Recent advances in on-line multidimensional liquid chromatography
Helle
Malerod
*,
Elsa
Lundanes
and
Tyge
Greibrokk
Department of Chemistry, University of Oslo, P.O. Box 1033, Blindern, NO-0315, Oslo, Norway. E-mail: hellema@kjemi.uio.no; Fax: +47 228 55441; Tel: +47 228 55573
First published on 23rd December 2009
Abstract
Multidimensional liquid chromatography (MD LC) has, due to enhanced peak capacity compared to one-dimensional LC, become an important analytical tool for separating components in complex samples, e.g. in proteomics. MD LC can be performed both on-line and off-line. Because of the advantages like possible automation and minimal sample loss, on-line MD LC has appeared to be very attractive also for high throughput analysis. This review includes only on-line coupled MD LC. Different aspects related to the compatibility of separation principles and column dimensions are highlighted and recent applications using on-line mainly two-dimensional (2D) LC have been included.
 Helle Malerod | Helle Malerod received her cand.scient degree in analytical chemistry at the Department of Chemistry, University of Oslo (UiO) in 2005. She started her PhD degree at UiO under supervision of Prof. Elsa Lundanes and Prof. Tyge Greibrokk in 2006. In her PhD, she is developing multidimensional liquid chromatographic methods coupled with mass spectrometry for identification of peptides and proteins in various biological samples. She is now in her last year as a PhD student. |
 Elsa Lundanes | Elsa Lundanes is Professor of Analytical Chemistry at the Department of Chemistry, University of Oslo, Norway. Her research interests are development of analytical methods for organic compounds based on miniaturized liquid chromatography and mass spectrometry. Together with Prof. Tyge Greibrokk, Lundanes is a core member of Bioanalytics@UiO, the center of analytical chemistry and separation science in Norway. |
 Tyge Greibrokk | Tyge Greibrokk is Professor of Analytical Chemistry at the Department of Chemistry, University of Oslo, Norway. During the last 35 years he has worked on the development of new methods and techniques in chromatography, mainly HPLC but formerly also supercritical fluid chromatography, electrochromatography as well as microplasma GC-MS. He is one of the editors of the Journal of Separation Science and has served on the editorial boards of half a dozen other journals. Currently his main research is connected to miniaturizing HPLC columns with particular emphasis on 2D systems. Greibrokk is also a core member of Bioanalytics@UiO. |
1. Introduction
Multidimensional liquid chromatography (MD LC) methods, with enhanced peak capacity compared to one-dimensional (1D) LC have in recent years become more frequently used for routine analyses, and in more explorative applications e.g. proteomics. In MD LC, components are subjected to separation by two or more separation mechanisms before detection. An alternative is a combination of a chromatographic separation and a subsequent detection by mass spectrometry (MS) or photodiode array detection (PDA) serving as the second dimension. MS provides more information than PDA and is often the detector of choice in MD LC. Furthermore, due to the requirement of increased peak capacity, MS or PDA is more commonly applied as the third dimension. The total theoretical peak capacity of the method is the product of the peak capacity of each dimension; Evans et al. reported a peak capacity of 3200 for a two-dimensional (2D) 15-fraction multidimensional protein identification technology (MudPIT) analysis, but when incorporating MS as a third dimension, the total peak capacity increased to 23 000.1The term MD LC has been used for combinations of different LC separation principles as well as methods including electrophoresis.2,3 Several reviews on different aspects of MD LC have been published the last years.4–14 In this review, we will only focus on on-line MD LC applying LC in two or more dimensions. Compared to off-line MD LC, on-line MD LC methods are more complicated, require more technical skills and demand a higher compatibility between the applied separation systems. The advantages are minimal sample loss and contamination, high repeatability and possibility of automation, which make the on-line methods very attractive, also for high throughput analysis. In this review, the term on-line MD LC is used for instrument systems where the eluent from the first dimension is automatically transferred to the second dimension via tubing and/or a valve with connecting storage loops or trap columns.15 Both heart-cutting (LC - LC) and comprehensive fractionation (LC × LC) can be performed by on-line MD LC. In the former approach, only a part of the first dimension eluent is transferred to the second dimension for subsequent separation.16–18 This technique is mostly used for determination of one or few target analytes. In comprehensive MD LC, the entire sample, divided in several fractions is transferred from the first to the second dimension for further LC analysis.18 Most on-line MD LC systems reported apply 2D LC, we will therefore restrict our description of MD LC instrumentation to on-line 2D LC.
2. Multidimensional liquid chromatographic (MD LC) methodology
2.1 Peak capacity in MD LC
| nMD = n1·n2…nn | (1) |
However, if the number of collected fractions is low, separable peaks will be collected in the same fractions (undersampling) and the resolution is reduced.19,25 Undersampling occurs very often in separation of complex samples as in proteomics and peptidomics, when the sample amount is limited and sensitivity is often more crucial than increased resolution and peak capacity. Due to first dimension undersampling, the actual peak capacity in the first dimension cannot be higher than the number of fractions collected.28 Even though the peak capacity is increased with increasing fractionation frequency, peak overlap makes the identification of compounds more difficult, especially in shot gun analysis. Thus, the peak width and complexity of the sample should decide the number of fractions to be collected.2 Hence, when the peak width varies during the first dimension separation, the fractionation frequency should vary as a function of the peak width. Additionally, with complex samples, it may be impossible to divide each peak in three fractions and hence a compromise has to be made. The effect of undersampling on first dimension peak capacity as described by Li et al. should also be considered.23
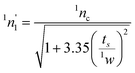 | (2) |
2.2 2D LC method development
When developing an on-line 2D LC method for comprehensive purposes, substantial knowledge as well as technical skills are needed due to the requirement of compatibility of LC dimensions. The analytes and sample type decide which separation mechanisms to apply and in order to achieve as much sample information as possible, an 2D LC system with high peak capacity is sought. In heart-cut analysis, it is more important to be able to separate the target compounds from all the matrix compounds, than to obtain high peak capacity.In both 2D LC techniques, column type and dimension, particle size, mobile phase composition and flow rate applied in both dimensions have to be compatible and optimized in order to maximize the peak capacity. In addition the dimensions of the tubing and valves/connectors used in the 2D LC system need to be compatible with the column dimensions and the flow rates in order to avoid flow inconsistency and extended dilution. Flow inconsistency can especially become a problem in column-switching 2D LC where two or more valves are included and can cause significantly changes in system-back pressure and consequently deviations in mobile phase flow rate and reduction in column lifetime.36 It is also important that the experimental conditions are optimized for minimum dilution in order to have maximum detector response.37 The dilution factor in a 2D LC system is the product of the dilution factor in each dimension and has been reported to be between 200 and 3000.37 However, when trap columns are incorporated for fraction collection, the analytes will be reconcentrated and the dilution factor will therefore be significantly decreased.
