on the identification of degradation products such as the release of the thiocyanate ion (SCN−) as proof of the presence of unstable isothiocyanates derived from 4-hydroxybenzyl and indol GLS40 has led to much of the early work being queried by subsequent workers. This brings into question the reliability of historical reports of glucosinolate content and the classifications of plants based on them. Following the lead of previous reviewers this review discounts reports of glucosinolates in mushroom, plantain and coca, and the reclassification of Pittosporaceae (e.g. Bursaria spinosa var. incana) and Phytolaccaceae (e.g. Phytolacca americana) from Capparales into Apiales and Caryophyllales, respectively. The 2001 review14 and earlier work (1991)40 would also appear to be in error when reporting 4-hydroxybenzyl GLS in Bursaria spinosa var. incana and in Phytolacca americana where the original work reported no detectable glucosinolates in either.14,40 The genus Drypetes had five reports of glucosinolates (1975–1991) and this had been regarded as reliable to date. Genetic sequencing has demonstrated a major mustard-oil clade (Brassicales) and one outlier Drypetes,41 bringing this back into doubt. Drypetes genus was traditionally placed in the sub-family Phyllanthoideae in Euphorbiaceae, but is now in the family Putranjivaceae. Many botanists are now adopting the Angiosperm Phylogeny Group classification (APG) for the orders and families of flowering plants. The APG II system42 has been adopted in whole, or in part in a number of recent major works. There is some disagreement on the relative merits of the traditional morphological approach against chemotaxonomy and molecular phylogenetics. The system is rather controversial at the family level, splitting a number of long-established families and submerging a number of other families, as it does with the Brassicaceae. Under this system, the Brassicales are an order of flowering plants, belonging to the eurosids II clade of Angiosperms. This clade contains the order Brassicales, which in this system (APG II) includes families classified under Capparales in previous classifications. The sub-families Capparaceae and Cleomaceae, and a number of monotypic genera, are now elevated to familial status, and with the demotion of Setchellanthaceae there are now 16 glucosinolate containing families in the order Brassicales. To date the full classification of the Brassicales families is still in flux and no consensus has yet been reached. Such emerging views are summarised in Fig. 2 with the relationships between the chemotaxonomic marker species.
The detailed positioning of genera and species is therefore not well defined and varies by source and classification system. In overview, the Brassicales order is a biogeographical dispersed lineage with many small but distinct clades and a large Brassicaceae family containing 93% of species within the order. The Brassicales order and Brassica genus are remarkable in that they each contain more commercially important agricultural and food crops than any other. With the minor exceptions of capers, papaya, nasturtium and the horseradish tree (Moringa oleifera), which are spread through the other orders, all of the species of dietary importance are contained in the core Brassicaceae order. This order is then divided into four clades and 25 tribes. With horseradish and cresses in the Cardamineae and Lepideae tribes of Clade 4. All other food genera (Alliaria, Bunias, Crambe, Diplotaxis, Euruca, Raphanaus, Sinapis, Wasabi) are now contained within the Brassiceae tribe of Clade 2. The Brassica genus evolved from three ancestral Brassica species with diploid genomes (with 10, 9 and 8) chromosomes (Brassica rapa AA, Brassica nigra BB, Brassica oleracea CC). These then interbred producing the three tetraploid species Brassica juncea (AABB), Brassica napus (AACC) and Brassica carinata (BBCC). These have further interbred, or been hybridized, producing the wide range of species and cultivars that form a large part of our dietary intake of vegetables (Table 1). Some major food uses are: roots (swedes B. neobrassica and turnips B. rapa), stems (Kohl rabi B. oleracea var. gongylodes), leaves (cabbages B. oleracea var. capitata, Brussels sprouts B. oleracea var. gemmifera), flowers (broccoli calabrese Brassica oleracea var. italica, cauliflower B. oleracea var. botrytis), seeds (mustards Sinapis alba) and oil (oil seed rape B. napus). In the UK the consumption of the individual varieties is somewhat limited (Supplemental material Table S1†), where by definition varieties must be readily available to both commercial producers and domestic gardeners for there to be any appreciable population based intake. In 2002 the reported mean consumption of six key Brassica vegetables was 10–15 g/day of each vegetable for persons that consume these foods (consumer mean), with an overall population mean intake of 20 g Brassicas/day/person (Table 2) and a maximum intake of 160 g/day.43
| Genera, species, group and forma | Common name |
|---|---|
| Brassica carinata | Ethiopian mustard |
| Brassica juncea | Indian mustard |
| Brassica juncea var. crispifolia | Chinese mustard |
| Brassica juncea var. integlifolia | Red giant mustard |
| Brassica juncea rugosa | Wrapped heart mustard cabbage |
| Brassica hirta | Yellow mustard |
| Brassica napus | Canola/rape seeds |
| Brassica napus var. pabularia | Siberian kale |
| Brassica napobrassica | Rutabaga (swede) |
| Brassica nigra | Black mustard |
| Brassica oleracea var. acephala | Kale |
| Brassica oleracea var. alboglabra | Kai-lan (Chinese broccoli) |
| Brassica oleracea var. botrytis | Cauliflower |
| Brassica oleracea var. botrytis f. romanesco | Romanesco broccoli |
| Brassica oleracea var. capitata f. alba | White cabbage (drum) |
| Brassica oleracea var. capitata f. conica | Pointed cabbage |
| Brassica oleracea var. capitata f. ruba | Red cabbage |
| Brassica oleracea var. capitata f. sabauda | Savoy cabbage |
| Brassica oleracea var. gemmifera | Brussels sprouts |
| Brassica oleracea var. gongylodes | Kohl rabi |
| Brassica oleracea var. italica | Broccoli |
| Brassica oleracea var. italica × botrytis | Broccoflower |
| Brassica oleracea var. komatsuna | Komatsuna |
| Brassica oleracea var. viridis | Collard greens |
| Brassica rapa | Mustard spinach |
| Brassica rapa var. chinensis | Pak Choi (Cantonese) |
| Brassica rapa var. narinosa | Broad beak mustard |
| Brassica rapa var. japonica | Mibuna |
| Brassica rapa var. parachinensis | Choy Sum (false Pak Choi) |
| Brassica rapa var. pekinensis | Chinese cabbage |
| Brassica rapa var. perviridis | Komatsuna |
| Brassica rapa var. perviridis × pekinensis | Senposai |
| Brassica rapa var. purpuraria | Purple stem mustard |
| Brassica rapa var. rapifera | Turnip |
| Brassica rapa var. rosularis | Tatsoi (rosette Pak Choi) |
| Brassica rapa var. ruvo | Rapini (broccoli raab) |
| Common name | Number of consumers | Population mean | Consumer mean | Consumer max | Brassica species |
|---|---|---|---|---|---|
| a Reproduced and adapted from: Henderson L., Gregory J. Swan G. National Diet and Nutrition Survey: adults aged 19–64 years. Volume 1: types and quantities of foods consumed, The Stationery Office 2002.43 | |||||
| Broccoli | 743 | 6.4 | 14.7 | 80.7 | Brassica oleracea var. italica |
| Head cabbage | 737 | 5.9 | 13.7 | 112.1 | Brassica oleracea var. capitata f. alba |
| Cauliflower | 767 | 5.6 | 12.6 | 158.7 | Brassica oleracea var. botrytis |
| Brussels sprouts | 212 | 1.9 | 15.9 | 72.7 | Brassica oleracea var. gemmifera |
| Kohl rabi | 0 | — | — | — | Brassica oleracea var. gongylodes |
| Chinese cabbage | 26 | 0.2 | 10.1 | 66.4 | Brassica rapa var. pekinensis |
| Totals | 1341 | 19.9 | 158.7 |
4. Glucosinolate content of plants
Historically a sense-mediated adaptive mechanism to avoid consumption of poisons has selected for low-glucosinolate content in vegetables.45 Recent sensory trials have shown typical rocket salad flavour and pungency are perceived as positive sensory traits, while bitter notes, characterized by high glucosinolate content (sinalbin/gluconapin-herbaceous; sinigrin-pungency), were much less acceptable.44 The reverse is true in condiments such as Sinapis alba seeds (condiment mustard) which were bred for piquancy and now contain one of the highest reported concentrations (250 μmol/g sinalbin).15,45 ‘Superbroccoli’ has been produced by traditional plant breeding including wild Sicilian broccoli, to produce a cross with a glucoraphanin content 3–4 times higher than that of normal varieties. This has been shown to elevate plasma sulforaphane the putative anticancer active principle and metabolites 3-fold.46 Since this is a larger non-volatile glucosinolate the acceptability of the flavour of the broccoli is not adversely affected.Glucosinolate concentrations in plants, although highly variable, are around 1% dry weight in some Brassica vegetables.47 There are a number of reports of amounts exceeding 10% in the seeds of some species15,45 and as high as 26% of rhamnose-benzyl-GLS in the seeds of Moringa oleifera.22 The young leaves and buds of the desert cabbage Schouwia purpurea contain unusually high levels of gluconapin, up to 10% dry weight.48 Glucosinolates are very stable water-soluble precursors of isothiocyanates and some fresh plants have been show to contain almost exclusively glucosinolates and no isothiocyanates. Glucosinolates are therefore considered the storage form of their biologically active aglycones (isothiocyanates). Reproductive tissues (florets, flowers and seeds) often contain as much as 10–40 times higher concentrations of GLS than is found in vegetative tissues.15,22,49,50 Seeds are therefore the best bulk source of quantities of glucosinolates. Roots can often be an equally high source, but are a less practical material to harvest. There are many practical advantages in using seeds rather than plant tissues in preparing a glucosinolate extract. Seeds are stable and can be readily stored. The three rapeseed certified reference materials (CRMs) have now been in use an astonishing 20 years, without degradation.51 Because of the low moisture content, ground seed-meal does not activate the moisture sensitive myrosinases, thereby preventing conversion to isothiocyanates and other degradates.
