A novel multicommutation stopped-flow system for the simultaneous determination of sulfamethoxazole and trimethoprim by differential pulse voltammetry on a boron-doped diamond electrode
Leonardo
Santos Andrade†
,
Romeu
Cardozo Rocha-Filho
,
Quezia
Bezerra Cass
and
Orlando
Fatibello-Filho
*
Departamento de Química, Universidade Federal de São Carlos, C. P. 676, 13560-970, São Carlos, SP, Brazil. E-mail: bello@ufscar.br; Fax: +55 16 3351-8350; Tel: +55 16 3351-8098
First published on 9th February 2010
Abstract
A method based on a multicommutation stopped-flow system was developed and applied in the simultaneous determination of sulfamethoxazole (SMX) and trimethoprim (TMP) in pharmaceutical formulations by differential pulse voltammetry (DPV) using a cathodically pre-treated hydrogen-terminated boron-doped diamond (HT-BDD) electrode. The obtained analytical curves were linear from 1.0 to 8.0 mg L−1 and 0.20 to 1.6 mg L−1 for SMX and TMP, respectively. The calculated LOD and LOQ values for SMX were 16.5 μg L−1 and 55.1 μg L−1 (or 65.1 nmol L−1 and 218 nmol L−1), respectively; for TMP, they were 18.3 μg L−1 and 61.1 μg L−1 (or 63.0 nmol L−1 and 210 nmol L−1), respectively. The optimization and application of the multicommutation stopped-flow system in the simultaneous electrochemical determination of SMX and TMP by DPV on the HT-BDD electrode yielded an excellent sample throughput (30 h−1). Addition and recovery studies indicated that the matrix effect did not present any significant interference in the determinations. Thus, the proposed method was successfully applied in the determination of SMX and TMP in two different commercial pharmaceutical formulations with results similar (at 95% confidence level) to those obtained using the USP method (HPLC). The obtained performance allowed concluding that the proposed method is quite advantageous and that its total automation can lead to rapid, sensitive, precise, and accurate results, with an excellent sample throughput.
Introduction
The search for analytical systems suitable for use in routine analyses involving pharmaceutical quality control includes simple, high-analytical-frequency, and low-reagent-consumption methods. There are several analytical procedures reported in the literature for drugs determination in pharmaceutical products, as described previously.1 However, specifically for sulfa drugs there is still need for rapid and reliable methods, although a number of methods for their quantification have been reported, which include titrimetric-assay,2 photometric,3 potentiometric,4 HPLC-UV5–8 and HPLC-MS9 methods.There are only a few reports available on the electrochemical detection of sulfa drugs,1,10–20 but in some of them the oxidation/reduction products can be strongly adsorbed on the surface of the electrodes, which thus tend to become fouled and deactivated.10–12 However, when a boron-doped diamond electrode (BDD) was used, no deactivation was found during the electrochemical detection of sulfa drugs.1,13,18,20 This is one of the reasons why BDD electrodes seem promising for electroanalytical purposes. Other reasons are their wide potential window (up to 3.5 V) in aqueous solutions, very low and stable voltammetric background current, long-term response stability, and low sensitivity to dissolved oxygen. These are important characteristics that allow the detection of many electroactive species on BDD, which otherwise would be masked by the water decomposition reactions.21
Sulfonamides are commonly used in the treatment of urinary-tract infections, pneumocystis pneumonia, chronic bronchitis, meningococcal meningitis, acute otitis, and toxoplasmosis. In all these cases, the corresponding pharmaceutical products usually consist of a sulfonamide mixed with another drug that potentiates its pharmacological action (in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio sulfonamide:potentiator), as for example the sulfamethoxazole (SMX) and trimethoprim (TMP) mixture,22 which is commonly known as SMX-TMP; trimethoprim is also available as a single-drug formulation.
:1 ratio sulfonamide:potentiator), as for example the sulfamethoxazole (SMX) and trimethoprim (TMP) mixture,22 which is commonly known as SMX-TMP; trimethoprim is also available as a single-drug formulation.