In an on-line 2D LC the analytes can be focused on the second dimension column by having a suitable mobile phase composition, volume of the fraction and column dimension. If the mobile phases differ in viscosity, like mixing ACN–water and MeOH–water mobile phases, considerable band broadening may occur and mobile phases with different viscosity should be avoided.16 The second (or last) dimension usually requires compatibility with MS, which is most often used for detection in 2D LC, and with electrospray ionization (ESI) the problem of ion suppression must be handled. When developing an on-line 2D LC method, it can therefore be useful to optimize the last dimension before designing the first dimension instrumental set up by choosing the correct flow rate, mobile phase composition, column dimension and particle size.38
2.3 On-line 2D LC instrumentation
On-line 2D LC can be performed by directly coupled columns or by column switching 2D LC. In column-switching 2D LC, a valve is used to connect the dimensions. With a valve interface, greater flexibility and repeatability is gained and it is easier to achieve compatibility with different separation mechanisms than directly coupled columns.1 | ||
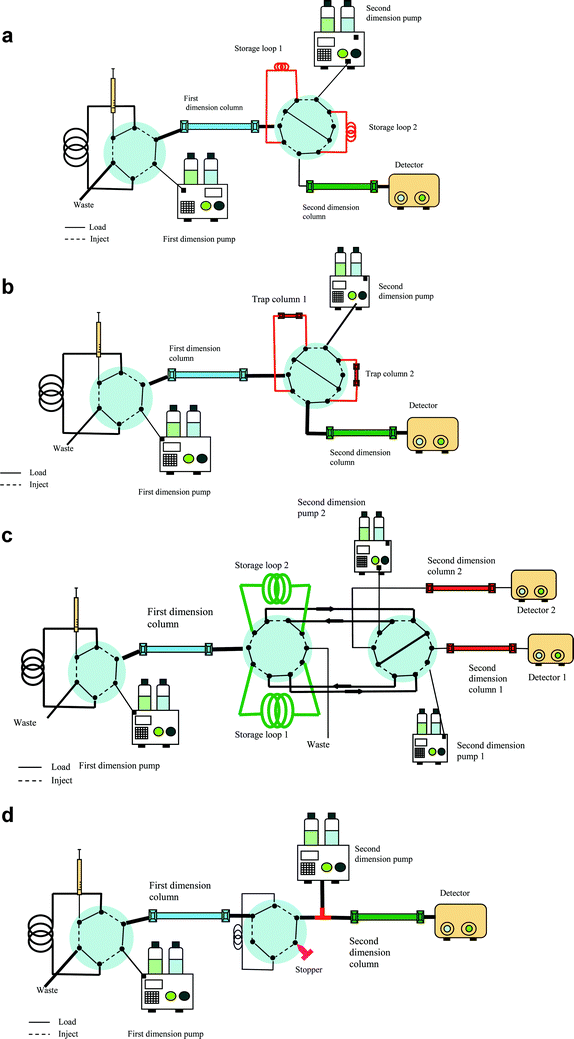
| Fig. 1 A biphasic column containing segments of SCX and RP for use in an on-line 2D LC system. | ||
 | ||
| Fig. 2 Column-switching 2D LC instrumentation using a) storage loops b) trap columns c) parallel analytical columns in the second dimension or the d) stop-flow approach for collecting first dimension fractions on-line. | ||
Storage loops. When using multiple storage loops, the loop volumes should be identical.8,16 The storage volume should at least be equal to the volume of each fraction, but due to the band broadening in the connecting tubing, a higher volume is preferred.19 In a 2D LC system with two storage loops only, the content of one loop is transferred for second dimension separation, while the other loop is filled with first dimension eluent.8 With this approach multiple fractionations can be performed. However, the fractionation frequency governs the second dimension separation time; with frequent (less than 1–2 min) fractionation, the second dimension separation including the re-equilibration of the column needs to be very fast.8 Fast second dimension separation can be accomplished by having a short analytical column packed with small particles, elevated temperature or monolithic columns.24,42,43When using higher temperature, the peak shape can be improved and the backpressure of the column will additionally be reduced, allowing faster separation. Fast gradients can be applied on monolithic columns without affecting the resolution, and the columns require short conditioning times.44
Trap columns and parallel analytical columns. The second dimension peak capacity has a large impact on the total peak capacity and if improved separation is required using longer separation time, storage loops can be replaced by trap columns enabling reconcentration of the components prior to the subsequent separation.8,45 When trap columns are applied for fractionation, the analytes can be focused on the trap column prior to the second dimension separation allowing narrow sample bands to be introduced to the second dimension column. The number of trap columns depends on the number of first dimension fractions and the second dimension separation time, but two trap columns are often applied, as shown in Fig. 2b. In a column-switching 2D LC system with only two trap columns, the analytes retained on the first trap column is transferred (by backflushing) to the second dimension for subsequent analysis, while the next fraction is diverted on the second trap column. When a longer second dimension separation is required two trap columns might, however, not be sufficient and the number has to be increased. Wilson et al. demonstrated the use of a column-switching MD LC system with 18 trap columns.46,47 With this on-line 2D LC instrumentation, several first dimension fractions can be collected on individual trap columns during the separation. The second dimension separation can therefore be longer. However, the total analysis time will be increased. Alternatively, the trap columns can be replaced by two analytical columns that are connected in parallel in the second dimension with individual sample loops (Fig. 2c). With such an on-line 2D LC system, one column is being loaded with a fraction collected from the first dimension, while the components retained on the second column are separated.2
Stop-flow. A longer second dimension separation can also be accomplished by the stop-flow technique with step gradients.48 In the stop-flow technique, two analytical columns are connected via a valve without using storage loops or trap columns (Fig. 2d).16,19,48,49 By switching the valve, the desired fraction is transferred to the second dimension, the primary flow is stopped and hence the first dimension separation, in order to carry out separation on the second column.16,19,48 After the second dimension separation, the primary flow is restarted to provide another fraction. When using this approach, the second dimension column needs to be longer to obtain a higher plate number compared to on-line 2D LC with continuous flow.16 This column-switching 2D LC approach can be very time demanding compared to the other 2D LC techniques and only few first dimension fractions are therefore collected, but the stop-flow technique with refocusing does not result in additional band broadening.49
2.4 Column dimensions
The column dimensions depend on the application, but also on the mobile phase composition, flow rate and MS compatibility. LC columns with capillary/microflow and nanoflow inner diameters are dominating today due to the high MS compatibility and many life science applications.As previously mentioned, the mobile phases applied in the different dimensions of on-line MD LC need to be sufficient compatible. The first dimension eluent is required to have a weak elution strength in the second LC dimension to avoid band broadening and to preserve the first dimension separation on the second LC dimension.10,52 Some mobile phase incompatibility can to a certain degree be compensated for by choosing the appropriate column dimensions. For instance, when a nanoflow column is used in the first dimension and a conventional sized column in the second dimension, only a small volume of the first dimension mobile phase is introduced on the second column and with no extra band broadening even when introducing a solvent with high elution strength. Finding optimal combinations of mobile phases in the different dimensions is often the most challenging part in developing MD LC methods.