One of the key drivers for food-based analytical method development is generating human exposure data, in this case the dietary intake of glucosinolates obtained through the consumption of [cruciferous] vegetables.13 A summary of the available glucosinolate content data collated from 18 studies was used to produce single values for the total glucosinolate content of each food.52 A further database has been prepared of 26 glucosinolates in 18 vegetables and a dietary intake of 14 mg/day estimated in Germany,53 an estimate of 6 mg/day has been calculated for Spain.54
5. Analysis
5.1 Stability
There appears to be little quantitative data documenting the stability of glucosinolates during processing and extraction. Generally extractions are conducted at temperatures of 65–100 °C, close to the solvent or mixtures boiling point, on the assumption that the overarching concern should be the inactivation of myrosinase.55–59 The benefit of this assumption is brought into question by data showing 80% degradation of glucobrassicin (3-indolylmethyl-GLS) within 5 min at 100 °C and 120 bar when extracting into 70% methanol in water in a pressurized liquid extractor. The optimal yield was obtained at 50 °C.60 Processing in the presence of a denaturing agent such as methanol should be sufficient to ensure GLS are not hydrolysed by myrosinases.61 Current procedures to minimize degradation include, harvesting into liquid nitrogen, microwave induced deactivation and freeze drying before homogenisation.50,62 There is a lack of clear validation data available on the effects of avoiding high temperatures and the significance of myrosinase inactivation. Thermal degradation has been studied in red cabbage, where cooking reduced indole (38%) and alkyl (8%) content. Canning was the most severe heat treatment studied (40 min, 120 °C) and reduced total-GLS by 73%.55 The vegetable matrix itself has an effect on thermal stability, after microwave inactivation of myrosinase, with the cellular environment of Brussels sprouts being one of the least favourable with glucobrassicin content halving within 10 min at 100 °C and gluconapin halving within 35 min.63 Cold storage of seed-sprouts for 3-weeks at 4 °C suggested that of the species studied only rocket showed decline in glucoerucin and glucoraphenin content.64 Gluconasturtiin has been shown to undergo a non-enzymatic, iron-dependent degradation to a simple nitrile. On heating the seeds to 120 °C, thermal degradation of this heat-labile glucosinolate increased simple nitrile levels many fold.65Myrosinase can be deactivated in wet tissue by microwaving, then cooling on ice.55 Microwave inactivation does not work as well on dry materials, and an optimum moisture content of 14–16% was needed in Crambe abyssinica seed.66 Canola seed at the lowest moisture content, 6%, required 485 s at 1500 W for deactivation.67 Microwaving resulted in an increase in the extracted GLS content relative to the uncooked cabbage.62
5.2 Extraction
Extraction of GLS from plant material is best achieved using protic solvents. This has been largely restricted to the use of methanol-water.57,59 Both ethanol-water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), or methanol-water (7:3) are recommended for freeze-dried green tissues.20 Water or phosphate buffer (20 mM, pH = 7, 20 min, 100 °C) was more effective than alcoholic solvents for extracting sinigrin from black mustard and horseradish.68 Phosphoric acid (2%) has been use to extract GLS from Brussels sprouts.69 Since methanol ruptures cell walls (where the aim was simply to remove glucosinolates from oilseed protein products), methanol-water-ammonia has been successfully employed; 10% ammonia in methanol containing 5% water, at a solvent-to-seed ratio of 6.7 and 2 min of blending with 10–15 min quiescent period being sufficient to lower GLS content to below the limit of detection.70,71 Large scale solvent based extractions are disfavored by time, energy and safety concerns and have therefore not become established. The noxious weed Cardaria draba is reported to give optimum extractability of glucoraphanin into 20% ethanol in water at 70 °C, 50 g dm−3 and pH 3 over 20 min.72 Given the diversity of structures and the range of seed and plant matrices, it is recommended that 70% methanol is used as the default extractant unless validation proves alternative solvents are required.
:1), or methanol-water (7:3) are recommended for freeze-dried green tissues.20 Water or phosphate buffer (20 mM, pH = 7, 20 min, 100 °C) was more effective than alcoholic solvents for extracting sinigrin from black mustard and horseradish.68 Phosphoric acid (2%) has been use to extract GLS from Brussels sprouts.69 Since methanol ruptures cell walls (where the aim was simply to remove glucosinolates from oilseed protein products), methanol-water-ammonia has been successfully employed; 10% ammonia in methanol containing 5% water, at a solvent-to-seed ratio of 6.7 and 2 min of blending with 10–15 min quiescent period being sufficient to lower GLS content to below the limit of detection.70,71 Large scale solvent based extractions are disfavored by time, energy and safety concerns and have therefore not become established. The noxious weed Cardaria draba is reported to give optimum extractability of glucoraphanin into 20% ethanol in water at 70 °C, 50 g dm−3 and pH 3 over 20 min.72 Given the diversity of structures and the range of seed and plant matrices, it is recommended that 70% methanol is used as the default extractant unless validation proves alternative solvents are required.
5.3 Isolation
Analytical standards of the individual GLS are isolated from specific plants, preferably those containing either high concentrations or less complex mixtures of GLS.15,20,50 Aqueous extraction may also include Pb(OAc)2 and Ba(OAc)2 to precipitate protein and free sulfate, respectively.50 After centrifugation, purification of GLS generally involves an anion exchange step, in most cases on the same DEAE-Sephadex A-25 resin used for the later enzymatic desulfation and analysis prescribed in method ISO9167-1.73 A typical procedure for intact GLS extraction is elution from the resin with 0.5 M K2SO4, followed by evaporation and dissolution in hot methanol to leave insoluble salts, then recrystallisation from cold ethanol and drying over P2O5.20 Florisil solid phase extraction is used to clean up intact GLS, with application in methanol/dichloromethane/hexane, washing with dichloromethane/hexane and elution with methanol/ethyl acetate.74,75 Anion exchange on a styrene-divinylbenzene copolymeric anion exchanger gives an additional dimension for separation, with high selectivity and elution in order of GLS hydrophobicity, using the inorganic anions SO42−, NO3−, ClO3− and ClO4− in sequence.76 High-speed counter-current chromatography relies solely on the partition coefficient of the solute between the stationary and mobile phases and can be used to separate gram quantities of structurally and chromatographically similar glucosinolates. Using a propanol-acetonitrile saturated aqueous ammonium sulfate-water, biphasic system, with the organic phase as the mobile phase and the aqueous as the stationary phase, the homologues glucoraphanin [methylsulfinylbutyl-GLS] and glucoiberin [methylsulfinylpropyl-GLS] are separated by their partition coefficients of 0.63 and 1.03.775.4 Detection methods
The detection of glucosinolates may be considered in three parts: as degradative totals by colourimetric techniques, as non-destructive totals, and as individual components by chromatographic separation and detection. The collection of degradative methods and chemical workflows are summarized in Fig. 3. The majority of these: thiourea,78 thymol,79 benzenedithiol cyclocondensation,80 palladium chloride,81 ferricyanide assays82,83 and sulfate ion release84 continue to be used without modification. Various glucose assays85 are now available, primarily as hexokinase coupled to NADH production and glucose oxidase and peroxidase coupled to various coloured dyes such as quinoneimine and dianisidine. An official method for glucosinolate content following glucose release has recently been published (ISO 9167-3 2007).86 All colourimetric methods have strengths and weaknesses and it has been recommended not to rely on any single method, but to conduct a number of assays to ensure a consensus is reached for a given sample type in order to avoid bias. The only technical advancement away from colourimetric detection has been in automation and technology. The cyclo-condensation of isothiocyanates and benzenedithiol gives a single chromatographically stable product 1,3-benzodithiole-2-thione for any GLS, and with a 3-fold increase in molar extinction to ε365 23,000 M−1 cm−1 it is well suited for the more sensitive HPLC-UV detection format.80 Biosensing is a second such example, where the enzymatic cascade from GLS through isothiocyanate to glucose and on to hydrogen peroxide has also been automated. Biosensing using amperometric enzyme electrodes based on glucose oxidase and tyrosinase were utilized after conversion to glucose and thiourea.87 Biosensing with a myrosinase-immobilized eggshell membrane, with glucose oxidase activity and an oxygen-sensitive optode membrane, measuring depletion of dissolved oxygen has also been reported.88,89 The first amperometric flow analyzer based on the biosensor concept was described in 2003, using myrosinase and glucose oxidase.90 A gold nanoparticle-carbon nanotube composite electrode using myrosinase and glucose oxidase with Teflon as the non-conducting binding material is also under development, but is as yet unpublished.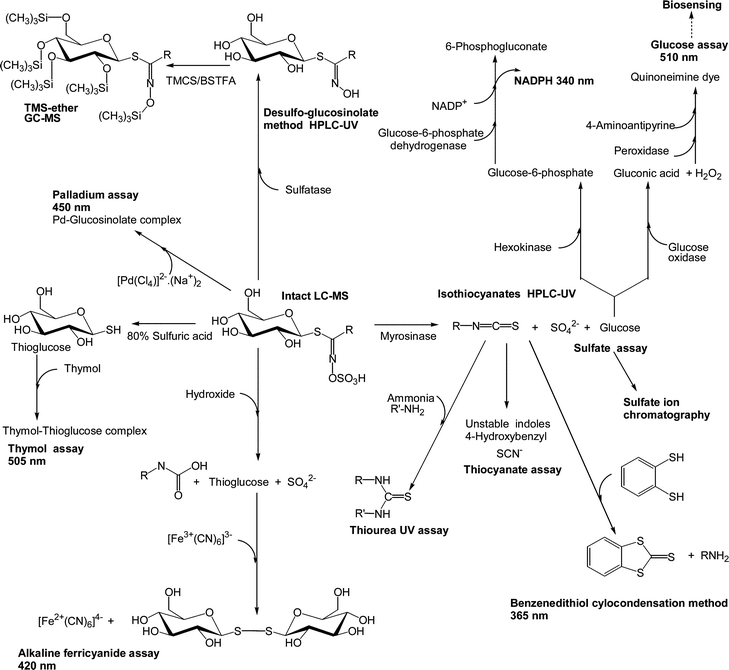 | ||
| Fig. 3 Chemical workflow for degradative methods of glucosinolate analysis. | ||
Near infrared reflectance spectroscopy (NIRS) is a validated non-destructive technique, in which O–H, C–H and N–H groups are associated with total glucosinolate content.91 This simple method continues in use, since protein and oil content are also determined simultaneously along with the GLS content.92 The official X-ray fluorescence method determines total sulfur (ISO 9167-2 1994).93 ELISA assays have been investigated, but recent reports are lacking.69
In the past, analysis of the intact glucosinolates was not possible. This was overcome by using hydrolysis to produce more chromatographically amenable forms. The original work was published in 1982,94 describing the conversion via sulfatase to desulfo-glucosinolates (ds-GLS) and was harmonized into an EU official method ISO 9167-1 (1992)57 and an AOCS method Ak 1–92 (AOCS 1998).59 These remain in widespread use,95,96 with suggested improvements,20,97,98 but have not yet been issued as official methods. The guidelines for analysis of green tissues used for biofumigation is the best attempt at a generic method for plant tissues e.g. food.20 In parallel, conversion via myrosinase and GC-MS of the more volatile isothiocyanates (epithioalkane-nitriles, nitriles, oxazolidine-2-thiones etc.) or derivatisation of the desulfo-GLS to trimethylsilyl (TMS)-ethers and GC-MS have now largely been dropped, but remain in use for plant volatiles and biofumigation work where the isothiocyanates rather than the parent GLS are the key form. Limited data is available for the myrosinase degradates, isothiocyanates, indoles and oxazolidinethiones by HPLC-UV at 240 nm.99
Analytical determinations can now be undertaken on the desulfo-GLS or intact-GLS forms, but it is uncommon to perform UV quantification on the intact forms.100 The desulfation process has been miniaturized and adapted to a 96-well filter plate format for 5 mg seed and 10 mg of leaf samples.17 The desulfation process typically acts as the combined sample extraction and clean-up step.