Recently, we found that the performance of a hydrogen-terminated boron-doped diamond (HT-BDD) electrode on the simultaneous differential pulse voltammetric determination of SMX and TMP in pharmaceutical products was very good.1 However, although it has been considered a simple, rapid, sensitive, precise, and accurate method for analytical purposes, the analytical frequency for this determination was not so high since the measurements were carried out in a batch mode. Thus, the development of an automatic analytical method based on a flow technique has some advantages compared to the batch method, such as easiness of automation, determination reproducibility, short analysis time, high sample throughput, and low reagents consumption.
Multicommutation is one of the most recent flow techniques, based on the use of multiple flow-commutation devices, usually three-way solenoid valves that control the analytical pathway of the solution toward the detector.23 Thus, the combination of a multicommutation flow system with electrochemical detection for the determination of SMX and TMP can be considered an interesting alternative. However, as the corresponding pharmaceutical products consist of both SMX and TMP, this simultaneous determination using a multicommutated flow system coupled with classical voltammetric techniques is only possible in static systems, i.e., if the flow is interrupted. Usually, stopped-flow measurements are applied to systems where the chemical reactions are quite slow, in order to increase the residence time of the sample zone in the analytical pathway.
In some instances, the stopped-flow technique has also been used for the detection of multiple analytes, such as phenols24 or heavy metals.25 In these cases, when the flow is interrupted, analyses by voltammetric techniques are initiated and the analytes are determined inside the detector, under static conditions. This mode of analysis allows total automation of the system, significant increase in the analytical frequency, and reduction in the analyzed sample volume. From the viewpoint of electrochemical detection, this type of analysis is quite attractive because it allows the simultaneous determination of various compounds, provided that the compounds have sufficiently different anodic/cathodic peak potentials.
Therefore, in this paper we report on the simultaneous electrochemical determination of SMX and TMP in pharmaceutical products in a multicommutation stopped-flow system coupled with differential pulse voltammetry (DPV) using a HT-BDD electrode (cathodically pre-treated). The results are compared with those obtained with the USP method (HPLC).26
Experimental
Reagents and standards
All reagents were of analytical grade; all solutions were prepared using deionized water. SMX (99.85%) and TMP (98.88%) samples were supplied by Laboratório Teuto-Brasileiro Ltda. (Anápolis-GO, Brazil). The analyzed commercial tablets (Bactrim®), containing SMX (400 mg or 800 mg per tablet) and TMP (80 mg or 160 mg per tablet), were acquired at a local drugstore. A Britton-Robinson (B-R) buffer solution (0.2 mol L−1 H3BO3, 0.2 mol L−1 H3PO4, and 0.2 mol L−1 CH3COOH; pH 7.0) was used as supporting electrolyte.Sample preparation
SMX and TMP stock solutions (300 mg L−1 and 60 mg L−1, respectively) were prepared by dissolving these compounds in methanol. Then, these stock solutions were diluted in the B-R buffer solution in order to obtain the working standard solutions.The stock solutions of the commercial-tablets were prepared by dissolving 10 finely powdered tablets in methanol, followed by centrifuging and filtering to separate any insoluble matter.