2.5 Combination of different separation mechanisms in 2D LC
Columns with different separation mechanisms should be selected to achieve the needed separation orthogonality in 2D LC. Mechanisms could include reversed phase (RP), ion-exchange chromatography (IEC) including cation exchange chromatography (CX) and anion exchange chromatography (AX), size exclusion chromatography (SEC) and normal phase chromatography (NP) depending on the analytes and type of sample. All combinations provide high selectivity as well as peak capacity compared to 1D LC. Due to high compatibility with ESI-MS, RP is usually used in the second dimension.In IEC-RP, components are mainly separated by electrostatic interactions in the first dimension and hydrophobic interactions in the second RP dimension. Ions are eluted from the IEC column by a salt and/or pH gradient.6,55–58 When IEC is used in the first dimension in combination with a salt gradient, the RP column or a trap column will also function as a desalting column prior to MS detection.6,55 With a pH gradient, proteins are eluted according to their isoelectric point (pI).56–58 During pH gradient elution, the components can be focused in narrow bands providing better peak shape than with the salt gradient. Negatively charged components are retained on an AX column when the pH is higher than their pI and will elute when the pH is lower or the same as their pI.53,54,59 The scarcity of MS-compatible buffers represents a problem when IEC is used in the second dimension.53,54,60
SCX-RP is used in on-line 2D LC combinations for many different applications, for instance in peptidomics/proteomics using MudPIT,61–72 for alkaloids,73 traditional Chinese medicine74 and B-vitamins in food analysis.75 The orthogonality between SCX and RP is high, but in addition to electrostatic interactions in the SCX, hydrophobic interactions may take place due to the polymeric backbones of the stationary phase and the orthogonality is slightly reduced.4 Thus for SCX elution, addition of an organic modifier to the mobile phase will decrease the hydrophobic interactions.4 Dependent on the application, the amount of organic modifier may however be restricted. In order to maintain weak elution strength into the second RP dimension, only 5–10% acetonitrile (ACN) or methanol (MeOH) can normally be included in the mobile phase for peptides at a low pH.4
An AX pH gradient usually contains a low concentration of salts and organic modifier and is therefore highly compatible with on-line RP LC. AX-RP is used for separation of macromolecules like proteins,56–58 but also for smaller anions like benzoic acids.76 SAX-RP has also been applied for protein fractionation with on-line transfer to an immobilized trypsin reactor for on-line trypsination and the resulting peptides were subsequently separated on an RP analytical column.58 Ren et al. recently also demonstrated the use of SAX-RP MS for determination of tryptic digested E. coli expressed human IgG Fc fusion protein.77
A variant of NP chromatography, hydrophilic interaction liquid chromatography (HILIC) appears to be more compatible with RP than the ordinary NP. HILIC can be characterized as NP, with hydrophilic functional groups as the stationary phase, applying aqueous mobile phases, most often with ACN as the organic component.90 Because of different separation mechanisms, the orthogonality of a HILIC-RP 2D LC system is high resulting in high theoretical peak capacity. Gilar et al. determined the peak capacity of a HILIC-RP system to be 4616, approximately twice as high as SCX-RP.22 Improved resolution and peak capacity compared to SCX-RP were also reported by Mihailova et al. for separation of neuropeptides in rat brain tissue.46 The HILIC mobile phase contains a high percentage of organic modifier, and analyte focusing on the second dimension might be difficult in an on-line coupling. However, by applying a rather narrow first dimension column and low flow rate, while introducing a larger inner diameter column in the second dimension and higher flow rate, the eluent is diluted and only a small percentage of organic solvent is introduced into the second dimension. Alternatively, a diluting pump can be placed between the two dimensions in order to decrease the percentage of organic modifier before transfer to the RP-column with the same dimensions as the HILIC column, as shown in Fig. 3.46,47,91 When applying an instrumental set-up like this, the volumetric flow rate of the diluting pump must be considered and must not be too high in order to avoid breakthrough on the trap columns serving as the interface in column-switching 2D LC.
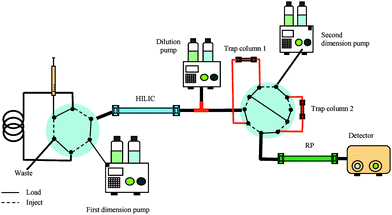 | ||
| Fig. 3 An on-line HILIC-RP column-switching system with two trap columns, using a diluter pump between the first and second dimension in order to dilute the HILIC eluate containing a high percentage with ACN. | ||
To obtain phase focusing on a HILIC column in order to increase sample load, the analytes have to be dissolved in a high percentage of organic modifier, like acetonitrile. Nevertheless, polar compounds like peptides have limited solubility in acetonitrile and prefer more aqueous solvents. This can be arranged by connecting a small RP SPE column connected to the 2D LC system prior to the HILIC column for injecting aqueous samples.46,47 On-line HILIC-RP have recently been applied for separation of samples containing peptides46,47,55 and drugs.91,92 Due to the compatibility with MS, HILIC can also be applied in the second dimension for determination of e.g. peptides.93,94
3. Three-dimensional liquid chromatography (3D LC)
Even though 2D LC provides higher peak capacity than 1D LC, the resolution might still be too limited for highly complex samples. Thus, a third dimension can be incorporated to increase the peak capacity even more.The 3D LC separation requires the choice of three independent and mutually orthogonal separation dimensions. Unfortunately, the selection of three types of LC modes with sufficient orthogonal selectivity and mobile phase compatibility is a difficult task.110 It will also be more complicated to obtain compatibility between column dimensions, mobile phase compositions and flow rates. Additionally, studies have shown that when incorporating a third LC dimension, the peak capacity does not necessarily improve significantly compared to 2D LC. This may be the case when storage loops are used to collect fractions from the first dimension applying fast second dimension separations. Then, the third separation has to be carried out even faster than the second dimension separation and consequently the peak capacity for each dimension will decrease.110 The amount of data that will be generated with 3D LC will be extensive and problematic to handle even though new and improved software for data acquisition and processing likely will be developed in the future.110 Only a few studies have been reported to be on-line 3D LC.111,112 In two studies the reported 3D LC separation was performed using an triphasic column with segments of RP, SCX and RP.111,112 With this on-line 3D LC system, no additional valves and pumps are incorporated making the system simpler and easier to handle.
4. Recent applications
As mentioned above, on-line 2D LC is widely used for separation in life science applications like peptidomics/proteomics as well as for food and industrial applications. Some of the recently published applications (from 2007 to September 2009) are included in this review.4.1 Proteins
Two-dimensional gel electrophoresis is widely used for separating proteins but this method is not always suited e.g. not for membrane proteins. LC has therefore become an alternative method to 2D gel electrophoresis in proteomics. However, proteins require special conditions for solubilization to prevent denaturation. Hence in most cases, proteins are digested prior to on-line MD LC analysis, since peptides are easier to handle, in a bottom-up approach. Alternatively, the proteins can be separated in the first dimension before on-line digestion and subsequent second dimension peptide separation, as reported by Tran et al.58,113 Far more studies using MD LC for separation of digested proteins (peptides) compared to separation of intact proteins have been published.The traditional MudPIT strategy with SCX-RP for peptide separation can be used for protein separation as well.72,114–116 Gao et al. used this approach to deplete high-abundance proteins in human liver tissue.72 After protein depletion of the 53 most abundant proteins, the remaining proteins were digested before subsequent peptide separation.72 As previously mentioned, AX is often preferred for protein separation due to high capacity and almost non-denaturating elution conditions.53,54 AX has been combined with RP for identification and quantification of degradation products in E. coli and determination of apolipoproteins in human plasma.113,57,56,77 Tran et al. demonstrated the use of an SAX-RP system with on-line trypsination.113 Native proteins were separated on a SAX column using a pH gradient, before fractionation on C4 trap columns with on-column reduction and alkylation. Nine fractions were collected in total. The reduced and alkylated proteins were subsequently separated on a RP column followed by on-line digestion and ESI-MS detection.113 The reduction, alkylation and tryptic digestion step were carried out using the stop-flow approach. Even though this on-line instrumentation is fairly complicated, both protein separation and digestion can be performed on-line minimizing the sample handling. The total time for each analysis was reported to be 320 min for one fraction and with additional 60 min for each fraction. Although, this is a quite time consuming MD LC method, the total time is still reduced compared to the traditional off-line methods including protein digestion procedure which is commonly done overnight.