Rationalization of detection of glucosinolate degradates as a means of verifying the (implied) presence of the parent GLS is still in general use,18 but there are clear limitations to the specificity and accuracy of these degradative approaches. In this review it is accepted that the presence of an isothiocyanate or a desulfo-GLS is proof of the parent GLS.
The HPLC-UV and GC-FID (flame ionisation detector) methods of measuring desulfo-glucosinolates were rigorously validated with no reported difference between the two detection techniques, when measuring eleven analytes in rapeseed by HPLC and seven by GC.101 GC-based techniques are fraught with difficulties and hence are mostly considered unsuitable for identification and quantification.61
5.5 Response factors for desulfated glucosinolates
The accuracy of the HPLC-UV desulfo-method clearly rests on a correct approach to numerical quantification, i.e. it relies on relative response factors (RRF) of the desulfo-glucosinolates (ds-GLS). These are calculated from individual purified standards relative to the response of sinigrin in the thymol-sulfuric acid UV assay. The majority of response factors have been continuously transcribed from the original work,102 from which three official methods were derived (EEC 1864/90 1990,103 ISO 9167-1 1992,57 Ak 1–92 AOCS59 1998). New data have been published2,20,50 where the list of relative response factors, also known as relative proportionality factors (RPF), has now grown to 28 entries (Table 3). The source of response factors presented by Vinjamoori104 is unreferenced and these are not normalized to sinigrin (1.05) and therefore appear to have an upwards bias of 0.05. It is recommended that the values published by Brown,50 Wathelet20 and Haughn2 are used. While response factors may vary up to 10-fold (0.2–2.1), the majority of determinations lie within a very narrow range, indicating that chain elongation and the sulfur oxidation state produce insignificant effects. Hydroxylalkyl groups have the highest values, while indoles, hydroxybenzyl and benzoate esters have the lowest. The value for 4-methylthio-3-butenyl ds-GLS is somewhat anomalous with respect to other alkenes. The values reported by Brown appear to have been obtained using an unspecified HPLC based flow-injection-analysis approach, a departure from the thymol-sulfuric acid assay.50 Data for minor components ethyl, propyl, isopropyl, 3-methylthiopropyl, butyl, 4-methylsulfonylbutyl, 5-methylthiopentyl, 7-methylsulfinylheptyl and (2R)-2-hydroxy-2-phenethyl-GLS are still required to encompass all of the individual GLS that can be encountered in foods (Table 4). The default of assigning the value of 1.00 to any unassigned component has been shown to generate inaccuracies. Current data generated with derived values for glucoraphasatin (0.40) and glucosinalbin (0.50) will be a factor of 2-times higher than data generated with the default values. A more rigorous approach, identifying the structural features of ds-GLS and assigning a class specific value e.g. 0.3 for indoles, 0.4 for benzoyl ester is recommended. It is therefore crucial to ensure that updated values are used when comparing data. A more rigorous approach to the documentation of experimental procedures is needed, including the listing of all RPF values used in each work. It is unacceptable to refer to the official methods and the limited range of factors therein. All individual ds-GLS components in a sample should be separated chromatographically and then integrated down to the 1% level. This process however then relies on the attainment of precisely reproducible retention times. Assigning the correct name-peak combinations in each new plant material requires careful comparison or the use of LC-UV-MS. While all researchers quote the official methods, a major problem in accuracy and cross comparison is the failure to indicate exactly which factor from Table 3 is used for each analyte. It is noted that the desulfation protocol was optimised for the analysis of gluconapin, epi- and progoitrin in rapeseed, and that velocity of desulfation and feedback inhibition were critical parameters. Other GLS such as glucoiberin require removal of hydrolysed ds-GLS and a second incubation.20 A flow through bioreactor with nylon-immobilised sulfatase has been used for large scale desulfation.105| Chain length | Trivial name | R Side chain | Buchner 1987102 | EC 1990103 | Haughn 19912 | ISO 199257 | AOCS 199859 | Brown 200350 | Vinjam 2004104 | Wathelet 200420 | Recommended value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Thymol-sulfuric acid assay derived UV response factors [relative proportionality factors (RPF)] for desulfoglucosinolates. EC 1990 = method EEC 1864/90 = 1990.103 ND = retention time provided without a response factor. No data available for: ethyl, propyl, butyl, 4-methylsulfonylbutyl, 5-methylthiopentyl, 7-methylsulfinylheptyl, (2R)-2-hydroxy-2-phenethyl. | |||||||||||
| C1 | Glucocapparin | Methyl | 1.0 | 1.25 | 1.25 | ||||||
| C3 | Sinigrin | 2-Propenyl | 1.00 | 1.00 | 1.00 | 1.00 | 1.0 | 1.05 | 1.00 | 1.00 | |
| Glucoibervirin | 3-Methylthiopropyl | 0.8 | 0.8 | ||||||||
| Glucoiberin | 3-Methylsulfinylpropyl | 1.07 | 1.07 | 1.07 | 1.07 | 1.2 | 1.13 | 1.07 | 1.07 | ||
| Glucocheirolin | 3-Methylsulfonylpropyl | 0.9 | 1.26 | 1.26 | |||||||
| Glucoputranjivin | 1-Methylethyl | 1.0 | 1.0 | ||||||||
| Glucosisymbrin | 2-Hydroxy-1-methylethyl | 1.32 | 1.32 | ||||||||
| Glucoerysimumhieracifolium | 3-Hydroxypropyl | 2.1 | 2.1 | ||||||||
| C4 | Gluconapin | 3-Butenyl | 1.00 | 1.11 | 1.11 | 1.11 | 1.0 | 1.17 | 1.11 | 1.11 | |
| Progoitrin | (2R)-2-Hydroxy-3-butenyl | 1.09 | 1.09 | 1.09 | 1.09 | 1.15 | 1.09 | 1.09 | |||
| Epiprogoitrin | (2S)-2-Hydroxy-3-butenyl | 1.09 | 1.09 | 1.09 | 1.15 | 1.09 | 1.09 | ||||
| Glucoerucin | 4-Methylthiobutyl | 1.0 | 0.9 | 1.04 | 1.04 | ||||||
| Glucoraphasatin | 4-Methylthio-3-butenyl | 0.40 | 0.40 | ||||||||
| Glucoraphanin | 4-Methylsulfinylbutyl | 1.07 | 1.07 | 1.07 | 0.9 | 1.13 | 1.07 | 1.07 | |||
| Glucoraphenin | 4-Methylsulfinyl-3-butenyl | 0.9 | 0.9 | ||||||||
| Glucoarabidopsithalianain | 4-Hydroxylbutyl | 1.48 | 1.4 | 1.4 | |||||||
| Glucoconringiin | 2-Hydroxy-2-methylpropyl | 1.00 | 1.00 | ||||||||
| C5 | Glucoalyssin | 5-Methylsulfinylpentyl | 1.07 | 1.07 | 1.07 | 0.9 | 1.13 | 1.07 | 1.07 | ||
| Glucobrassicanapin | Pent-4-enyl | 1.15 | 1.15 | 1.15 | 1.15 | ||||||
| Gluconapoleiferin | 2-Hydroxy-pent-4-enyl | 1.00 | 1.00 | 1.00 | 1.05 | 1.00 | 1.00 | ||||
| Glucocleomin | 2-Hydroxy-2-methylbutyl | 1.07 | 1.07 | ||||||||
| C6 | Glucolesquerellin | 6-Methylthiohexyl | 1.0 | 1.0 | |||||||
| Glucohesperin | 6-Methylsulfinylhexyl | 1.0 | 1.0 | ||||||||
| C7 | Glucoarabishirsutain | 7-Methylthioheptyl | 1.0 | 1.0 | |||||||
| C8 | Glucoarabishirsuin | 8-Methylthiooctyl | 1.0 | 1.1 | 1.1 | ||||||
| Glucohirsutin | 8-Methylsulfinyloctyl | 1.1 | 1.1 | ||||||||
| Ind | Glucobrassicin | 3-Indolylmethyl | 0.29 | 0.29 | 0.25 | 0.29 | 0.29 | 0.31 | 0.29 | 0.29 | |
| 4-Hydroxyglucobrassicin | 4-Hydroxy-3-indolylmethyl | 0.28 | 0.28 | 0.28 | 0.28 | 0.29 | 0.28 | 0.28 | |||
| 4-Methoxyglucobrassicin | 4-Methoxy-3-indolylmethyl | 0.25 | 0.25 | 0.25 | 0.26 | 0.25 | 0.25 | ||||