Analytical curves
Using previously optimized experimental parameters for the DPV technique,1 the analytical curves were constructed using different standard solutions. All measurements were carried out in triplicate for each concentration. The limits of detection (LOD) and quantification (LOQ) were estimated as LOD = 3 SB/b and LOQ = 10 SB/b, where SB is the standard deviation of the blank (n = 10) and b the slope of the calibration curve.Electrode and apparatus
The BDD film (8000-ppm doping level) used as working electrode (1.2 cm × 1.2 cm) was prepared on a silicon wafer at the Centre Suisse de Electronique et de Microtechnique SA (CSEM), Neuchatêl, Switzerland, as described elsewhere.27 The stopped-flow electrochemical experiments were carried out in a home-made electrochemical detector, which was made out of two acrylic blocks, as shown in Fig. 1. In one of them, three holes were drilled. The first hole (a) allows the introduction of the solution into the detector, the second one (b) the exit of the solution, and the third one the insertion of a miniaturized Ag/AgCl (3.0 mol L−1 KCl) reference electrode28 (c), which was used in all measurements. A stainless-steel tube (d), used as counter electrode, was embedded on the solution exit hole (b). A silicone rubber O-ring (e) and the BDD electrode (f) were fixed between the two acrylic blocks, leaving a 0.64 cm2 exposed area. The electrical contact for the BDD electrode was made through a stainless-steel screw (g) at the gold-plated bottom of the silicon wafer.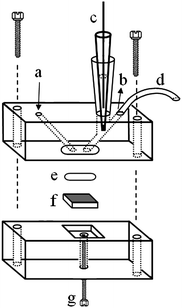 | ||
| Fig. 1 Schematic representation of the electrochemical detector used in the multicommutation stopped-flow measurements: (a) solution inlet; (b) solution outlet; (c) reference electrode; (d) stainless-steel tube (counter electrode); (e) silicone-rubber O-ring; (f) HT-BDD electrode, and (g) screw (electrical contact for the HT-BDD electrode). | ||
Prior to use, the BDD electrode surface was cleaned with isopropanol and rinsed with ultra-pure water. Then, in order to obtain a HT-BDD,27 the electrode was cathodically pre-treated in a 0.5 mol L−1 H2SO4 solution by applying −0.5 A cm−2, during 60 s. The DPV measurements were carried out using an Autolab PGSTAT-30 (Ecochemie) potentiostat/galvanostat controlled with the GPES 4.0 software.
The simultaneous determinations of SMX and TMP by the HPLC USP method26 were carried out using an SIL-10A auto-injector, an SPD-20A UV detector operated at 254 nm, a CBM-20A control system, and an LC-10AT pump. The data acquisition was carried out using the Lab-Solution software (Shimatzu). The mobile phase was an 80:20 mixture of triethylamine (pH adjusted to 5 with 1% v/v acetic acid) and acetonitrile. A flow rate of 2.0 mL min−1 was used and the UV detector was set up at 254 nm. The analytical column (150 mm length × 4.6 mm diameter) was packed with C18 nucleosil silica (5 μm, 100 Å; Macherey-Nagel, Germany) by the ascending-slurry method, using methanol for both the preparation of the slurry (50 mL) and the packing. The packing was carried out at a pressure of 7500 psi and then the column was conditioned for about 4 h with methanol at a flow rate of 1.0 mL min−1.
Multicommutation stopped-flow procedure
The multicommutation flow module (Fig. 2) consisted of two solenoid valves (represented by V1 and V2) that controlled the injection of the carrier (C) and sample (S) solutions, respectively. In the figures, each solenoid valve is depicted as a circle with two lines that indicate the possible pathways of the sample and/or carrier solution. The full lines depict the carrier solution pathway toward the electrochemical detector and the recirculation of the sample solution back to the reservoir when the V1 and V2 valves are switched off, respectively. On the other hand, the dashed lines indicate the solutions pathways when these valves are switched-on; at this condition, the carrier solution C is recycling while the sample solution S moves toward the electrochemical detector. After a certain time, the flow is stopped for 60 s and the DPV measurement is initiated for the oxidation of the compounds. After, the valves are switched off and the flow is restarted for 30 s, in order to clean the detector with the carrier solution.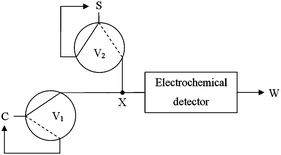 | ||
| Fig. 2 Diagram of the multicommutation stopped-flow system employed for the simultaneous determination of SMX and TMP. V1 and V2 = solenoid valves; X = confluence point; C = carrier solution; S = sample solution; W = waste. (Here the valves are shown switched off.) | ||
Results and discussion
The only physical parameter optimized in the multicommutation stopped-flow system was the sample volume injected in the detector. For this, the maximum analytical signals (current response) obtained from DPV voltammograms in the simultaneous determination of SMX and TMP for different injected sample volumes (from 250 μL to 580 μL) were considered, as shown in Fig. 3. As the flow rate used was 1.0 mL min−1, the time used for the injections was in the range 15–35 s. The optimizations of the DPV parameters as well as of the chemical conditions and the type of BDD pre-treatment (anodic or cathodic) to obtain the maximum analytical signal are described and discussed in detail elsewhere.1 The values of these parameters were chosen to attain maximum sensitivity and selectivity, i.e., highest values of the peak current and lowest values of the half-peak width, respectively.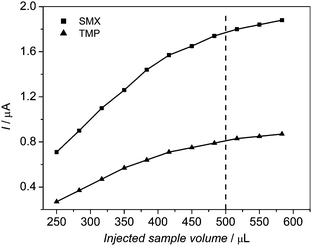 | ||
| Fig. 3 Anodic-peak-current responses for SMX and TMP oxidation obtained from differential pulse voltammetry (DPV) measurements on a HT-BDD electrode using a mixture of 5.0 mg L−1 SMX and 1.0 mg L−1 TMP in a 0.2 mol L−1 Britton-Robinson buffer solution (pH 7.0) versus injected sample volume. DPV conditions: scan rate, 50 mV s−1; pulse amplitude, 60 mV; pulse width, 10 ms. | ||
Using the optimized conditions, the anodic peak current values increased with the injected sample volume, for both SMX and TMP, as it can be seen in Fig. 3. However, less-intense increments of these values were observed for volumes greater than ∼500 μL. Therefore, a 500 μL sample volume (or 30 s injection time) was chosen for the SMX/TMP determinations, considering that this volume is suitable for any type of analysis, being a good compromise between analytical signal magnitude and sample throughput. Consequently, a sample throughput of 30 h−1 could be attained, compared to about 6 h−1 for the batch mode.1
Fig. 4 shows the DPV voltammogram series obtained by carrying out triplicate 2 min steps in the optimized multicommutation stopped-flow system using the HT-BDD electrode at different concentrations of SMX/TMP (from 1.0 to 8.0 mg L−1 and 0.20 to 1.6 mg L−1 for SMX and TMP, respectively) in the B-R buffer solution. As it can be seen in this figure, two well-defined oxidation waves were obtained: the first one (Ep = 900 mV vs. Ag/AgCl) corresponds to SMX oxidation and the second (Ep = 1070 mV vs. Ag/AgCl) to TMP oxidation. An excellent sample throughput (30 h−1) was achieved, especially if one takes into account that each analysis by the official USP method26 (HPLC) for the determination of sulfonamides takes about 10 min to be carried out. Therefore, the same analysis could be done five times faster than by the official method; this performance might be further improved, if desirable, by either decreasing the time of the cleaning step or increasing the flow rate, thus diminishing the total time accordingly.
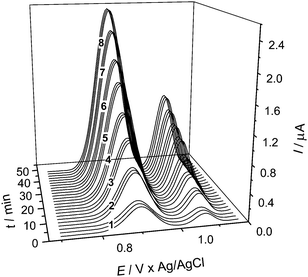 | ||
| Fig. 4 Differential pulse voltammetry (DPV) responses (simultaneous oxidation) obtained on a HT-BDD electrode at different concentrations (triplicates) of SMX/TMP in a 0.2 mol L−1 Britton-Robinson buffer solution (pH 7.0). SMX/TMP concentrations, in mg L−1: (1) 1.0/0.20; (2) 2.0/0.40; (3) 3.0/0.60; (4) 4.0/0.80; (5) 5.0/1.0; (6) 6.0/1.2; (7) 7.0/1.4, and (8) 8.0/1.6. DPV conditions as indicated in Fig. 3. | ||
The analytical curves (r2 = 0.9998, for both SMX and TMP) obtained from the DPV voltammograms (Fig. 4) for SMX and TMP are presented in Fig. 5a and 5b. The respective linear dynamic ranges (determined according to IUPAC) were 1.0–8.0 mg L−1 for SMX and 0.20–1.6 mg L−1 for TMP. The LOD and LOQ values calculated (according to IUPAC) for SMX were 16.5 μg L−1 (65.1 nmol L−1) and 55.1 μg L−1 (218 nmol L−1), respectively; for TMP, these values were 18.3 μg L−1 (63.0 nmol L−1) and 61.1 μg L−1 (210 nmol L−1), respectively. The sensitivity (slope of the analytical curve) was 323 μA g−1 L for SMX and 730 μA g−1 L for TMP. It should be pointed out that these values are about five times higher than the ones previously reported for a batch method;1 the reason for this difference is most probably the unsymmetrical design and placement of the working and counter electrodes in the flow cell then used.