AX has also been combined with HILIC for separating derivatives of N-glycans.117 The AX and HILIC columns were directly coupled and a mobile phase containing water, ACN and ammonium acetate was used.117 With a step-gradient, four fractions in total were transferred to the HILIC column.117
Depletion of high abundant proteins has also been carried out using RP in combination with columns packed with restricted access material (RAM) or single or tandem immunoaffinity columns.118–120 Different immunoaffinity columns can be combined depending on which proteins are most abundant. The depletion is never complete, but is still reported to be very high (99.7%).120
Since the concentration of most proteins is usually low and necessitates a compatible MS detection, capillary/microflow and nanoflow columns have become a requirement in proteomics. However, peptides are easier to determine and proteins can therefore be digested after the on-line 2D LC separation with an additional LC separation and MS detection. Because of high complexity it is also important to use an MD LC method with high peak capacity. Trap columns are often used to collect fractions in order to have an additional preconcentration prior to the second dimension separation.
4.2 Peptides
Peptides, either as digested proteins in proteomics or endogenous as in peptidomics, are frequently separated by on-line MD LC applying different combinations of separation mechanisms. However, the method of choice is often the MudPIT strategy using SCX in the first dimension and RP in the second dimension.40,51,61–72,78,121–128As an alternative to MudPIT, Motoyama et al. demonstrated the use of a mixed-bed ion exchange column with a combination of AX and CX to improve peptide recovery and detection compared to only SCX-RP.4 The reported increased recovery is due to the added retention of acidic peptides in addition to the basic and neutral peptides, but the retention of basic and neutral peptides was slightly reduced.4 Even though AX is more used for protein analysis, a fully automated AX-RP system was used to separate and identify phosphopeptides in HeLa cells using a pH gradient in the first dimension.129 More than 1800 phosphopeptides were identified and when comparing the AX-RP method with another using combined cation exchange and titanium dioxide, the AX-RP system identified more acidic and multiphosphorylated peptides.129 As mentioned earlier, RP in combination with HILIC has demonstrated increased peak capacity compared to SCX-RP and has therefore also been applied for separation of peptides.46,47,93
Francois et al. applied an RP-RP LC system with different mobile phase pH using the same RP analytical columns for separation of tryptic digested BSA and human blood serum.97 The pH in the first dimension was 1.8, while the pH in the second dimension was 10.97 Acidic peptides were retained more on the first RP dimension at lower pH, while the basic peptides have stronger retention on the second RP dimension at higher mobile phase pH.97 The selectivity of neutral peptides also altered with changing pH.97 The authors reported this 2D LC system to be robust and could also be applied with high pH in the first RP dimension mobile phase and low pH in the second dimension.97 The peak capacity of peptides from digested BSA was determined to be 4677.97 Peptide analysis can also be performed using RP-RP LC with different analytical columns, like a combination of a C5 column and a C18 column.104 This RP-RP 2D LC combination was applied for determination of C-peptide, a biomarker for insulin secretion and can be sufficient for target analysis, but the peak capacity will not be sufficient for comprehensive analysis.104 Thus even though the MudPIT strategy is most often used for peptides, alternative combinations of separation mechanisms have been introduced showing similar as well as improved peptide separation and identification as compared to SCX-RP.
Capillary/microflow and nanoflow columns are most often used for peptides, this is because the concentration of the peptides is often low and the flow rate has to be compatible with MS which has become a necessity for peptide identification. Both storage loops and trap columns are used for collecting first dimension fractions prior to the second dimension separation. The number of collected fractions differs and depends on the complexity of the sample.
4.3 Food
Food constitutes a complex matrix and contains many different components like lipids, proteins, vitamins, antioxidants, water and minerals.7 Due to the content and direct human impact, food analyses are important and most often comprehensive approaches are applied rather than heart-cut, particularly in the area of lipids.7Saturated, unsaturated and trans fatty acids have a large impact on blood serum and cholesterol levels. Thus, it has become a requirement that the trans fat content must be listed on nutrition fact labels for packaged food.130 One such type of trans fat is triacylglycerols (TAGs).130 Recently, TAGs separation in different plant oils have been carried out using a silver-ion coated CX column as the primary column and a RP column in the second dimension.131 This method has become one of the most common MD LC techniques for quantification of TAGs.131,130 A MeOH–ACN containing gradient was used to elute the TAGs from the first dimension column.131 The second dimension mobile phase contained MeOH and tert-butyl methyl ether (70/30, v/v) obtaining high compatibility with the first dimension mobile phase. The authors determined more than 40 TAGs in corn oil.131
Carotenoids found in plant-derived food and products have been described to have beneficial health properties like antioxidant activity and prevention of cancer and have therefore attracted increased attention recently.32,33 Dugo et al. determined the carotenoids composition in red orange and mandarin oil using on-line NP-RP comprehensive LC.32,33 In the first dimension, the carotenoids were separated by NP LC using either a cyano based column or a silica column. When using the cyano column, the mobile phase contained n-hexane, butylacetate and acetone, while a mixture of n-hexane and ethyl alcohol was used in combination with the silica column. A two-minute second dimension gradient was accomplished using a monolithic RP column.32,33 The authors detected more than 20 and 40 carotenoids in mandarin and red orange oil, respectively.32,33 A similar comprehensive NP-RP LC system was used to analyze lemon oil extracts.48 The first dimension separation was carried out with a gradient of n-hexane and ethyl acetate using a diol-based NP column, while two C18-analytical columns using a mobile phase containing ACN/water were used for the second dimension separation.48
Polyphenols present in skins and seeds in grapes and berries, have also shown antioxidant effects.18,99,102 Recently, samples containing phenolic compounds have been analyzed using RP-RP LC with different RP stationary phases and either UV or MS for detection.18,102 Column-switching MD LC with storage loops, trap columns and the stop-flow approach has been applied to determine polyphenols in juice and wines.99,102,18,132 When using the stop-flow technique, after separation on the first dimension C18 column (250 × 4.6 mm ID), ten fractions were subsequently transferred to the second dimension carbon based column.18 The first dimension mobile phase contained MeOH and 10 mM ammonium acetate, while MeOH and phosphate buffer was used in the second dimension.18 As demonstrated above, different combinations of separation mechanisms are used in food analysis depending on the properties of the analytes. In most cases, mobile phase compatibility is obtained having a larger second dimension column compared to the first dimension analytical column. Storage loops are often used to connect the two dimensions and the number of collected fractions varies from 30 to 120.
4.4 Plant extracts analysis
Plant extracts contain hundreds of different components at various concentration levels with different pharmaceutical and toxic properties. Different approaches have been applied, but due to the improved peak capacity of MD LC, this is frequently used. Depending on the properties of the analytes, different MD LC combinations have been reported e.g. NP-RP,87,88,133 RP-RP48,134,135 and RP in combination with IEC.74,136,137 The main problem with NP-RP MD LC is the mobile phase compatibility between dimensions. However, Wei et al. added 1,4-dioxane, which is a water soluble and non-polar solvent, to n-hexane (99.5/0.5, v/v) in the first dimension mobile phase to overcome the compatibility problem with the RP mobile phase for analysis of plant extracts using a stop-flow NP-RP LC system.133 A silica-based analytical column was applied for the first dimension separation. In this MD LC system, the inner diameter of the analytical columns were the same in both dimensions, but two RP columns, 25 cm each, were connected in series to improve the peak capacity. A linear gradient of isopropanol/water and MeOH was applied in the second separation.133 Thirty fractions were collected in storage loops before the subsequent second dimension separation.Kivilompolo et al. explored different stationary phases for analyzing the components in a herb matrix.135 The most promising comprehensive MD LC combination appeared to be RP-RP interfaced to MS applying different RP stationary phases, C18 and CN in the first and second dimension, respectively.135 Chen et al. applied a comprehensive SAX-RP UV-MS method for identification of compounds in a Chinese medicine.137 Fifty-eight components were identified using a gradient of MeOH and phosphate buffer in the first dimension and a fast gradient with ACN and 0.1% acetic acid in the second.137
Typically, when analyzing plant extracts the number of collected fractions varies from 10 to 60, depending on the complexity of the sample. If the separation requires an extended second dimension separation, storage loops can be replaced with trap columns or the stop-flow approach. In most applications RP is used in one or both LC dimensions.