| Neoglucobrassicin | N-Methoxy-3-indolylmethyl | 0.20 | 0.20 | 0.20 | 0.20 | 0.21 | 0.20 | 0.20 | |||
| Ar | Glucotropaeolin | Benzyl | 0.95 | 0.95 | 0.95 | 0.95 | 0.8 | 1.00 | 0.95 | 0.95 | |
| Glucosinalbin | p-Hydroxybenzyl | 0.4 | 0.50 | 0.50 | |||||||
| Gluconasturtiin | 2-Phenethyl | 0.95 | 0.95 | 0.95 | 1.0 | 1.00 | 0.95 | 0.95 | |||
| Glucobarbarin | (2S)-2-Hydroxy-2-phenethyl | 1.09 | 1.09 | ||||||||
| Glucomalcomiin | 3-Benzoyloxypropyl | 0.4 | 0.4 | ||||||||
| — | 4-Benzoyloxybutyl | 0.41 | 0.3 | 0.3 |
| No | Chain length | Trivial name | R Side chain | Food source |
|---|---|---|---|---|
| 1 | C1 | Glucocapparin | Methyl | Capers |
| 2 | C2 | Glucolepidiin | Ethyl | Radish |
| 3 | C3 | — | Propyl | Cabbage |
| 4 | Glucoputranjivin | Isopropyl | Radish | |
| 5 | Sinigrin | 2-Propenyl | Cabbage | |
| 6 | Glucoiberin | 3-Methylsulfinylpropyl | Cabbage | |
| 7 | Glucoibervirin | 3-Methylthiopropyl | Cabbage | |
| 8 | Glucocheirolin | 3-Methylsulfonylpropyl | Cow's milk | |
| 9 | C4 | Glucocapparisflexuosain | Butyl | Cabbage |
| 10 | Gluconapin | 3-Butenyl | Cabbage | |
| 11 | Progoitrin | (2R)-2-Hydroxy-3-butenyl | Cabbage | |
| 12 | Epiprogoitrin | (2S)-2-Hydroxy-3-butenyl | Sea kale | |
| 13 | Glucoerucin | 4-Methylthiobutyl | Cabbage | |
| 14 | Glucoraphanin | 4-Methylsulfinylbutyl | Broccoli | |
| 15 | Glucoerysolin | 4-Methylsulfonylbutyl | Cabbage | |
| 16 | Dehydroerucin | 4-Methylthiobut-3-enyl | Daikon radish | |
| 17 | Glucoraphenin | 4-Methylsulfinylbut-3-enyl | Radish | |
| 18 | C5 | Glucobrassicanapin | 4-Pentenyl | Chinese cabbage |
| 19 | Glucoberteroin | 5-Methylthiopentyl | Cabbage | |
| 20 | Glucoalyssin | 5-Methylsulfinylpentyl | Rocket | |
| 21 | Gluconapoleiferin | 2-Hydroxy-pent-4-enyl | Swede | |
| 22 | C7 | Glucosiberin | 7-Methylsulfinylheptyl | Watercress |
| 23 | C8 | Glucohirsutin | 8-Methylsulfinyloctyl | Watercress |
| 24 | Ind | 4-Hydroxyglucobrassicin | 4-Hydroxy-3-indolylmethyl | Cabbage |
| 25 | Glucobrassicin | 3-Indolylmethyl | Cabbage | |
| 26 | 4-Methoxyglucobrassicin | 4-Methoxy-3-indolylmethyl | Cabbage | |
| 27 | Neoglucobrassicin | N-Methoxy-3-indolylmethyl | Cabbage | |
| 28 | Ar | Glucotropaeolin | Benzyl | Cabbage |
| 29 | Glucosinalbin | p-Hydroxybenzyl | Mustard | |
| 30 | Gluconasturtiin | 2-Phenethyl | Cabbage | |
| 31 | Glucobarbarin | (2S)-2-Hydroxy-2-phenethyl | Land cress | |
| 32 | Glucosibarin | (2R)-2-Hydroxy-2-phenethyl | White mustard |
5.6 Chromatography
The current state-of-the-art in the analytical measurement of GLS is for HPLC-MS analysis of the intact glucosinolates reconstituted in water.15,22,106,107 This approach has yet to be cross-validated against any of the validated official methods. While the change of detection systems from UV to mass spectrometry (MS) detection has been a natural progression, the most important change is arguably in the choice of the chromatographic stationary phase. Novel approaches, such as supercritical fluid chromatography,108 micellar electrokinetic109,110 and capillary zone electrophoresis have found use.111 Hydrophilic interaction liquid chromatography (HILIC) has been investigated,112 employing second-generation HILIC phases based on silica zwitterions, which are reportedly more robust and reproducible than the original polyhydroxylethyl aspartamide columns.113 Earlier applications of anion exchange and porous graphite phases have not progressed to date.114,115The use of octadecyl (C18) reverse phase remains the preferred chromatographic approach. When not constrained to a MS compatible buffering system, ion-pairing chromatography with 5 mM tetraoctylammonium bromide,56 tetrapentylammonium bromide and triethylamine/formate24,116 remain viable. The strong acid modifier trifluoroacetic acid (0.1–0.5% TFA) still finds regular use as buffer, despite clear incompatibility issues with MS/MS detection.100,114,116–118 These TFA based chromatographic separations however remain the benchmark for analysis of intact glucosinolates.15,116 Separations of intact GLS is difficult to achieve without ion-pair buffers, and the use of an acetonitrile/water mixture without any buffer has been reported,32 as well as the use of 30 mM ammonium acetate pH 5.0 (formic acid),75 10 mM ammonium formate (formic acid),60 and 5 mm NH4·acetate.107,119 The use of formic acid mobile phase modifier coupled with 100% aqueous compatible columns, shows great promise as a viable alternative without the involvement of nonvolatile ion-pair agents or TFA, and modern separations are now directly comparable with the earlier separations; e.g. water (0.1% HCOOH)/acetonitrile, with Luna C18 column,21 water/acetonitrile each with 0.1% formic acid.34,120,121
Early claims of simultaneous analysis of intact and desulfated glucosinolates were unsubstantiated.114 Ion-pair reagents function by neutralizing the charge on the sulfate group and the most appropriate modern stationary phases function by minimizing this effect almost solely by hydrophobic interactions, hence analysis of both intact and desulfo-glucosinolates can now be readily achieved with surprisingly small retention time shifts in the same chromatographic run without the need to change the mobile phase. It is therefore recommended that this approach be considered for assessing desulfation efficiency.
5.7 Mass spectrometry
The majority of the currently available mass spectrometry ionization techniques and detector configurations have been reported for GLS detection. This includes fast atom bombardment (FAB)122 and matrix assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF),123,124 atmospheric pressure chemical ionization (APCI) and electrospray ionization (ESI). Ion traps,21,75,125 single quadrupole (LC-MS)15,74 and tandem quadrupole (LC-MS/MS) instruments118 have all been utilised. Quantification of known target analytes in single plant varieties is more commonly undertaken using quadrupole instruments.15,117 The preferred configuration for rapid identification of glucosinolates in crude plant extracts is ESI-LC-TOF.34,60,74,120,125 However, LC-MS cannot discriminate between the numerous GLS isomers. As an illustration, all three of the possible isomers for the 2′′, 3′′ and 4′′-acetylation of rhamnose-benzyl-GLS were readily observed, but the isomeric positions could not be assigned.22Precursor ion scanning can be used to locate all masses that produce the ions m/z 75 [S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CNOH]−, 80 [SO3H]−, 96 [SO4]− and 97 [SO4H]−,107 which is an advance over selected ion monitoring (SIM) for the same ions.15 Fragmentation patterns have been studied (Fig. 4), and match well with those data acquired by LC-MS/MS),119 and by Q-TOF.,125 whilst differing from those by ion-trap,106 an extension of the fragment naming system of Fabre is proposed (Fig. 4).
CNOH]−, 80 [SO3H]−, 96 [SO4]− and 97 [SO4H]−,107 which is an advance over selected ion monitoring (SIM) for the same ions.15 Fragmentation patterns have been studied (Fig. 4), and match well with those data acquired by LC-MS/MS),119 and by Q-TOF.,125 whilst differing from those by ion-trap,106 an extension of the fragment naming system of Fabre is proposed (Fig. 4).