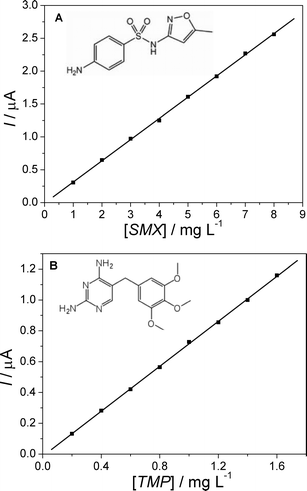 | ||
| Fig. 5 Analytical curves obtained in a 0.2 mol L−1 Britton-Robinson buffer solution (pH 7.0) for (a) SMX and (b) TMP. Differential pulse voltammetry conditions as indicated in Fig. 3. | ||
The reproducibility was 0.29% for SMX and 0.71% for TMP, for 10 determinations (n = 10). On the other hand, the short and long-term response stabilities expressed as RSD were 0.18% (first 4 injections) and 0.17% (5th to 10th injections), for SMX, and 0.18% (first 4 injections) and 0.90% (5th to 10th injections), for TMP.
The results obtained when using the batch system and carrying out the analysis in a conventional electrochemical cell1 yielded LOD and LOQ values ∼4.5 times lower than those here reported. This difference can be considered normal, especially if one takes into account that the positioning of the electrodes and the distance between them are very different in each system. Anyway, despite the fact that LOD and LOQ are not considered so relevant in drugs determination (because of their high concentration in commercial formulations29), their obtained values clearly indicate that quite low concentrations of SMX and TMP could also be detected on the HT-BDD electrode using the proposed method. Besides, this mode of analysis allowed a significant increase of the analytical frequency with a concomitant decrease of the analyzed sample volume. It should be mentioned that, as far as we could verify, the results here reported are superior to most of the ones reported by other authors on the electroanalytical determination of SMX and/or TMP. For example, You et al.11 investigated the determination of SMX and sulfadiazine (SDZ) on a carbon-fiber electrode (microdisk) after separation by capillary electrophoresis and obtained an LOD for SMX of 100 nmol L−1. Kotoucek et al.16 investigated the reduction of SMX and TMP on a mercury electrode and the detection limits obtained using DPV were 221 nmol L−1 and 255 nmol L−1, respectively. Msagati & Ngila17 investigated the simultaneous determination (by reduction) of seven sulfonamides by square-wave voltammetry (SWV) using a poly(3-methylthiophene)-coated glassy carbon electrode and obtained an LOD determined individually for SMX of 40 nmol L−1. More recently, Özkorucuklu et al.19 found an LOD of 359 nmol L−1 in the SMX determination using an overoxidized polypyrrole electrode (molecularly imprinted polymer), while Souza et al.20 quantified SMX and SDZ independently in pharmaceutical products by SWV using a HT-BDD electrode and achieved an LOD for SMX of 1.15 μmol L−1.
In order to evaluate the selectivity of the proposed method, some possible excipients (sodium stearate, povidone, soluble starch, and succinic acid) were added in a standard solution containing 1.0 mg L−1 SMX and 1.0 mg L−1 TMP at the concentrations ratios (standard solution:excipient) 1:1, 1:5, and 5:1; the current signals obtained were compared with those in the absence of each excipient. The obtained results showed that only povidone led to a significant deviation (more than ±5%) and only in the concentrations ratios 1:1 and 1:5. However, it should be mentioned that the povidone content in tablets is much lower than those here investigated; thus, its interference is not expected at all when determining SMX/TMP in commercial tablets using the method here proposed.