4.5 Polymers and industrial products
Polymers, like various acrylates and methacrylates, are frequently used in cosmetics such as mascara, nail enamels and hair dyes.138 Such polymers have large distributions in molecular mass and chemical composition.14,138 The increased selectivity obtained with MD LC is therefore advantageous when analyzing polymers and other complex industrial products, and SEC is often used in combination with NP or RP.16 In heart-cut MD LC, SEC is mostly applied in the first dimension whereas NP or RP is applied in the second dimension.16 When SEC is used in the first dimension, the eluent which often contains tetrahydrofuran (THF), has high elution strength and may affect the second separation.14 On the other hand, in comprehensive MD LC, SEC can be advantageously used in the second dimension, and NP or RP in the first dimension to improve the separation according to the composition of the polymers.16 With such an instrument set-up, higher sample capacity of the first dimension is obtained.14Raust et al. used a fully automated on-line RP-SEC 2D LC system with storage loops when analyzing a mixture of acrylate and methacrylate esters.138 The first dimension separation was carried out using a RP column (150 × 4.6 mm ID) with an ACN–THF gradient.138 The eluate was collected in two storage loops, and in total 109 fractions were transferred to the SEC column (150 × 7.5 mm ID) for subsequent separation. An on-line SEC-NP-UV system with storage loops was used for quantitative characterization of solid epoxy resins.83 The mobile phase in both dimensions contained MeOH and dichloromethane.83
When analyzing polymer samples using on-line MD LC, SEC is generally the separation mechanism of choice in combination with either RP or NP. Usually, large amounts of sample are available and the inner diameter of the columns is therefore often large (4.6–7.5 mm). Mobile phase compatibility is accomplished using columns with different inner diameters. Storage loops are most often used for on-line fractionation and combining the LC dimensions.
4.6 Other applications
Prostaglandins and metabolites in human plasma have been separated using an MD LC system with two different RP columns with MS detection.106 In addition to different stationary phases, increased selectivity and resolution were obtained using MeOH in the first dimension and ACN for the second dimension separation.106 One RP trap column was used to preconcentrate the analytes prior to the second dimension. A 2D LC system with two different RP columns was also used to analyze a mixture of phenolic compounds and flavones.42 The analytical column in the first dimension was 2.1 × 150 mm, while the second dimension analytical column was 4 × 30 mm.42 The flow rates were 50 μL/min and 2 mL/min, respectively. Matching parallel gradients were used in both dimensions and hence no second dimension column re-equilibration was necessary. A ten-port valve with two storage loops was used to combine the two dimensions and 70 fractions were collected in total.42On-line MD LC can also be used for determination of drugs. The activity of a drug component can be dependent of the component configuration, ketorolac is a non-steroidal anti-inflammatory drug where the (−)S enantiomer is responsible for the anti-inflammatory and analgesic activity, while the (+)R enantiomer has little or no activity.101,128 Thus, in order to determine the different isomers of ketorolac, Ing-Lorenzini et al. used a heart-cut MD LC-MS method with a RP-C18 column in the first dimension and a chiral column in the second dimension.101 The whole analysis was carried out in less than 20 min, using a fast first dimension gradient and isocratic elution in the second dimension. A similar MD LC approach, only comprehensive, was applied to determine carvedilol, a non-selective beta blocker, in plasma samples.139 Alternatively, drug samples can be analyzed using RP-RP with two different stationary phases.100
5. Future aspects of on-line MD LC
On-line MD LC will most probably become more frequently utilized in the future, due to the increasing need for automatic, high throughput comprehensive and target analysis of complex samples. At present, miniaturized on-line MD LC systems are becoming more common with capillary/microflow and nanoflow columns due to the increasing need of detecting different components at very low concentration levels and such systems will be more frequently applied also in routine analyses. Miniaturization is especially important in proteomics, peptidomics and possibly metabonomics where the available sample amount is limited, and the target analytes are present in low concentration. On the other hand, for food and polymer analyses where there are no sample limitations, second dimension columns with narrow inner diameter are not that crucial. Hence, in these fields standard size column will continue to be used unless environmental requirements force laboratories to minimize the columns (and use split-free pumping systems to spend less solvent).Many on-line MD LC methods are reported using fast second dimension separation reducing the total analysis time. Thus, monolithic columns or UPLC systems will be more frequently incorporated in the second dimension. Even though the availability of monolithic columns so far has been limited, these columns have gained much attention due to the potential higher performance compared to particle-packed columns. Thus, we believe that the quest for monolithic columns will increase, leading to more commercial products. UPLC, on the other hand suffers from lack of suitable hardware like low dead volume high pressure valves, but when such valves become more easily available there is no reason why UPLC could not be included more frequently in on-line MD LC. However, depending on the sample and the goal, it is not possible to substantially decrease the time of analysis and still obtain high total peak capacity. The total run time will always be dependent on the first and second dimension elution as well as the fractionation frequency. Thus, a compromise has to be made between the peak capacity and time.
Even though some have started to use 3D LC, due to the limitations with orthogonality, compatibility between dimensions, data processing and instrumental complexity compared to 2D LC, 3D LC is presently not widely used. 2D LC also still suffers from limitations which have to be overcome. One such problem is data processing and presentation. At present, software with statistical and data-visualizing tools is often incorporated with the data acquisition using MS for detection but not that often for other detection methods like UV. Statistical calculations have become a necessity in comparison-studies when e.g. searching for potential biomarkers for cancer or other diseases in a large number of samples, but the user friendliness of such software should in some cases be improved.
When analyzing a large number of samples, it is advantageous to use a fully automated on-line MD LC system with limited manual sample handling. Hence, in the future we will see that many off-line MD LC methods will be replaced with on-line MD LC methods, also in order to save time. Presently, only a few commercial MD LC instrumentations are reported to be fully automated. However, we believe that more fully automated on-line 2D LC methods will be reported in the near future and more effort will be made to overcome the present limitations with on-line 2D LC in order to perform high throughput analyses.
6. References
- C. R. Evans and J. W. Jorgenson, Anal. Bioanal. Chem., 2004, 378, 1952–1961 CrossRef CAS.
- K. Sandra, M. Moshir, F. D'Hondt, R. Tuytten, K. Verleysen, K. Kas, I. Francois and P. Sandra, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1019–1039 CrossRef CAS.
- L. N. Waller, K. Shores and D. R. Knapp, J. Proteome Res., 2008, 7, 4577–4584 CrossRef CAS.
- A. Motoyama and J. R. Yates, Anal. Chem., 2008, 80, 7187–7193 CrossRef CAS.
- R. E. Majors, LCGC North Am., 2008, 70–73 CAS.
- R. Tomas, K. Kleparnik and F. Foret, J. Sep. Sci., 2008, 31, 1964–1979 CrossRef CAS.
- P. Dugo, T. Kumm, F. Cacciola, G. Dugo and L. Mondello, J. Liq. Chromatogr. Relat. Technol., 2008, 31, 1758–1807 CrossRef CAS.
- P. Jandera, J. Sep. Sci., 2008, 31, 1421–1437 CrossRef CAS.
- J. Tang, M. Gao, C. Deng and X. Zhang, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 866, 123–132 CrossRef CAS.
- P. Dugo, F. Cacciola, T. Kumm, G. Dugo and L. Mondello, J. Chromatogr., A, 2008, 1184, 353–368 CrossRef CAS.