![MS/MS fragmentation pattern of glucosinolates. Illustrated with sinigrin (2-propenyl-glucosinolate). The glucosinolate molecule fragments about the central isothiocyanate group, with cleavage of the alkyl, glucose and sulfate chains resulting in major ions for hydrogen sulfate m/z 97, sulfate radical anion m/z 96 and N-hydroxy-isothiocyanate m/z 75. Minor ubiquitous ions based on cleavage of the thioglucose and transfer of the sulfate group Glc1-5 m/z 195, 241, 259, 275 and 291 are present in most glucosinolate spectra. Other diagnostic ions are dependent on the R-group and are M-SO3 [M-80]−, M-glucose [M-162]−, M-thioglucose (+hydroxyl) [M-178]− and M-thioglucose-SO3 [M-242]−.21,106,107,119,125](/image/article/2010/AY/b9ay00280d/b9ay00280d-f4.gif) | ||
| Fig. 4 MS/MS fragmentation pattern of glucosinolates. Illustrated with sinigrin (2-propenyl-glucosinolate). The glucosinolate molecule fragments about the central isothiocyanate group, with cleavage of the alkyl, glucose and sulfate chains resulting in major ions for hydrogen sulfate m/z 97, sulfate radical anion m/z 96 and N-hydroxy-isothiocyanate m/z 75. Minor ubiquitous ions based on cleavage of the thioglucose and transfer of the sulfate group Glc1-5 m/z 195, 241, 259, 275 and 291 are present in most glucosinolate spectra. Other diagnostic ions are dependent on the R-group and are M-SO3 [M-80]−, M-glucose [M-162]−, M-thioglucose (+hydroxyl) [M-178]− and M-thioglucose-SO3 [M-242]−.21,106,107,119,125 | ||
Regulation of the biosynthetic pathways to glucosinolate production is central to Brassica metabolomics. Qualitative identification has progressed to become a sensitive tool for focused metabolomic analysis.106 One approach is based on a tandem quadrupole mass spectrometry, by multiple reaction monitoring (MRM) as the RIKEN database.121,126 A simpler approach is LC-TOF.34,120
5.8 Quantification
The most modern mass spectrometry studies on GLS analysis are able to report glucosinolate contents using semi-quantitative methods, whereby concentrations of other GLS are calculated using the response of sinigrin as a single calibration standard.126 While linear in response, the individual analyte calibration lines have been shown to be offset in slope 3-fold. The variation in absolute response makes semiquantitation (using one standard in place of another) inaccurate.117 CRMs and the corresponding indicative values have been used to construct LC-MS calibration curves to quantify unknowns.75 It is reported that ionisation is significantly influenced by the vegetable matrix, and standard addition and internal standardisation with isotopomers must be used for accurate quantification.118 Accurate quantification in LC-MS is completely reliant on having a pure standard of each target analyte.5.9 Purity of analytical standards: water content by NMR
The isolation of GLS and their elution from ion-exchange resin with potassium sulfate produces potassium salts, which while often presenting as white crystals, may not be pure. The organic content is measured by various procedures, generally HPLC-UV and acceptably high purities >95% are often quoted. Given the highly hygroscopic nature of GLS salts, it is unclear how much water of crystallisation is present in each standard. Water content in standards can be assessed by quantitative proton NMR (qHNMR). Trimethoxybenzene (0.333 mM) internal standard in 10% d4-methanol in D2O was used to dissolve 1000 ± 4 μg of solid glucosinolate (3 mM) in the NMR tube.60 For non-aromatic GLS, the integral area of the anomeric hydrogen at 4.5 ppm was compared to the area of nine methyl protons of the trimethoxybenzene. For aromatic GLS the region 7–8 ppm was integrated and then divided by the number of originating protons. True content ranged from 99% sinigrin/glucotropaeolin, 77% gluconapin, to 17% for 4-hydroxyglucobrassicin. This appears to be the most appropriate and accurate method for determining the absolute amount of a given GLS in a hitherto “chromatographically pure” standard.605.10 Extinction coefficients
Molar extinction coefficients are a more accessible indication of organic substances purity. One of the common correction procedures used in quantification with natural products standards is calculation of actual purity from a reference extinction coefficient. At the λmax of 224 nm, the N-hydroxysulfate moiety of glucosinolates has an extinction ε224 of around 7,000 M−1 cm−1. Indole, aryl and alkenyl side chains contribute absorbances with relatively lower intensity at 250–280 nm.112 The determination of definitive literature extinction values relies heavily on achieving consensus. Extinction coefficient measurements are generally of high accuracy and precision. An interlaboratory trial (n = 5) for the isoflavone genistein, with each participant independently sourcing the standard produced an extinction coefficient with a c.v. of 1.2%.127 The measurement precision ε235 for progoitrin and glucotropaeolin with three determinations at three concentrations (n = 9) was 3.2% c.v.117 For the most readily available and best characterised GLS sinigrin, the reported extinction coefficient values are 6,780 (ε235), 6,784, 6,950, 7,273 (ε227), 7,369 (ε227) 7,800 (ε227) and 8,000 (ε227) M−1 cm−1.77,109,117,128−131 The average of these values is 7,279 ± 483, with 6.6% c.v.Extinction values (ε235) for glucoiberin (6,234), glucoerucin (6,531), progoitrin (4,130), glucotropaeolin (8,312, 8,870 M−1 cm−1) are available.81,117 A much earlier value ε230.5 = 6,720 M−1 cm−1 is available for glucoconringiin (1959).132 Note that these extinction coefficients have no direct correlation to the desulfoglucosinolate response factors from the thymol assay. A decrease in extinction (ε227) from 7,800 to 5,700 M−1 cm−1 for desulfosinigrin suggests removal of the sulfate has a substantial effect on the central chromophore.131 The isothiocyanate sulforaphane derived from glucoraphanin [methylsulfinylbutyl-GLS ε235 = 6,872], has a more pronounced decrease in extinction coefficient, with ε238 of just 910 M−1 cm−1.77,133
In a single study, the ε227 values for purified sinigrin ranged from 7,140–7,662 M−1 cm−1, (average 7,379 ± 194, 2.6% c.v.). The standard error associated with the individual values was minimal (<0.4% c.v.), and the extraction solvent was considered to have influenced the extinction outcome.68 This variation can only be accounted for by the presence of impurities, such as water, organic solvent residues and salt. Temperature and solvent composition may also affect the accuracy of these determinations. Wathelet reports that with poor HPLC column temperature regulation, increasing the column temperature above 30 °C reduces the response of ds-4-hydroxyglucobrassicin.20 Linear regression calculations made at various concentrations have been used to establish if the UV response is linear in any given solvent or mixture. For synthetic 2,2-diphenylethyl-GLS ε227 decreased from 11,282 to 9,481 when acetonitrile was added (1:1) to phosphate buffer (0.1 M, pH 7.4), while ε227 of the isomer (biphenyl-2-yl)methyl-GLS remained similar (14,922 to 14,632). The measurements failed in pure acetonitrile. For the derived isothiocyanates the converse held and measurements failed in the aqueous buffer, but were similar in the 1:1 mixture and pure acetonitrile (10,131 to 10,901 diphenylethyl-ITC and 5,008 to 4,649 for (biphenyl)methyl-ITC).134
It appears that robust consensus values for extinction coefficient values of GLS have not yet been achieved, hence considerable further effort is required before these can be of use to the analytical chemist in correcting purity. It is recommended therefore that measurements are standardised as being conducted as ε227 in water at ambient (20–25 °C) temperature, using 6-point calibration lines with absorbance <1 (e.g. 5–60 μmol/l sinigrin A = 0.1–1.0), with r2 > 0.995, and where a plot of absorbance vs. molar concentration (M.L−1) has a slope equal to the extinction coefficient. For best accuracy this protocol should be conducted with replicate (n = 6) weighings, each of 20 mg, into wide mouthed vials to avoid static dispersion of the powder, followed by quantitative transfer into 250 ml volumetric flasks, giving 80 μg/ml. Individual solutions should be measured in triplicate, with the spectrometer precision <0.5% c.v., and overall measurement should achieve a precision of <1.0% c.v.
A (ε227), value as the mono-hydrated potassium salt ([Sinigrin-H]−·K+·H2O MW 415.48096) of 7,299 ± 28 (0.4% c.v.) was determined in this work. The 95% confidence interval of 7,271–7,327 overlaps with and is therefore statistically the same to the literature average value.