For the recovery studies, three determinations were carried out for each sample using the standard addition method and calculating the standard deviations. For that, known amounts of SMX and TMP were added in each sample, followed by DPV analysis with the proposed flow method. The recovery values for the two different pharmaceutical commercial formulations, ranging from 94.9% to 98.4% for SMX and from 99.2% to 102% for TMP (see Table 1), indicate that the matrix effect does not present any significant interference.
Finally, aiming at demonstrating the practical application of the proposed method, simultaneous analyses of SMX and TMP in two different commercial pharmaceutical formulations were comparatively carried out. The results obtained for the two commercial formulations, employing the proposed method and the USP (HPLC) method,26 are in satisfactory agreement with the predicted values (see Table 2). Besides, the paired t-test statistical treatment applied for the results obtained by the two methods showed that the calculated t-values (2.36 for SMX and 2.06 for TMP) were smaller than the critical t-value (tcritical = 2.57, α = 0.05). Hence, it can be concluded that there is no difference between the obtained results at a confidence level of 95%, indicating that the proposed method is quite accurate.
| Samples | SMX/TMP (mg L−1) | Relative error 1c (%) | Relative error 2d (%) | ||
|---|---|---|---|---|---|
| Label valuesa | HPLC methodb | Proposed voltammetric methodb | |||
| a Taking into account the dilutions. b Average of three measurements. c Relative error 1 (%): 100 × [(voltammetric value − label value)/label value]. d Relative error 2 (%): 100 × [(voltammetric value − HPLC value)/HPLC value]. | |||||
| Bactrin® tablets | 4.00/0.80 | 4.55 ± 0.04/0.89 ± 0.08 | 4.12 ± 0.01/0.85 ± 0.01 | 3.0/6.2 | −9.4/−4.5 |
| Bactrin F® tablets | 4.00/0.80 | 4.13 ± 0.03/0.85 ± 0.04 | 4.02 ± 0.03/0.82 ± 0.02 | 0.50/2.5 | −2.7/−3.5 |
Conclusions
The optimization and application of a multicommutation stopped-flow system in the simultaneous electrochemical determination of SMX and TMP in pharmaceutical products on a HT-BDD electrode by DPV were demonstrated. An excellent sample throughput (30 h−1) was achieved and the addition and recovery studies indicated that the matrix effect did not present any significant interference. Furthermore, the results obtained for two commercial-tablet formulations showed a satisfactory agreement between the predicted values and the proposed method. The LOD and LOQ values calculated for SMX were only 16.5 μg L−1 and 55.1 μg L−1 (or 65.1 nmol L−1 and 218 nmol L−1), respectively; for TMP these values were 18.3 μg L−1 and 61.1 μg L−1 (or 63.0 nmol L−1 and 210 nmol L−1), respectively.Therefore, the results here reported demonstrate that the use of a multicommutation stopped-flow system coupled to electrochemical detection using a HT-BDD electrode for the simultaneous determination of SMX and TMP by DPV can be considered a quite advantageous method for analytical purposes, since it can be totally automated, leading to rapid, sensitive, precise, and accurate results, with an excellent sample throughput.
Acknowledgements
Financial support and scholarships from the Brazilian funding agencies CNPq, CAPES, and FAPESP (Brazil) are gratefully acknowledged.References
- L. S. Andrade, R. C. Rocha-Filho, Q. B. Cass and O. Fatibello-Filho, Electroanalysis, 2009, 21, 1475–1480 CrossRef CAS.
- S. S. Tseng and T. J. Smith, Anal. Lett., 1994, 27, 1507–1515.
- M. T. Tena, M. D. Luque de Castro and M. Valcárcel, Analyst, 1994, 119, 1625–1628 RSC.
- M. M. A. K. Nazer, T. K. Shaber and P. Riyazuddin, Chem. Pharm. Bull., 2001, 49, 278–281 CrossRef CAS.
- E. J. Simeonidou, N. A. Botsoglou, I. E. Psomas and D. J. Fletouris, J. Liq. Chromatogr. Relat. Technol., 1996, 19, 2349–2364 CAS.
- A. V. Pereira and Q. B. Cass, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2005, 826, 139–146 CrossRef CAS.
- F. C. C. R. de Paula, A. C. de Pietro and Q. B. Cass, J. Chromatogr., A, 2008, 1189, 221–226 CrossRef CAS.
- K. Kishida and N. Furusawa, Talanta, 2005, 67, 54–58 CrossRef CAS.
- K. Heinig and J. Henion, J. Chromatogr., B: Biomed. Sci. Appl., 1999, 732, 445–458 CrossRef CAS.
- A. Momberg, M. E. Carrera, D. V. Baer, C. F. Bruhn and M. R. Smith, Anal. Chim. Acta, 1984, 159, 119–127 CrossRef.
- T. Y. You, X. R. Yang and E. K. Wang, Analyst, 1998, 123, 2357–2360 RSC.
- J. M. P. Carrazon, A. D. Recio and L. M. P. Diez, Talanta, 1992, 39, 631–635 CrossRef CAS.
- T. N. Rao, B. V. Sarada, D. A. Tryk and A. Fujishima, J. Electroanal. Chem., 2000, 491, 175–181 CrossRef CAS.
- T. G. Diaz, A. G. Cabanillas, M. I. A. Valenzuela and F. Salinas, Analyst, 1996, 121, 547–552 RSC.
- J. J. Berzas, J. Rodriguez and G. Castañeda, Anal. Chim. Acta, 1997, 349, 303–311 CrossRef CAS.
- M. Kotoucek, J. Skopalova and D. Michalkova, Anal. Chim. Acta, 1997, 353, 61–69 CrossRef CAS.
- T. A. M. Msagati and J. C. Ngila, Talanta, 2002, 58, 605–610 CrossRef CAS.
- A. Preechaworapun, S. Chuanuwatanakul, Y. Einaga, K. Grudpan, S. Motomizu and O. Chailapakul, Talanta, 2006, 68, 1726–1731 CrossRef CAS.
- S. P. Özkorucuklu, Y. Sahin and G. Alsancak, Sensors, 2008, 8, 8463–8478 CrossRef CAS.
- C. D. Souza, O. C. Braga, I. C. Vieira and A. Spinelli, Sens. Actuators, B, 2008, 135, 66–73 CrossRef.
- A. Fujishima, Y. Einaga, T. N. Rao and D. A. Tryk (Eds.), Diamond Electrochemistry, Elsevier, Amsterdam, 2005 Search PubMed.
- P. Walsh (Ed.), Physicians Desk Reference (P D R), 54th Edition, Medical Economics Company, Montvale, NJ, 2000, p. 2614 Search PubMed.
- B. F. Reis, M. F. Giné, E. A. G. Zagatto, J. L. F. C. Lima and R. A. Lapa, Anal. Chim. Acta, 1994, 293, 129–138 CrossRef CAS.
- I. N. Rodriguez, J. A. M. Leyva and J. L. H. H. de Cisneros, Analyst, 1997, 122, 601–604 RSC.
- J. Jakmunee, S. Suteerapataranon, Y. Vaneesorn and K. Grudpan, Proc. IUPAC Internat. Congress Anal. Sci., 2001, 17, i399–i401 Search PubMed.
- United States Pharmacopeia 30-National Formulary 25, USP, 2007, 3247 Search PubMed.
- G. R. Salazar-Banda, L. S. Andrade, P. A. P. Nascente, P. S. Pizani, R. C. Rocha-Filho and L. A. Avaca, Electrochim. Acta, 2006, 51, 4612–4619 CrossRef CAS.
- J. J. Pedrotti, L. Angnes and I. G. R. Gutz, Electroanalysis, 1996, 8, 673–675.
- G. A. Shabir, J. Chromatogr., A, 2003, 987, 57–66 CrossRef CAS.
Footnote |
| † Current address: Departamento de Química, Universidade Federal de Goiás, Campus de Catalão, Avenida Lamartine P. Avelar 1120, 75704-020 Catalão - GO, Brazil |
| This journal is © The Royal Society of Chemistry 2010 |