- M. L. Fournier, J. M. Gilmore, S. A. Martin-Brown and M. P. Washburn, Chem. Rev., 2007, 107, 3654–3686 CrossRef CAS.
- F. David, G. Vanhoenacker, B. Tienpont, I. Francois and P. Sandra, LC-GC Eur., 2007, 20, 154–158 CAS 160–162.
- C. R. Evans and J. W. Jorgenson, in Multidimensional liquid chromatography theory and applications in industrial chemistry and the life sciences, ed. S. A. Cohen and M. R. Schure, John Wiley & Sons, Inc., Hoboken, Editon edn, 2008, pp. 177–204 Search PubMed.
- F. Rittig and H. Pasch, in Multidimensional liquid chromatography theory and applications in industrial chemistry and the life sciences, ed. S. A. Cohen and M. R. Schure, John Wiley & Sons, Inc., Hoboken, Editon edn, 2008, pp. 387, 394, 387–423 Search PubMed.
- T. Stroink, M. C. Ortiz, A. Bult, H. Lingeman, G. J. de Jong and W. J. M. Underberg, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2005, 817, 49–66 CrossRef CAS.
- P. Jandera, J. Sep. Sci., 2006, 29, 1763–1783 CrossRef CAS.
- L. Blumberg and M. S. Klee, J. Chromatogr., A, 2010, 1217, 99–103 CrossRef CAS.
- F. Cacciola, P. Jandera and L. Mondello, Chromatographia, 2007, 66, 661–667 CrossRef CAS.
- I. Francois, K. Sandra and P. Sandra, Anal. Chim. Acta, 2009, 641, 14–31 CrossRef CAS.
- J. M. Davis, D. R. Stoll and P. W. Carr, Anal. Chem., 2008, 80, 8122–8134 CrossRef CAS.
- G. Guiochon, L. A. Beaver, M. F. Gonnord, A. M. Siouffi and M. Zakaria, J. Chromatogr., A, 1983, 255, 415–437 CrossRef CAS.
- M. Gilar, P. Olivova, A. E. Daly and J. C. Gebler, Anal. Chem., 2005, 77, 6426–6434 CrossRef CAS.
- X. Li, D. R. Stoll and P. W. Carr, Anal. Chem., 2009, 81, 845–850 CrossRef CAS.
- R. A. Shellie and P. R. Haddad, Anal. Bioanal. Chem., 2006, 386, 405–415 CrossRef CAS.
- R. E. Murphy, M. R. Schure and J. P. Foley, Anal. Chem., 1998, 70, 1585–1594 CrossRef CAS.
- K. Horvath, J. N. Fairchild and G. Guiochon, Anal. Chem., 2009, 81, 3879–3888 CrossRef CAS.
- D. R. Stoll, J. D. Cohen and P. W. Carr, J. Chromatogr., A, 2006, 1122, 123–137 CrossRef CAS.
- S. Eeltink, S. Dolman, R. Swart, M. Ursem and P. J. Schoenmakers, J. Chromatogr., A, 2009, 1216, 7368–7374 CrossRef CAS.
- J. M. Davis, D. R. Stoll and P. W. Carr, Anal. Chem., 2008, 80, 461–473 CrossRef CAS.
- K. Horvath, J. Fairchild and G. Guiochon, J. Chromatogr., A, 2009, 1216, 2511–2518 CrossRef CAS.
- Y. Saito, K. Jinno and T. Greibrokk, J. Sep. Sci., 2004, 27, 1379–1390 CrossRef CAS.
- P. Dugo, M. Herrero, D. Giuffrida, T. Kumm, G. Dugo and L. Mondello, J. Agric. Food Chem., 2008, 56, 3478–3485 CrossRef CAS.
- P. Dugo, M. Herrero, T. Kumm, D. Giuffrida, G. Dugo and L. Mondello, J. Chromatogr., A, 2008, 1189, 196–206 CrossRef CAS.
- R. E. Murphy and M. R. Schure, in Multidimensional liquid chromatography theory and applications in industrial chemistry and the life sciences ed. S. A. Cohen and M. R. Schure, John Wiley & Sons, Inc., Hoboken, Editon edn, 2008, p. 137 Search PubMed.
- J. M. Davis, in Multidimensional liquid chromatography theory and applications in industrial chemistry and the life sciences, ed. S. A. Cohen and M. R. Schure, John Wiley & Sons, Inc., Hoboken, Editon edn, 2008, pp. 49–50 Search PubMed.
- E. Rogatsky, K. Braaten, G. Cruikshank, H. Jayatillake, B. Zheng and D. Stein, J. Chromatogr., A, 2009, 1216, 7721–7727 CrossRef CAS.
- K. Horvath, J. N. Fairchild and G. Guiochon, J. Chromatogr., A, 2009, 1216, 7785–7792 CrossRef CAS.
- R. E. Murphy and M. R. Schure, in Multidimensional liquid chromatography theory and applications in industrial chemistry and the life sciences, ed. S. A. Cohen and M. R. Schure, John Wiley & Sons, Inc., Hoboken, Editon edn, 2008, p. 132 Search PubMed.
- J. N. Fairchild, K. Horvath and G. Guiochon, J. Chromatogr., A, 2009, 1216, 6210–6217 CrossRef CAS.
- P. Taylor, P. A. Nielsen, M. B. Trelle, O. B. Horning, M. B. Andersen, O. Vorm, M. F. Moran and T. Kislinger, J. Proteome Res., 2009, 8, 1610–1616 CrossRef CAS.
- M. P. Washburn, D. Wolters and J. R. Yates, 3rd, Nat. Biotechnol., 2001, 19, 242–247 CrossRef CAS.
- P. Jandera, P. Cesla, T. Hajek, G. Vohralik, K. Vynuchalova and J. Fischer, J. Chromatogr., A, 2008, 1189, 207–220 CrossRef CAS.
- T. Ikegami, H. Aoki, H. Kimura, K. Hosoya and N. Tanaka, in Multidimensional liquid chromatography theory and applications in industrial chemistry and the life sciences, ed. S. A. Cohen and M. R. Schure, John Wiley & Sons, Inc., Hoboken, Editon edn, 2008, pp. 147, 160, 163 Search PubMed.
- F. Cacciola, P. Jandera, Z. Hajdu, P. Cesla and L. Mondello, J. Chromatogr., A, 2007, 1149, 73–87 CrossRef CAS.
- K. Sandra, M. Moshir, F. D'Hondt, K. Verleysen, K. Kas and P. Sandra, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 866, 48–63 CrossRef CAS.
- A. Mihailova, H. Malerod, R. Wilson Steven, B. Karaszewski, R. Hauser, E. Lundanes and T. Greibrokk, J. Sep. Sci., 2008, 31, 459–467 CrossRef CAS.
- S. R. Wilson, M. Jankowski, M. Pepaj, A. Mihailova, F. Boix, G. V. Truyols, E. Lundanes and T. Greibrokk, Chromatographia, 2007, 66, 469–474 CrossRef CAS.
- I. Francois, A. de Villiers, B. Tienpont, F. David and P. Sandra, J. Chromatogr., A, 2008, 1178, 33–42 CrossRef CAS.
- F. Bedani, W. T. Kok and H.-G. Janssen, J. Chromatogr., A, 2006, 1133, 126–134 CrossRef CAS.
- S. Mirabaud, C. Rolando and M. Regert, Anal. Chem., 2007, 79, 6182–6192 CrossRef CAS.
- Q. Luo, Y. Gu, S.-L. Wu, T. Rejtar and B. L. Karger, Electrophoresis, 2008, 29, 1604–1611 CrossRef CAS.