5.11 Reference materials
Certified reference materials (CRM) with total glucosinolate levels of 12.1 ± 0.8 μmol/g (CRM 366), 25.5 ± 0.9 μmol/g (CRM 190) and 102.4 ± 3 μmol/g (CRM 367) were prepared between 1988 and 1991.51 As stocks of CRM 190 were depleted, a further batch was prepared from the original refrigerated material and the other two materials were re-certified as 11.9 ± 1.3 μmol/g (BCR-366R), 23 ± 4 μmol/g (BCR-190R) and 99 ± 9 μmol/g (BCRM –367R).135 Now available as ERM-BC190, 366 and 367, these materials remain in use for assessing method performance, especially accuracy, some 20 years after preparation. Despite their availability and a modest price, these are used surprising infrequently.21,55,56,58,75 Indicative values of individual glucosinolate contents are given and portions of these CRMs and the corresponding indicative values have been used to construct calibration curves to quantify these known GLS.755.12 Labelled internal standards
Concurrently with obtaining pure individual analytical standards, stable isotope labelled glucosinolates are required for use as internal standards (IS) in mass spectrometric determination by isotope dilution mass spectrometry (IDMS). To reduce interference due to the natural abundance isotopic distribution in the mass spectrum, a suitable internal standard should have at least a +3 mass unit difference from the native analyte. Labels must be both stable and non-exchangeable and hence the use of deuterated standards is therefore discouraged. Most of the existing methods for isotopic labelling of GLS have incorporated either unstable deuterium136 or the isotopes into the side chain, which are more appropriate for metabolism studies, for example, [2,3,4,5,6-2H5]-phenylglucosinolate.137 The drawback of this approach is that a separate synthesis is required for each glucosinolate. In a greatly simplified approach, specifically to produce IDMS internal standards, our collaborators have prepared the 13C6-labelled building block 2,3,4,6-tetra-O-acetyl-1-thio-β-D-[13C6]glucopyranose. Coupling of this intermediate with various hydroximoyl chlorides affords the synthesis of any GLS in another three-step sequence. Three typical 13C6-labelled GLS were synthesised [glucose-13C6]-gluconasturtiin, [glucose-13C6]-sinigrin and [glucose-13C6]-glucoerucin.138 An isotopic purity assessment of [13C6]-sinigrin relative to unlabelled sinigrin is shown in Fig. 5 and these will be included in an IDMS-LC-MS validation against the official methods in due course.![Isotopic purity determination of 13C6-labelled sinigrin analytical standard, as full scan mass spectra by infusion of 10 μg/ml solutions in aqueous 0.1% formic acid. The labelled form has no measurable isotopic impurities from M–H to M + 4 (grey box), and is >99% isotopically pure with a <1% trace of [12C113C5]-glucose]sinigrin. The unlabelled form has no isotopic contribution form M + 3 to M + 7 (grey box). There is no overlap in the respective measurement channels (grey boxes), indicating suitability for use in isotope dilution standardisation.](/image/article/2010/AY/b9ay00280d/b9ay00280d-f5.gif) | ||
| Fig. 5 Isotopic purity determination of 13C6-labelled sinigrin analytical standard, as full scan mass spectra by infusion of 10 μg/ml solutions in aqueous 0.1% formic acid. The labelled form has no measurable isotopic impurities from M–H to M + 4 (grey box), and is >99% isotopically pure with a <1% trace of [12C113C5]-glucose]sinigrin. The unlabelled form has no isotopic contribution form M + 3 to M + 7 (grey box). There is no overlap in the respective measurement channels (grey boxes), indicating suitability for use in isotope dilution standardisation. | ||
5.13 Workplan for future research
This review highlights the basic aspects of glucosinolates research that are of importance to analytical chemists, and there is considerable scope for further progress. Readers are initially invited to add to the list of identified glucosinolates with any omissions, or new structures and other data, such as response factors and other relevant comments. An updated list would then be published as a technical note. Availability of the required range of individual analytical standards, together with their purity measurements is still hindering progress, and perhaps this will be the most difficult issue to resolve. Very little work has been carried out on enhancing stability with “stabilisers and keepers” and formal stability data are needed, especially in aqueous solution. An integrated programme is required where for example after ensuring the removal of ionic species such as free sulfate by ion exchange SPE, the water and glucosinolate ratios are established using quantitative NMR. A comprehensive table of assigned consensus extinction coefficients of monohydrated-GLS is required before any meaningful purity correction factors can be applied. Further work is needed on the optimisation of extraction conditions with the view to documenting enhanced chemical and thermal stability by processes such as microwave inactivation of enzymes prior to room temperature extractions. Chromatographic separation techniques and mass spectrometric assays continue to improve and it is no longer essential to desulfate GLS prior to analysis, providing that a robust quantification method is utilised. It is essential to fully harmonise, document and validate the next generation of glucosinolates methods and this has not yet been attempted. A comprehensive intercomparison study that uses these new approaches, including analysis of intact-GLC by LC-MS/MS vs. desulfation and by HPLC-UV, NMR purity measurement of standards, including the analysis of certified reference materials is required to raise this field of analysis to an acceptable standard.6. Discussion
Glucosinolate research continues to progress, with new technologies being applied to many well established problems. Enzymatic desulfation and the use of relative response factors remains the favoured route to the quantification of GLS in more complex samples. However, there remains a need for simple, sensitive, and robust, automated methods for the determination of intact glucosinolates that can be readily transferred into the wider analytical community. A mobile phase gradient with water/methanol and formic acid (0.1%) on a 100% aqueous compatible reverse phase support provides the optimal chromatographic condition for the separation of intact glucosinolates. Whilst new chromatographic phases and mass spectrometer configurations are facilitating the separation, detection and identification issues, quantification still remains the most problematic issue. Supply of analytical standards and measurement of their absolute purity against literature benchmarking values continues to be the single largest issue in quantification, and is the one where least progress has been made. Available official methods have been well validated, but are specific to the small niche of rape seed analysis. All subsequent work, such as on green-tissues, has not yet been validated.The standard of quality control (QC) in GLS analysis is poor relative to other areas of analytical chemistry. It is essential that published work in the area provides expanded experimental details and associated QC. Further data are needed on the absolute purity of standards, especially with respect to water and salt content, stability of standard mixtures in solution, extraction recoveries, myrosinase deactivation, enzymatic desulfation efficiencies, and most importantly, the analysis of reference materials. Provision of within and between batch reproducibility and precision measurements is requisite, as is thorough validation of new methods and a benchmarking comparison to the official methods is also required.
Acknowledgements
Financial support was provided by the UK Food Standards Agency (FSA) under contract E01086. The conclusions and opinions expressed are the views of the author alone.References
- D. A. Moreno, M. Carvajal, C. Lopez-Berenguer and C. Garcia-Viguera, J. Pharm. Biomed. Anal., 2006, 41, 1508–1522 CrossRef CAS.
- G. W. Haughn, L. Davin, M. Giblin and E. W. Underhill, Plant Physiol., 1991, 97, 217–226 CrossRef CAS.
- K. R. Anilakumar, F. Khanum and A. S. Bawa, J. Food Sci. Technol., 2006, 43, 8–17 Search PubMed.
- H. Zukalova and J. Vasak, Rostlinna Vyrobay, 2002, 48, 175–180 Search PubMed.
- A. M. Bones and J. T. Rossiter, Phytochemistry, 2006, 67, 1053–1067 CrossRef CAS.
- B. A. Halkier and J. Gershenzon, Annu. Rev. Plant Biol., 2006, 57, 303–333 CrossRef CAS.
- R. J. Hopkins, N. M. van Dam and J. J. A. van Loon, Annu. Rev. Entomol., 2009, 54, 57–83 CrossRef CAS.
- B. Holst and G. Williamson, Nat. Prod. Rep., 2004, 21, 425–47 RSC.
- M. E. Cartea and P. Velasco, Phytochem. Rev., 2008, 7, 213–229 CrossRef CAS.
- A. P. Vig, G. Rampal, T. S. Thind and S. Arora, LWT–Food Sci. Technol., 2009, 42, 1561–1572 CrossRef CAS.
- M. Traka and R. Mithen, Phytochem. Rev., 2009, 8, 269–282 CrossRef CAS.
- S. Das, A. K. Tyagi and H. Kaur, Curr. Sci., 2000, 79, 1665–1671 CAS.
- R. Verkerk, M. Schreiner, A. Krumbein, E. Ciska, B. Holst, I. Rowland, R. De Schrijver, M. Hansen, C. Gerhauser, R. Mithen and M. Dekker, Mol. Nutr. Food Res., 2009, 53, S219–S265.
- J. W. Fahey, A. T. Zalcmann and P. Talalay, Phytochemistry, 2001, 56, 5–51 CrossRef CAS.
- R. N. Bennett, F. A. Mellon and P. A. Kroon, J. Agric. Food Chem., 2004, 52, 428–438 CrossRef CAS.
- N. Bellostas, A. D. Sorensen, J. C. Sorensen and H. Sorensen, Adv. Bot. Res., 2007, 45, 369–415 Search PubMed.
- D. J. Kliebenstein, J. Kroyman, P. Brown, A. Figuth, D. Pedersen, J. Gershenzon and T. Mitchell-Olds, Plant Physiol., 2001, 126, 811–825 CrossRef.
- H. M. Radwan, M. M. El-Missiry, W. M. Al-Said, A. S. Ismail, K. A. Abdel Shafeek and M. M. Seif-El-Nasr, Res. J. Med. Med. Sci., 2007, 2, 127–132 Search PubMed.
- M. Kjaer, M. Ohashi and D. Carl, Acta Chem. Scand., 1963, 17, 2143–2154 CrossRef.
- J. P. Wathelet, R. Iori, O. Leoni, P. Rollin, A. Quinsac and S. Palmieri, Agroindustria, 2004, 3, 257–266 Search PubMed.
- T. R. I. Cataldi, A. Rubino, F. Lelario and S. A. Bufo, Rapid Commun. Mass Spectrom., 2007, 21, 2374–2388 CrossRef CAS.
- R. N. Bennett, F. A. Mellon, N. Foidl, J. H. Pratt, M. S. DuPont, L. Perkins and P. A. Kroon, J. Agric. Food Chem., 2003, 51, 3546–3553 CrossRef CAS.
- T. Songsak and G. B. Lockwood, Fitoterapia, 2002, 73, 209–216 CrossRef CAS.
- S. Montaut, J. Grandbois, L. Righetti, J. Barilarri, R. Iori and P. Rollin, J. Nat. Prod., 2009, 72, 889–893 CrossRef CAS.
- N. Agerbirk, C. E. Olsen and J. K. Nielsen, Phytochemistry, 2001, 58, 91–100 CrossRef CAS.
- A. A. M. Andersson, A. Merker, P. Nilsson, H. Sorensen and P. Aman, J. Sci. Food Agric., 1999, 79, 179–186 CrossRef CAS.
- M. Reichelt, P. D. Brown, B. Schneider, N. J. Oldham, E. Stauber, J. Tokuhisa, D. J. Kliebenstein, T. Mitchell-Olds and J. Gershenzon, Phytochemistry, 2002, 59, 663–671 CrossRef CAS.
- A. R. Hamed, K. A. Abdel-Shafeek, N. S. Abdel-Azim, S. I. Ismail and F. M. Hammouda, eCAM, 2007, 4, 25–28 Search PubMed.
- A. Frechard, N. Fabre, C. Pean, S. Moutaut, M.-T. Fauvel, P. Rollin and I. Fouraste, Tetrahedron Lett., 2001, 42, 9015–9017 CrossRef CAS.
- S.-J. Kim, C. Kawaharada, S. Jin, M. Hashimoto, G. Ishii and H. Yamauchi, Biosci., Biotechnol., Biochem., 2007, 71, 114–121 CrossRef CAS.
- S.-J. Kim, S. Jin and G. Ishii, Biosci., Biotechnol., Biochem., 2004, 68, 2444–2450 CrossRef CAS.
- R. N. Bennett, R. Carvalho, F. A. Mellon, J. Eagles and E. A. S. Rosa, J. Agric. Food Chem., 2007, 55, 67–74 CrossRef CAS.
- R. N. Bennett, F. A. Mellon, N. P. Botting, J. Eagles, E. A. S Rosa and G. Williamson, Phytochemistry, 2002, 61, 25–30 CrossRef CAS.
- J. J. B. Keurentjes, J. Fu, C. H. R. de Vos, A. Lommen, R. D. Hall, R. J. Bino, L. H. W. van der Plas, R. C. Jansen, D. Vreugdenhil and M. Koornneef, Nat. Genet., 2006, 38, 842–849 CrossRef CAS.
- D. Gueyrard, J. Barillari, R. Iori, S. Palmieri and P. Rollin, Proc. Phytochem. Soc. Eur., 2002, 47, 415–420 Search PubMed.
- J. C. Hall, K. J. Sytsma and H. H. Iltis, Am. J. Bot., 2002, 89, 1826–1842 CrossRef CAS.
- J. C. Hall, H. H. Iltis and K. J. Sytsma, Syst. Bot., 2004, 29, 654–669 Search PubMed.
- J. E. Rodman, K. G. Karol, R. A. Price and K. J. Sytsma, Syst. Bot., 1996, 21, 289–307 Search PubMed.
- J. E. Rodman, P. S. Soltis, D. E. Soltis, K. J. Sytsma and K. G. Karol, Am. J. Bot., 1998, 85, 997–1006 CrossRef CAS.
- M. E. Daxenbichler, G. F. Spencer, D. G. Carlson, G. B. Rose, A. M. Brinker and R. G. Powell, Phytochemistry, 1991, 30, 2623–2638 CrossRef CAS.
- D. Bailey, M. A. Koch, M. Mayer, K. Mummenhoff, S. L. O'Kane, S. I. Warwick, M. D. Windham and I. A. Al-Shehbaz, Mol. Biol. Evol., 2006, 23, 2142–2160 CrossRef.
- P. F. Stevens. (2001 onwards). Angiosperm Phylogeny Website. Version 9, June 2008http://www.mobot.org/MOBOT/research/APweb/.
- L. Henderson, J. Gregory and G. Swan. National Diet and Nutrition Survey: adults aged 19–64 years. Volume 1: types and quantities of foods consumed, The Stationery Office 2002 Search PubMed.
- L. F. D'Antuono, S. Elementi and R. Neri, J. Sci. Food Agric., 2009, 89, 713–722 CrossRef CAS.
- T. J. O'Hare and L. S. Wong, Acta Hortic., 2005, 694, 457–462 Search PubMed.
- A. V. Gasper, A. Al-Janobi, J. A. Smith, J. R. Bacon, P. Fortun, C. Atherton, M. A. Taylor, C. J. Hawkey, D. A. Barrett and R. F. Mithen, Am. J. Clin. Nutr., 2005, 82, 1283–1291 CAS.
- A. S. Rosa, R. Heaney, G. Fenwick and C. Portas, Hortic. Rev., 1997, 19, 99–215 Search PubMed.
- K. L. Falk and J. Gershenzon, J. Chem. Ecol., 2007, 33, 1542–1555 CrossRef CAS.
- T. J. O'Hare, L. S. Wong and L. E. Force, Acta Hortic., 2007, 744, 181–187 Search PubMed.
- P. D. Brown, J. G. Tokuhisa, M. Reichelt and J. Gershenzon, Phytochemistry, 2003, 62, 471–481 CrossRef CAS.
- J. P. Wathelet, P. J. Wagstaffe, A. Boenke, M. Marlier and M. Severin, Fresenius J. Anal. Chem., 1993, 347, 396–399 CAS.
- S. A. McNaughton and G. C. Marks, Br. J. Nutr., 2003, 90, 687–697 CrossRef CAS.
- A. Steinbrecher and J. Linseisen, Ann. Nutr. Metab., 2009, 54, 87–96 CrossRef CAS.
- A. Agudo, R. Ibáñez, P. Amiano, E. Ardanaz, A. Barricarte, A. Berenguer, M. D. Chirlaque, M. Dorronsoro, P. Jakszyn, N. Larrañaga, C. Martinez, C. Navarro, G. Pera, J. R. Quirós, M. J. Sanchéz, M. J. Tormo and C. A. González, Eur. J. Clin. Nutr., 2008, 62, 324–331 CrossRef CAS.
- K. Oerlemans, D. M. Barrett, C. B. Suades, R. Verkerk and M. Dekker, Food Chem., 2006, 95, 19–29 CrossRef CAS.
- S. Chuanphongpanich, D. Buddhasukh, P. Pirakitikuir and S. Phanichphant, Chiang Mai J. Sci., 2006, 33, 223–230 Search PubMed.
- ISO 1992. Rapeseed - Determination of glucosinolates content - Part1: Method using high-performance liquid chromatography. ISO 9167-1, 1–9 Search PubMed.
- M. Schreiner, P. Peters and A. Krumbein, J. Food Sci., 2007, 72, S585–S589 CrossRef CAS.
- AOCS. 1998. Official methods and recommended practices of the American Oil Chemists Society, 5th ed. AOCS, Champaign IL. Method American Oil Chemists Society. AK1–92 Determination of glucosinolate content in rapeseed and canola by HPLC. Revised 1993 Search PubMed.
- T. Mohn, B. Cutting, B. Ernst and M. Hamburger, J. Chromatogr., A, 2007, 1166, 142–151 CrossRef CAS.
- G. Kiddle, R. N. Bennett, N. P. Botting, N. E. Davidson, A. A. B. Robertson and R. M. Wallsgrove, Phytochem. Anal., 2001, 12, 226–242 CrossRef CAS.
- R. Verkerk and M. Dekker, J. Agric. Food Chem., 2004, 52, 7318–7323 CrossRef CAS.
- M. Dekker, K. Hennig and R. Verkerk, Czech. J. Food Sci., 2009, 27, S85–S88 Search PubMed.
- L. E. Force, T. J. O'Hare, L. S. Wong and D. E. Irving, Postharvest Biol. Technol., 2007, 44, 175–178 CrossRef CAS.
- D. J. Williams, C. Critchley, S. Pun, M. Chaliha and T. J. O'Hare, Phytochemistry, 2009, 70, 1401–1409 CrossRef CAS.
- B. Medeiros, A. W. Kirleis and R. J. Vetter, J. Am. Oil Chem. Soc., 1978, 55, 679–682 CrossRef.
- Y. J. Owusu-Ansah and M. Marianchuk. J. Food Sci., 56, pp. 1372–1374 Search PubMed.
- D. F. Stoin and R. D. Dogaru, Bull. USAMV-CN, 2007, 63, 77–82 Search PubMed.
- H. E. Van Doorn, G.-J. van Holst, G. C. van der Kruk, N. C. M. E. Raaijmakers-Ruijs and E. Postmae, J. Agric. Food Chem., 1998, 46, 793–800 CrossRef CAS.
- L. J. Rubin, L. L. Diosady, M. Naczk and M. Halfani, Can. Inst. Food Sci. Technol. J., 1986, 19, 57–61 Search PubMed.
- J. Liu, M. Shi, L. L. Diosady and L. J. Rubin, J. Food Eng., 1995, 24, 35–45 CrossRef.
- E. E. Powell, G. A. Hill, B. H. J. Juurlink and D. J. Carrier, J. Chem. Technol. Biotechnol., 2005, 80, 985–991 CrossRef CAS.
- J. Barillari, R. Cervellati, A. T. Paolini, A. Tatibouet, P. Rollin and R. Iori, J. Agric. Food Chem., 2005, 53, 9890–9896 CrossRef CAS.
- K.-C. Lee, M.-W. Cheuk, W. Chan., A. W.-M. Lee, Z.-Z. Zhao, Z.-H. Jiang and Z. Cai, Anal. Bioanal. Chem., 2006, 386, 2225–2232 CrossRef CAS.
- S. Millán, M. C. Sampedro, P. Gallejones, A. Castellón, M. L. Ibargoitia, M. Aranzazu Goicolea and R. J. Barrio, Anal. Bioanal. Chem., 2009, 394, 1661–1669 CrossRef CAS.
- C. Elfakir, J. P. Mercier and M. Dreux, Chromatographia, 1994, 38, 585–590 CAS.
- J. W. Fahey, K. L. Wade, K. K. Stephenson and F. E. Chou, J. Chromatogr., A, 2003, 996, 85–93 CrossRef CAS.
- L. R. Wetter and C. G. Youngs, J. Am. Oil Chem. Soc., 1976, 53, 162–164 CrossRef CAS.
- D. R. DeClercq and J. K. Daun, J. Am. Oil Chem. Soc., 1989, 66, 788–791 CrossRef CAS.
- Y. Zhang, K. L. Wade, T. Prestera and P. Talalay, Anal. Biochem., 1996, 239, 160–167 CrossRef CAS.
- W. Thies, Fat Sci. Technol., 1988, 90, 311–314 Search PubMed.
- J. Jezek, B. G. D. Haggett, A. Atkinson and D. M Rawson, J. Agric. Food Chem., 1999, 47, 4669–4674 CrossRef CAS.
- H. P. S. Makkar, P. Siddhuraju and K. Becker. Plant secondary metabolites. Methods in Molecular Biology, 2007, 393, Chapter 11 Glucosinolates, 55–60, Humana Press Inc., Totowa, NJ Search PubMed.
- S. S. Goyal, Commun. Soil Sci. Plant Anal., 2002, 33, 15–18.
- J. T. Tholen, G. Buzza, D. I. McGregor and R. J. W. Truscoir, Plant Breed., 1993, 110, 137–143 CrossRef CAS.
- ISO 2007. Rapeseed - Determination of glucosinolates content - Part 3: Spectrometric method for total glucosinolates by glucose release. ISO 9167–3: 2007 (E) Search PubMed.
- L. Stancik, L. Macholan, I. Pluhacek and F. Scheller, Electroanalysis, 1995, 7, 726–730 CAS.
- B. Wu, G. Zhang, S. Shuang, C. Dong, M. M. F. Choi and A. W. M. Lee, Sens. Actuators, B, 2005, 2, 700–707.
- M. M. F. Choi, M. M. K. Liang and A. W. M. Lee, Enzyme Microb. Technol., 2005, 36, 91–99 CrossRef CAS.
- C. G. Tsiafoulis, M. I. Prodromidis and M. I. Karayanis, Anal. Chem., 2003, 75, 927–934 CrossRef CAS.
- R. Font, M. Del Rio-Celestino, E. Rosa, A. Aires and A. De Haro-Bailon, J. Agric. Sci., 2005, 143, 65–73 Search PubMed.
- F. Ul-Hassan, A. Manaf, G. Qadir and S. M. A. Basra, Int. J. Agric. Bot., 2007, 9, 504–508 Search PubMed.
- ISO 1994. Rapeseed - Determination of glucosinolates content - Part 2: Method using X-ray fluorescence spectrometry. ISO 9167–2: 1994 Search PubMed.
- I. Michinton, J. Sang, D. Burke and R. J. W. Truscott, J. Chromatogr., A, 1982, 247, 141–148 CrossRef CAS.
- B. Kusznierewicz, A. Bartoszek, L. Wolska, J. Drzewiecki, S. Gorinstein and J. Namiesnik, LWT–Food Sci. Technol., 2008, 41, 1–9 CrossRef CAS.
- M. E. Cartea, V. M. Rodríguez, A. de Haro, P. Velasco and A. Ordás, Euphytica, 2008, 159, 111–122 CAS.
- J. P. Wathelet, N. Mabon and M. Marlier. Determination of glucosinolates in rapeseed improvement of the official HPLC ISO method (precision and speed). Proceedings of the 10th International Rapeseed Congress, Canberra, Australia. 1999, p 185.
- H. J. Fiebig, Lipid Fett., 2006, 93, 264–267 Search PubMed.
- B. Matthäus and H.-J. Fiebig, J. Agric. Food Chem., 1996, 44, 3894–3899 CrossRef.
- M. Francisco, D. A. Moreno, M. E. Cartea, F. Ferreres, C. García-Viguera and P. Velasco, J. Chromatogr., A, 2009, 1216, 6611–6619 CrossRef CAS.
- K. Hrncirik, J. Velisek and J. Davidek, Z. Lebensm.-Unters. -Forsch. A, 1998, 206, 103–107 CrossRef CAS.
- R. Buchner. Approaches to determination of HPLC response factors for glucosinolates. Glucosinolates in rapeseeds: Analytical Aspects. J. P. Wathelet, ed, World Crops vol. 13: Production, Utilization, Description. Martinus Nijhoff Publishers, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1987, pp 50–58. ISBN 90-247-3525-4 Search PubMed.
- European Community ( 1990). European Economic Community, Commission Regulation, EEC No 1864/90. Oilseeds - determination of glucosinolates - high performance liquid chromatography. Official Journal of the European Community L, 170: 27–34 Search PubMed.
- D. V. Vinjamoori, J. R. Byrum, T. Hayes and P. K. Das, J. Anim. Sci., 2004, 82, 319–328 Search PubMed.
- O. Leoni, R. Iori, T. Haddoum, M. Marlier, J. P. Wathelet, P. Rollin and S. Palmieri, Ind. Crops Prod., 1998, 7, 335–343 CrossRef CAS.
- S. J. Rochfort, V. C. Trenerry, M. Imsic, J. Panozzo and R. Jones, Phytochemistry, 2008, 69, 1671–1679 CrossRef CAS.
- D. Skutlarek, H. Farber, F. Lippert, A. Ulbrich, A. Wawrzun and H. Buning-Pfaue, Eur. Food Res. Technol., 2004, 219, 643–649 CrossRef CAS.
- S. Buskov, J. Hasselstrom, C. E. Olsen, H. Sorensen, J. C. Sorensen and S. Sorensen, J. Biochem. Biophys. Methods, 2000, 43, 157–174 CrossRef CAS.
- N. Bellostas, J. C. Sorensen and H. Sorensen, J. Chromatogr., A, 2006, 1130, 246–252 CrossRef CAS.
- C. Bjergegaard, S. Michaelsen, P. Moller and H. Sorensen, J. Chromatogr., A, 1995, 717, 325–333 CrossRef CAS.
- G. Bringmann, I. Kajahn, C. Neusüss, M. Pelzing, S. Laug, M. Unger and U. Holzgrabe, Electrophoresis, 2005, 26, 1513–1522 CrossRef CAS.
- J. K. Troyer, K. K. Stephenson and J. W. Fahey, J. Chromatogr., A, 2001, 919, 299–304 CrossRef CAS.
- K. L. Wade, I. J. Garrard and J. W. Fahey, J. Chromatogr., A, 2007, 1154, 469–472 CrossRef CAS.
- C. Elfakir and M. Dreux, J. Chromatogr., A, 1996, 727, 71–82 CrossRef CAS.
- M. El-Haddad, S. Lazar, S. El-Antri, M. N. Benchekroun, M. Akssira and M. Dreux, J. Liq. Chromatogr. Relat. Technol., 2003, 26, 751–761 CrossRef CAS.
- F. A. Mellon, R. N. Bennett, B. Holst and G. Williamson, Anal. Biochem., 2002, 306, 83–91 CrossRef.
- Q. Tian, R. A. Rosselot and S. J. Schwartz, Anal. Biochem., 2005, 343, 93–99 CAS.
- L. Song, J. J. Morrison, N. P. Botting and P. J. Thornalley, Anal. Biochem., 2005, 347, 234–243 CrossRef CAS.
- K.-C. Lee, W. Chan, Z. Liang, N. Liu, Z. Zhao, A. W.-M. Lee and Z. Cai, Rapid Commun. Mass Spectrom., 2008, 22, 2825–2834 CrossRef CAS.
- J. J. Jansen, J. W. Allwood, E. Marsden-Edwards, W. H. van der Putten, R. Goodacre and N. M. van Dam, Metabolomics, 2008, 5, 150–161.
- Y. Sawada, A. Kuwahara, M. Nagano, T. Narisawa, A. Sakata, K. Saito and M. Y. Hirai, Plant Cell Physiol., 2009, 50, 1181–1190 CrossRef CAS.
- P. Kokkonen, J. van der Greef, W. M. A. Niessen, U. R. Tjaden, G. J. ten Hove and G. van de Werken, Rapid Commun. Mass Spectrom., 1989, 3, 102–106 CAS.
- C. H. Botting, N. E. Davidson, D. W. Griffiths, R. N. Bennett and N. P. Botting, J. Agric. Food Chem., 2002, 50, 983–988 CrossRef CAS.
- R. Shroff, F. Vergara, A. Muck, A. Svatos and J. Gershenzon, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6196–6201 CrossRef CAS.
- N. Fabre, V. Poinsot, L. Debrauwer, C. Vigor, J. Tulliez, I. Fouraste and C. Moulis, Phytochem. Anal., 2007, 18, 306–319 CrossRef CAS.
- Y. Sawada, K. Akiyama, A. Sakata, A. Kuwahara, H. Otsuki, T. Sakurai, K. Saito and M. Y. Hirai, Plant Cell Physiol., 2009, 50, 37–47 CrossRef CAS.
- H. Wiseman, K. Casey, D. B. Clarke, K. A. Barnes and E. Bowey, J. Agric. Food Chem., 2002, 50, 1404–1410 CrossRef CAS.
- S. Palmieri, O. Leoni and R. Iori, Anal. Biochem., 1982, 123, 320–324 CAS.
- P. Sakorn, N. Rakariyatham, H. Niamsup and P. Kovitaya, ScienceAsia, 1999, 25, 189–194 CrossRef CAS.
- C. Barth and G. Jander, Plant J., 2006, 46, 549–562 CrossRef CAS.
- W. Thies, Naturwissenschaften, 1979, 66, 364–365 CrossRef CAS.
- R. Gmelin and A. I. Virtanen, Acta Chem. Scand., 1959, 13, 1718–1719 CrossRef CAS.
- J. W. Fahey, X. Haristoy, P. M. Dolan, T. W. Kensler, I. Scholtus, K. K. Stephenson, P. Talalay and A. Lozniewski, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7610–7615 CrossRef CAS.
- J. R. Mays, R. L. Weller Roska, S. Sarfaraz, H. Mukhtar and S. R. Rajski, Chem. Biol. Chem., 2008, 9, 729–747 Search PubMed.
- T. Linsinger, N. Kristiansen, N. Beloufa, H. Schimmel and J. Pauwels. 2001. Certification report. European Commission Joint Research Centre, Institute for Reference Materials and Measurements, Geel, Belgium. Report EUR 19764 EN. ISBN92-894-0892-8.
- A. A. B. Robertson and N. P. Botting, J. Labelled Compd. Radiopharm., 2006, 49, 1201–1211 CrossRef CAS.
- J. B. Bialecki, J. Ruzicka and A. B. Attygalle, J. Labelled Compd. Radiopharm., 2007, 50, 711–715 CrossRef CAS.
- Q. Zhang, T. Lebl, A. Kulczynska and N. P. Botting, Tetrahedron, 2009, 65, 4871–4876 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: A database of structures, formulae and accurate masses of both the 200 known, and a further 180 predicted GLS, for use in mass spectrometry; Fig. S1. Further glucosinolates; Fig. S2. Screening for cinnamoyl and benzoyl esters; and Table S1. Some sources of Brassicale seeds in the UK. See DOI: 10.1039/b9ay00280d |
| This journal is © The Royal Society of Chemistry 2010 |