- E. Hogendoorn, P. Van Zoonen and F. Hernandez, LC-GC Eur., 2003, 16, 44–51 CAS.
- M. Pepaj, S. R. Wilson, K. Novotna, E. Lundanes and T. Greibrokk, J. Chromatogr., A, 2006, 1120, 132–141 CrossRef CAS.
- M. Pepaj, A. Holm, B. Fleckenstein, E. Lundanes and T. Grelbrokk, J. Sep. Sci., 2006, 29, 519–528 CrossRef CAS.
- P. J. Boersema, N. Divecha, A. J. R. Heck and S. Mohammed, J. Proteome Res., 2007, 6, 937–946 CrossRef CAS.
- M. Pepaj, E. Lundanes and T. Greibrokk, J. Liq. Chromatogr. Relat. Technol., 2007, 30, 1879–1894 CrossRef CAS.
- B. Q. Tran, M. Pepaj, E. Lundanes and T. Greibrokk, Chromatographia, 2007, 66, 709–715 CrossRef CAS.
- B. Q. Tran, M. Pepaj, E. Lundanes and T. Greibrokk, J. Liq. Chromatogr. Relat. Technol., 2008, 31, 1387–1411 CrossRef CAS.
- T. Andersen, M. Pepaj, R. Trones, E. Lundanes and T. Greibrokk, J. Chromatogr., A, 2004, 1025, 217–226 CrossRef CAS.
- L. Shan and D. J. Anderson, Anal. Chem., 2002, 74, 5641–5649 CrossRef CAS.
- C. I. Balog, P. J. Hensbergen, R. Derks, J. J. Verweij, G. J. van Dam, B. J. Vennervald, A. M. Deelder and O. A. Mayboroda, Clin. Chem., 2009, 55, 117–125 CAS.
- R. C. Dwivedi, V. Spicer, M. Harder, M. Antonovici, W. Ens, K. G. Standing, J. A. Wilkins and O. V. Krokhin, Anal. Chem., 2008, 80, 7036–7042 CrossRef CAS.
- F. F. Evans, M. J. Raftery, S. Egan and S. Kjelleberg, J. Proteome Res., 2007, 6, 967–975 CrossRef CAS.
- M. Gilar, Y.-Q. Yu, J. Ahn, J. Fournier and J. C. Gebler, J. Chromatogr., A, 2008, 1191, 162–170 CrossRef CAS.
- J. A. Dowell, D. C. Frost, J. Zhang and L. Li, Anal. Chem., 2008, 80, 6715–6723 CrossRef CAS.
- P. A. Kirkland, M. A. Humbard, C. J. Daniels and J. A. Maupin-Furlow, J. Proteome Res., 2008, 7, 5033–5039 CrossRef CAS.
- J. H. Prieto, S. Koncarevic, S. K. Park, J. Yates, III and K. Becker, PLoS One, 2008, 3 Search PubMed No pp given.
- E. P. Romijn and J. R. Yates, III, Methods Mol. Biol., 2008, 432, 1–16 CAS.
- H. Yuan, L. Zhang, W. Zhang, Z. Liang and Y. Zhang, Fenxi Ceshi Xuebao, 2008, 27, 227–230 Search PubMed.
- T. Umemura and H. Kobayshi, Chromatography, 2009, 30, 43–44 CAS.
- J. F. Kelly and W. Ding, Methods Mol. Biol., 2008, 439, 257–267 CAS.
- M. Gao, C. Deng, W. Yu, Y. Zhang, P. Yang and X. Zhang, Proteomics, 2008, 8, 939–947 CrossRef CAS.
- C. V. Hoffmann, M. Laemmerhofer and W. Lindner, Anal. Bioanal. Chem., 2009, 393, 1257–1265 CrossRef CAS.
- T. Tang, W.-B. Zhang, T. Li and F.-Y. Wang, Fenxi Huaxue, 2007, 35, 1767–1771 CAS.
- R. J. Goldschmidt and W. R. Wolf, J. AOAC Int., 2007, 90, 1084–1089 CAS.
- T. Leitner and C. W. Klampfl, J. Liq. Chromatogr. Relat. Technol., 2008, 31, 169–178 CrossRef CAS.
- D. Ren, G. Ratnaswamy, J. Beierle, M. J. Treuheit, D. N. Brems and P. V. Bondarenko, Int. J. Biol. Macromol., 2009, 44, 81–85 CrossRef CAS.
- L. Hu, K.-S. Boos, M. Ye, R. a. Wu and H. Zou, J. Chromatogr., A, 2009, 1216, 5377–5384 CrossRef CAS.
- K. Im, H.-w. Park, S. Lee and T. Chang, J. Chromatogr., A, 2009, 1216, 4606–4610 CrossRef CAS.
- V. Mass, V. Bellas and H. Pasch, Macromol. Chem. Phys., 2008, 209, 2026–2039 CrossRef CAS.
- B. Winther, J. Leon and E. Reubsaet, J. Sep. Sci., 2005, 28, 477–482 CrossRef CAS.
- L. M. de Souza, T. R. Cipriani, C. F. Sant'Ana, M. Iacomini, P. A. J. Gorin and G. L. Sassaki, J. Chromatogr., A, 2009, 1216, 99–105 CrossRef CAS.
- S. Julka, H. Cortes, R. Harfmann, B. Bell, A. Schweizer-Theobaldt, M. Pursch, L. Mondello, S. Maynard and D. West, Anal. Chem., 2009, 81, 4271–4279 CrossRef CAS.
- D. Boschmann, R. Edam, P. J. Schoenmakers and P. Vana, Macromol. Symp., 2009, 275–276, 184–196 CrossRef CAS.
- R. Edam, D. M. Meunier, E. P. C. Mes, F. A. Van Damme and P. J. Schoenmakers, J. Chromatogr., A, 2008, 1201, 208–214 CrossRef CAS.
- R. E. Murphy, M. R. Schure and J. P. Foley, Anal. Chem., 1998, 70, 4353–4360 CrossRef CAS.
- H.-z. Tian, J. Xu and Y.-f. Guan, Fenxi Huaxue, 2008, 36, 860–864 CAS.
- H.-Z. Tian, J. Xu and Y. Guan, J. Sep. Sci., 2008, 31, 1677–1685 CrossRef CAS.
- H.-Z. Tian, J. Xu and Y.-F. Guan, Gaodeng Xuexiao Huaxue Xuebao, 2007, 28, 630–634 CAS.
- P. Hemstroem and K. Irgum, J. Sep. Sci., 2006, 29, 1784–1821 CrossRef.
- C. Apostolou, C. Kousoulos, Y. Dotsikas and L. Loukas Yannis, Biomed. Chromatogr., 2008, 22, 1393–1402 CrossRef CAS.
- S. Louw, A. S. Pereira, F. Lynen, M. Hanna-Brown and P. Sandra, J. Chromatogr., A, 2008, 1208, 90–94 CrossRef CAS.
- A. Liu, J. Tweed and C. E. Wujcik, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1873–1881 CrossRef CAS.
- C. J. Platerink, H.-G. M. Janssen and J. Haverkamp, Anal. Bioanal. Chem., 2008, 391, 299–307 CrossRef CAS.
- T. Ikegami, T. Hara, H. Kimura, H. Kobayashi, K. Hosoya, K. Cabrera and N. Tanaka, J. Chromatogr., A, 2006, 1106, 112–117 CrossRef CAS.
- C. J. Venkatramani and A. Patel, J. Sep. Sci., 2006, 29, 510–518 CrossRef CAS.
- I. Francois, D. Cabooter, K. Sandra, F. Lynen, G. Desmet and P. Sandra, J. Sep. Sci., 2009, 32, 1137–1144 CrossRef CAS.
- J. Cong and B. Lin, J. Liq. Chromatogr. Relat. Technol., 2008, 31, 891–911 CrossRef CAS.
- P. Cesla, T. Hajek and P. Jandera, J. Chromatogr., A, 2009, 1216, 3443–3457 CrossRef CAS.
- A. J. Alexander and L. Ma, J. Chromatogr., A, 2009, 1216, 1338–1345 CrossRef CAS.
- K. R. Ing-Lorenzini, A. Desmeules Jules, M. Besson, J.-L. Veuthey, P. Dayer and Y. Daali, J. Chromatogr., A, 2009, 1216, 3851–3856 CrossRef CAS.
- M. Kivilompolo and T. Hyotylainen, J. Sep. Sci., 2008, 31, 3466–3472 CrossRef CAS.
- T. Hajek, V. Skerikova, P. Cesla, K. Vynuchalova and P. Jandera, J. Sep. Sci., 2008, 31, 3309–3328 CrossRef CAS.
- E. Rogatsky, V. Tomuta, H. Jayatillake, G. Cruikshank, L. Vele and D. T. Stein, J. Sep. Sci., 2007, 30, 226–233 CrossRef CAS.
- P. Jandera, K. Vynuchalova, T. Hajek, P. Cesla and G. Vohralik, J. Chemom., 2008, 22, 203–217 CrossRef CAS.
- J. Komaba, D. Matsuda, K. Shibakawa, S. Nakade, Y. Hashimoto, Y. Miyata and M. Ogawa, Biomed. Chromatogr., 2009, 23, 315–323 CrossRef CAS.
- J. Pol, B. Hohnova and T. Hyoetylaeinen, J. Chromatogr., A, 2007, 1150, 85–92 CrossRef CAS.
- A. Ghassempour, H. Rezadoost, M. Ahmadi and H. Y. Aboul-Enein, J. Liq. Chromatogr. Relat. Technol., 2009, 32, 1434–1447 CrossRef CAS.
- Y. Zhou, Y. Wang, R. Wang, F. Guo and C. Yan, J. Sep. Sci., 2008, 31, 2388–2394 CrossRef CAS.
- G. Guiochon, N. Marchetti, K. Mriziq and R. A. Shalliker, J. Chromatogr., A, 2008, 1189, 109–168 CrossRef CAS.
- J. Wei, J. Sun, W. Yu, A. Jones, P. Oeller, M. Keller, G. Woodnutt and M. Short Jay, J. Proteome Res., 2005, 4, 801–808 CrossRef CAS.
- W. H. McDonald, R. Ohi, D. T. Miyamoto, T. J. Mitchison and J. R. Yates, Int. J. Mass Spectrom., 2002, 219, 245–251 Search PubMed.
- B. Q. Tran, H. Loftheim, L. Reubsaet, E. Lundanes and T. Greibrokk, J. Sep. Sci., 2008, 31, 2913–2923 CrossRef CAS.
- J. A. Karty, W. E. Running and J. P. Reilly, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 847, 103–113 CrossRef CAS.
- J.-L. Zhou, J.-J. An, P. Li, H.-J. Li, Y. Jiang and J.-F. Cheng, J. Chromatogr., A, 2009, 1216, 2394–2403 CrossRef CAS.
- Z.-B. Ning, Q.-R. Li, J. Dai, R.-X. Li, C.-H. Shieh and R. Zeng, J. Proteome Res., 2008, 7, 4525–4537 CrossRef CAS.
- K. Deguchi, T. Keira, K. Yamada, H. Ito, Y. Takegawa, H. Nakagawa and S.-I. Nishimura, J. Chromatogr., A, 2008, 1189, 169–174 CrossRef CAS.
- Y. Jmeian and Z. El Rassi, Electrophoresis, 2008, 29, 2801–2811 CAS.
- N. A. Cellar, A. S. Karnoup, D. R. Albers, M. L. Langhorst and S. A. Young, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 79–85 CrossRef CAS.
- L. Rieux, R. Bischoff, E. Verpoorte and H. A. G. Niederlaender, J. Chromatogr., A, 2007, 1149, 169–177 CrossRef CAS.
- A. Mihailova, B. Karaszewski, E. M. Faergestad, R. Hauser, W. M. Nyka, E. Lundanes and T. Greibrokk, J. Sep. Sci., 2008, 31, 468–479 CrossRef CAS.
- A. Mihailova, B. Karaszewski, R. Hauser, E. Lundanes and T. Greibrokk, J. Sep. Sci., 2007, 30, 249–256 CrossRef CAS.
- C. Liu and X. Zhang, J. Chromatogr., A, 2007, 1139, 191–198 CrossRef CAS.
- F. Wang, J. Dong, X. Jiang, M. Ye and H. Zou, Anal. Chem., 2007, 79, 6599–6606 CrossRef CAS.
- F. Wang, J. Dong, M. Ye, X. Jiang, R. a. Wu and H. Zou, J. Proteome Res., 2008, 7, 306–310 CrossRef CAS.
- T. Kajdan, H. Cortes, K. Kuppannan and S. A. Young, J. Chromatogr., A, 2008, 1189, 183–195 CrossRef CAS.
- Q. Luo, G. Yue, G. A. Valaskovic, Y. Gu, S.-L. Wu and B. L. Karger, Anal. Chem., 2007, 79, 6174–6181 CrossRef.
- A.-M. Hesse, P. Marcelo, J. Rossier and J. Vinh, J. Chromatogr., A, 2008, 1189, 175–182 CrossRef CAS.
- J. Dai, L.-S. Wang, Y.-B. Wu, Q.-H. Sheng, J.-R. Wu, C.-H. Shieh and R. Zeng, J. Proteome Res., 2009, 8, 133–141 CrossRef CAS.
- R. G. Harfmann, S. Julka and H. J. Cortes, J. Sep. Sci., 2008, 31, 915–920 CrossRef CAS.
- E. J. C. van der Klift, G. Vivo-Truyols, F. W. Claassen, F. L. van Holthoon and T. A. van Beek, J. Chromatogr., A, 2008, 1178, 43–55 CrossRef CAS.
- F. Cacciola, P. Jandera and L. Mondello, J. Sep. Sci., 2007, 30, 462–474 CrossRef CAS.
- Y. Wei, T. Lan, T. Tang, L. Zhang, F. Wang, T. Li, Y. Du and W. Zhang, J. Chromatogr., A, 2009, 1216, 7466–7471 CrossRef CAS.
- C. Champmartin, P. Simon, P. Delsaut, M. Dorotte and B. Bianchi, J. Chromatogr., A, 2007, 1142, 164–171 CrossRef CAS.
- M. Kivilompolo and T. Hyoetylaeinen, J. Chromatogr., A, 2007, 1145, 155–164 CrossRef CAS.
- M. Eggink, M. Wijtmans, R. Ekkebus, H. Lingeman, I. J. P. de Esch, J. Kool, W. M. A. Niessen and H. Irth, Anal. Chem., 2008, 80, 9042–9051 CrossRef CAS.
- X. Chen, Z. Jiang, Y. Zhu and J. Tan, Chromatographia, 2007, 65, 141–147 CrossRef CAS.
- J.-A. Raust, A. Bruell, C. Moire, C. Farcet and H. Pasch, J. Chromatogr., A, 2008, 1203, 207–216 CrossRef CAS.
- A. Medvedovici, F. Albu, C. Georgita, D. I. Sora, T. Galaon, S. Udrescu and V. David, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 850, 327–335 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |