Analytical protocol for identification of BMAA and DAB in biological samples
Zdeněk
Spáčil†
bc,
Johan
Eriksson†
*a,
Sara
Jonasson
a,
Ulla
Rasmussen
a,
Leopold L.
Ilag
*b and
Birgitta
Bergman
a
aDepartment of Botany, Stockholm University, 10691, Stockholm, Sweden. E-mail: johan.eriksson@botan.su.se; Fax: +46 (0)8 165525; Tel: +46 (0)8 163793
bDepartment of Analytical Chemistry, Stockholm University, 10691, Stockholm, Sweden. E-mail: leopold.ilag@anchem.su.se; Fax: +46 (0)8 156391; Tel: +46 (0)8 162435
cFaculty of Pharmacy, Charles University in Prague, Czech Republic
First published on 26th November 2009
Abstract
β-N-methylamino-L-alanine (BMAA) is a non-protein amino acid, thought to be inflicting neurodegenerative diseases related to ALS/PDC in human beings. Due to conflicting data concerning the presence of BMAA in various biological matrixes, we present a robust and sensitive method for high confidence identification of BMAA after derivatization by 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC). The efficient sample pretreatment in combination with LC-MS/MS SRM enables chromatographic separation of BMAA from the isomer 2,3-diaminobutyric acid (DAB). The method is applicable for selective BMAA/DAB detection in various biological samples ranging from a prokaryotic cyanobacterium to eukaryotic fish.
Introduction
β-N-methylamino-L-alanine (BMAA) is a potential human neurotoxin, which was identified for the first time in 1967, in seeds of Cycas micronesica, a gymnosperm tree common on the island of Guam (Pacific Ocean).1–5 BMAA was hypothesized to cause the high frequency of incidences of amyotrophic lateral sclerosis/parkinsonism-dementia complex (ALS/PDC) found among the indigenous population (the Chamorro), who feed on cycad products.3 Interestingly, BMAA is also produced by most cyanobacterial taxa, photosynthetic microorganisms with a ubiquitous distribution worldwide3 and has been reported to be detected in human populations outside the island of Guam.6 As such, the presence of BMAA may have critical medical implications due to its proven neurotoxicity.5 However, conflicting data have been published with regards to the occurrence of BMAA in both cyanobacteria,7–11 in cerebrospinal fluid (CSF) and in brain samples of patients suffering from Alzheimer's disease, as well as in Chamorro people suffering from ALS/PDC.6,7,12,13 Recently, α-γ-diamino butyric acid (DAB), a structural isomer of BMAA, was found to co-occur with BMAA in a cyanobacterium.11 Like BMAA, DAB is known to be neurotoxic, although primarily hepatotoxic.14 The inherent difficulty in distinguishing BMAA from DAB and vice versa, together with their potential co-occurrence in various biological matrices may indeed have contributed to the inconsistencies seen in various BMAA studies. In addition, as the analytical methodology used to detect and quantify BMAA in biological matrices has varied substantially,3,7–11,15,16 a critical re-evaluation of the methodology used is a prerequisite, in particular with regards to interfering substances in such varied and complex matrices.Any BMAA analyses need to be based on a reliable and robust analytical method which is available to the broad scientific community. We here present such a straight forward method including multiple step sample pretreatment leading to a robust BMAA identification and semi-quantification. The procedure requires the following steps: i) cell lysis, ii) acid hydrolysis of proteins, iii) cation-exchange solid phase extraction (SPE), and iv) derivatization, using 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC); followed by v) liquid chromatographic (LC) separation from the isomer DAB and vi) tandem mass spectrometric (MS/MS) detection of analytes. A detailed scheme of the sample pre-treatment workflow is given in Fig. 1.
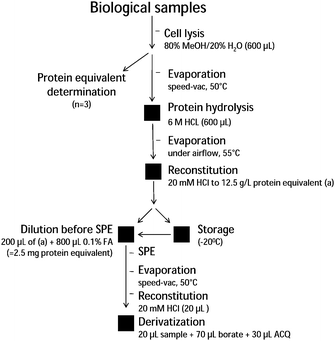 | ||
| Fig. 1 Schematic overview of the sample handling process from cell disruption to sample derivatization with ACQ prior to LC-MS/MS analysis. For detailed explanations see Experimental. | ||
The derivative 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (ACQ) is commonly used for the derivatization of amino acids, due to its quantitative reaction with both primary and secondary amines resulting in stable fluorescent derivates. The excess reagent is hydrolyzed within a minute into more polar products, which do not interfere with the analysis.17
Previously, a number of analytical approaches have been used with regards to BMAA identification, but so far, only one has included a simultaneous identification of BMAA and DAB.11 Moreover, it is also pivotal to address the sample complexity when selecting a sample pre-treatment procedure. Hence, we examined and optimized each component/step in detail, in order to obtain a reliable and reproducible method, suitable for BMAA analyses in complex biological matrices.
Experimental
Chemicals and reagents
The chemicals used in this study were purchased from Sigma-Aldrich and Fisher Scientific. A stock solutions of BMAA (Sigma-Aldrich, L-BMAA hydrochloride B-107) and DAB (Fluka, L-2-4-Diamobutyric acid hydrochloride #32830) were prepared to a concentration of ∼8 × 10−6 M by mixing individual standards in Millipore (18.2 MΩ) water.Samples and growth conditions
Axenic Leptolyngbya PCC 73110 was grown in batch cultures in BG11 media.18 The cultures were kept on a shaking table in a climate controlled chamber at +20 °C with continuous 80 μmol photons m−2 s−1 irradiance. After six days of growth, cells were harvested by centrifugation. Seeds of C. revoluta were harvested from vouchered specimen grown in the Bergianska Tropical Garden (Stockholm, Sweden). Field samples of Baltic Sea cyanobacteria were collected during the summer of 2008 in the southern part of the Stockholm archipelago using a planktonic net (90 μm). The mixture of cyanobacteria mainly consisted of the bloom-forming, heterocystous species Nodularia spumigena and Aphanizomenon sp. Fresh fish and oysters were purchased at the fish market in Stockholm, Sweden. All samples collected, harvested or purchased where immediately frozen at −80 °C for further extractions and analyses.Lysis of samples and acid hydrolysis of proteins
The extraction of the amino acids was performed according to Eriksson et al. (2009)19 with some minor changes. Cells were dissolved in 600 μl 80% methanol (80/20 methanol/water, v/v). Cell lysis was ensured by first freeze/thawing the suspension three times in liquid nitrogen followed by sonication (BANDELIN SONOPLUS, Model HP 2070) under 4 cycles of 30 s at 70% intensity. Between each cycle the sample was allowed to cool for at least 30 s on ice. During the sonication, the tube was kept in an ice-water bath, to prevent protein degradation.The protein concentration was measured after cell lysis in triplicates using the RC/DC kit (BioRad, Sweden). Each time 5 μL of sample after cell lysis were taken. Remaining volume was completely dried using a speed-vac (DNA Speedvac, Model DNA 100, Savant) at 50 °C. Then the sample was redissolved in 600 μL of 6 M HCl and hydrolysed for 24 h at 110 °C. The hydrolysates were filtered using a Centricon® spin-filter (Millipore) device at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for 5 min. HCl was evaporated under gentle airflow at 55 °C to complete dryness and thereafter the pellet was reconstituted in appropriate volume of 20 mM HCl to reach final concentration of 12.5 g L−1 of proteins and stored at −20 °C awaiting SPE clean-up.
000 g for 5 min. HCl was evaporated under gentle airflow at 55 °C to complete dryness and thereafter the pellet was reconstituted in appropriate volume of 20 mM HCl to reach final concentration of 12.5 g L−1 of proteins and stored at −20 °C awaiting SPE clean-up.
Solid phase extraction
The solid phase column (Isolute HCX-3, 100 mg, Sorbent AB, Sweden) was pre-conditioned with 1 mL methanol followed by 1 mL water and then conditioned with 1 mL 0.1% formic acid in water. 200 μl of the hydrolyzed sample (2.5 mg of protein equivalent) was diluted in 800 μL of 0.1% formic acid to a total volume of 1 mL and applied to the SPE column. The washing step was done using 1 mL 0.1% formic acid in water followed by a two step elution. The first elution step consisted of 1 mL 0.1% formic acid in 25% methanol and the second final elution step was 2% ammonium hydroxide in 100% methanol. Both eluates were evaporated using a speed-vac (DNA Speedvac, Model DNA 100, Savant) at 50 °C for approximately 3 h. The samples were then stored at −20 °C while awaiting further derivatization steps.Derivatization
Standards/samples were derivatized with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (Waters AccQ·Tag Ultra kit, Waters, Milford, MA, USA). All chemicals used for the reaction were supplied with the kit and solutions were applied as follows: 20 μL of a standard solution in required concentration was used directly or in case of real samples 20 μL of 20 mM HCl was added to redissolve the dried sample pellet from previous SPE procedure (2.5 mg of protein equivalents), then 70 μL of borate buffer was added followed by 30 μL ACQ tag. The sample was then vortexed and analyzed within 48 h.HPLC-MS/MS instrumentation
The HPLC system was an API 2000™ LC/MS/MS (Applied Biosystems, Foster City, CA, USA) consisting of binary pumps LC-10ADvp, autosampler SIL-10ADvp and bench top triple quadrupole mass analyzer API 2000. Separation was achieved on a Hypersil GOLD C18 100 × 2.1 mm, 3 μm analytical column (Thermo Scientific, San Jose, CA) injecting 50 μL of sample and using a gradient elution mode at a flow rate of 0.4 mL min−1. Eluent A was 0.1% formic acid in water and acetonitrile (93:7); eluent B was 0.1% formic acid in acetonitrile. The elution program was applied as follows: 0.0 min = 0% B; 10.0 min = 20% B; 10.1 min = 100% B; 12.0 min = 100%. ESI ionization was performed in positive ion detection mode. The mass analyzer used SRM scan type with recorded transitions: 459.1 > 119.1 (CE 30.0), 459.1 > 188.0 (CE 38.0), 459.1 > 258.0 (CE 30.0). Other parameters of the ESI source and the mass analyzer are listed below: capillary voltage (5500 V), source temperature (450 °C), declustering potential (50), focusing potential (350), entrance potential (6). Analyst 4.2 software was used to explore acquired chromatographic data.
Analytical procedures and validation
Detection limits of BMAA and DAB were evaluated experimentally by injecting dilution series of analyte stock solutions, signal to noise (S/N) ratios were calculated for the chromatographic peak at the lowest visible concentration. Standards from a stock solution were used to prepare a dilution series in order to make calibration curves used for semi-quantification. The other parameters such as resolution, peak capacity and peak width were calculated according to appropriate guidelines.20–22 The intraday and interday precisions were evaluated according to the RSD (%) of the peak area, retention time and SRM transitions ratio (n = 6).Result and discussion
HPLC-MS/MS method features
The LC separation with tandem mass spectrometric detection of BMAA and DAB combines efficient features ensuring confident and selective identification of the two analytes. These are: i) mutual chromatographic separation of BMAA (Rt = 3.5 min) and DAB (Rt = 4.3 min), ii) MS/MS detection using Selected Reaction Monitoring (SRM) of diagnostic transitions, and iii) calculated constant ratios between peak areas among observed SRM transitions.ACQ derivates provide excellent peak shape when analyzed on reversed phase column and also exhibit intense signals ionized by ESI. Considering the structural similarity of BMAA and DAB derivates appropriate elution conditions are of great importance. The gradient elution program established allowed baseline separation of BMAA and DAB derivates, with baseline resolution (typically 2.5); a peak capacity >30 and a 20 s peak width. Repeatability of retention time, peak area and SRM ratio are stated in Tab. 1.
| BMAA | DAB | ||||
|---|---|---|---|---|---|
| SRM Transition | SRM Transition | ||||
| 459.1/119.1 | 459.1/258.1 | 459.1/119.1 | 459.1/188.1 | ||
| Intraday RSD% (n = 6) | Peak Area | 7.11 | 7.27 | 6.04 | 6.45 |
| Retention Time | 0.39 | 0.39 | 0.43 | 0.50 | |
| SRM ratio | 1.75 | 1.02 | |||
| Interday RSD% (n = 6) | Peak Area | 9.42 | 9.89 | 7.75 | 8.21 |
| Retention Time | 0.54 | 0.60 | 0.52 | 0.53 | |
| SRM ratio | 1.71 | 1.02 | |||
| Inter work-up RSD% (n = 3) | Peak Area | 11.72 | 10.79 | 11.01 | 12.72 |
| Retention Time | 0.43 | 0.69 | 0.97 | 0.97 | |
| SRM ratio | 1.44 | 1.67 | |||
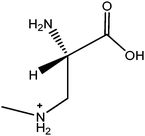
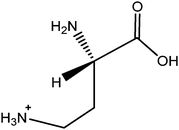
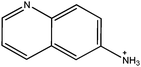
The ACQ derivates of BMAA and DAB show similar fluorescence, identical molecular weight and almost identical product ion spectra, thus fluorescent or inappropriate MS detection techniques might result in misleading identifications. Fig. 1a and 1b display spectra acquired under the same MS/MS conditions and hypothesized structures of fragments are given in Tab. 2. Practically all observed product ions (119.1 m/z, 145.1 m/z, 171.1 m/z, 289.1 m/z and 315.1 m/z) of precursor at 459.1 m/z originate from N–C bond cleavages in both BMAA and DAB. Ions at 145.1 m/z and 171.1 m/z represents [M + H]+ ions of the ACQ tag, arising from the cleavage of the bond between the amide nitrogen and the carbonyl carbon. As such, these lack any specificity. Their complementary ions at 315.1 m/z and 289.1 m/z incorporate structure of amino acids giving them more diagnostic relevance, but still insufficient for distinguishing between amino acids of the same molecular weight. The truly specific fragment ion for BMAA was observed at 258.1 m/z, likely originating from the cleavage of the bond between the secondary amine nitrogen and the β-carbon. Another diagnostic ion at 188.1 m/z is created after cleavage of the bond between the primary amine nitrogen and the α-carbon of DAB, as it is absent in the BMAA product ion spectra. These ions together with the product ion at 119.1 m/z, corresponding to the protonated intact amino acid, were suitable for SRM using the following diagnostic transitions: 459.1 > 119.1; 459.1 > 258.1 and 459.1 > 188.1. The detection sensitivity was approximately 70 fmol (S/N = 9.2) for BMAA and approximately 700 fmol (S/N = 8.0) for DAB injected on column. The constant ratio between peak areas of SRM transitions can also be used as diagnostic tools to confirm correct identities of the two analytes. These transition ratio are 8.52 ± 0.2 (459.1 > 119.1: 459.1 > 258.1) for BMAA standard and 3.07 ± 0.2 (459.1 > 119.1: 459.1 > 188.1) for DAB standard.
| m/z | BMAA | DAB |
|---|---|---|
| 119.1 |
|
|
| 145.1 |
|
|
| 171.1 |
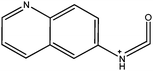
|
|
| 188.1 |
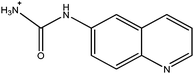
|
|
| 258.1 |
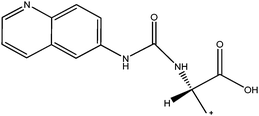
|
|
| 289.1 |
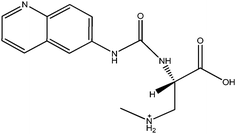
|
|
| 315.1 |
|
|
Biological samples analysis
The broad applicability of the BMAA/DAB identification method presented here is demonstrated for distinctly varying types of biological sample matrices, representing organisms from different kingdoms, such as prokaryotes (laboratory cultures and field samples of a cyanobacteria), and some eukaryotes, such as plants (the gymnosperm Cycas revoluta, the angiosperm Brassica oleracea, broccoli), a vertebrate (Coregonus lavaretus, white fish), an invertebrate (Ostrea edulis, oysters), all examined to validate the method. All samples were standardized by determination of protein concentration and material corresponding to 2.5 mg of protein equivalent was analyzed for each sample. Therefore the total amount of BMAA and DAB, both free and protein associated, was included as we do not distinguish between these forms. Resulting chromatograms from these experiments are presented in Fig. 2 and 3. We observed 0.2 min shift in retention time analyzing the biological samples. This was due to the sample matrix, as the shift was not visible in the runs with BMAA/DAB standard in the same sample batch and the shift was also consistent in terms of retention time and resolution between BMAA and DAB. The detected concentrations of BMAA and DAB are expressed as μg L−1 contained in the amount of samples corresponding to 2.5 mg of protein equivalent. The highest detected concentration of BMAA was found in the sample from seeds of the plant Cycas revoluta (Tab. 3). Also laboratory cultures of the filamentous cyanobacterium Leptolyngbya PCC 73110 exhibit fairly high content of BMAA, while the concentration in a field sample of mixed cyanobacteria was about 100 fold lower. Both white fish (Coregonus lavaretus) and oyster (Ostrea edulis) exhibited similar levels of BMAA, while broccoli (Brassica oleracea) and a standard of Bovine serum albumin (BSA) were negative. DAB was present in similar concentration as BMAA in the two cyanobacterial samples, in oyster and in broccoli, illustrating the common occurrence and the need for separating these two compounds (Tab. 3). For example, the broccoli sample was previously assayed using the HPLC-FD method,19 and only one peak with Rt corresponding to BMAA was detected (data not shown). In contrast, the procedure and methodology presented above revealed that BMAA was not present in broccoli, while DAB was present at clearly detectable concentration. These findings stress the need to introduce a method that is able to reliably distinguish between BMAA and DAB. In order to rule out the possibility that BMAA or DAB were artifacts from the protein hydrolysis step, potentially caused by side reactions during the acidic protein hydrolysis, 10.5 mg of BSA standard was hydrolyzed and pretreated in the same way as the biological samples. The hypothesis was rejected as neither a BMAA nor a DAB signal was detected (Fig. 4d).| Seeds of Cycas revoluta (*) | Brassica oleracea (*) | Leptolyngbya PCC 73110 (**) | Field sample of cyanobacteria (**) | Ostrea edulis (***) | Coregonus lavaretus (****) | |
|---|---|---|---|---|---|---|
| BMAA | ||||||
| μg L−1 | 933.4 | Not detected | 109.4 | 1.9 | 47.9 | 49.4 |
| 459/119 | Detected | Not detected | Detected | Detected | Detected | Detected |
| 459/258 | Detected | Not detected | Detected | Detected | Detected | Detected |
| Ratio | 8.58 | Not detected | 8.48 | 8.46 | 8.51 | 8.57 |
| Rt | 3.25 | Not detected | 3.44 | 3.18 | 3.25 | 3.26 |
| DAB | ||||||
| μg L−1 | Not detected | 171.7 | 148.0 | 175.9 | 201.7 | Not detected |
| 459/119 | Not detected | Detected | Detected | Detected | Detected | Not detected |
| 459/188 | Not detected | Detected | Detected | Detected | Detected | Not detected |
| Ratio | — | 2.94 | 2.88 | 2.81 | 3.01 | — |
| Rt | — | 4.28 | 4.22 | 3.89 | 4.07 | — |
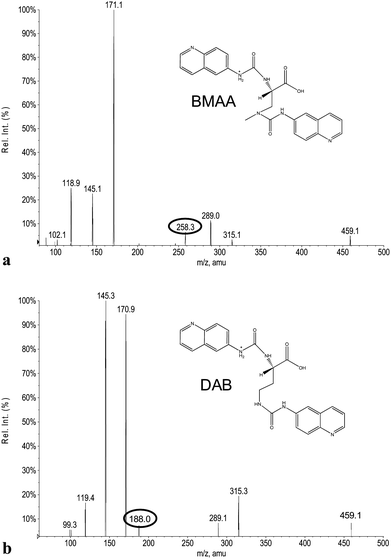 | ||
| Fig. 2 Tandem MS product ion spectra of (a) BMAA and (b) DAB. The spectra were acquired under the same conditions with the precursor ion at 459.1 m/z. Specific product ions: at 258.1 m/z (BMAA) and at 188.1 m/z (DAB) are encircled. | ||
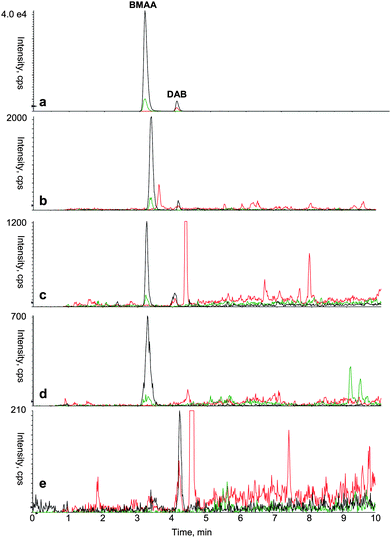 | ||
| Fig. 3 Typical mass spectrum acquired using HPLC-MS/MS based analytical protocol to identify BMAA and DAB in various types of samples. (a) Standard of derivatized BMAA (0.35 μg L−1) and DAB (0.35 μg L−1) (b)Leptolyngbya PCC 73110 (laboratory cyanobacterial culture) (c)Ostrea edulis (oyster) (d)Coreganus lavaretus (white fish) (e)Brassica oleracea (broccoli). Color legend for SRM transition: 459.1 > 119.1 (black); 459.1 > 258.1 (green); 459.1 > 188.1 (red). | ||
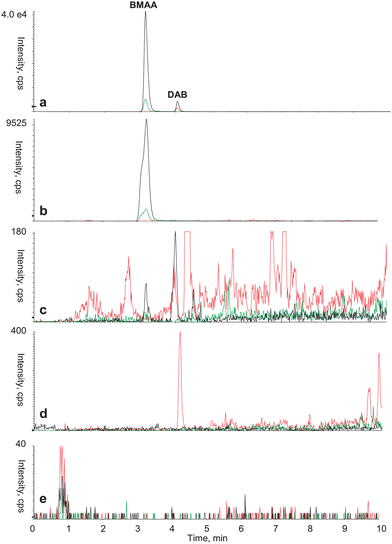 | ||
| Fig. 4 Typical mass spectrum acquired using HPLC-MS/MS based analytical protocol to identify BMAA and DAB in various biological matrixes. (a) Standard of derivatized BMAA (0.35 μg L−1) and DAB (0.35 μg L−1), (b) seeds from C. revoluta, (c) field sample of mixed cyanobacteria, (d) standard of Bovine Serum Albumin (BSA) and (e) procedural blank. Color legend for SRM transition: 459.1 > 119.1 (black); 459.1 > 258.1 (green); 459.1 > 188.1 (red). | ||
Conclusion
Here we present a robust analytical protocol including multiple step sample pretreatment combined with high confidence LC-MS/MS analysis in order to identify BMAA as well as distinguish between BMAA and its structural isomer DAB. This method provides qualitative and semi-quantitative information and enabled us to detect BMAA, and to separate BMAA from DAB in a range of organisms representing different kingdoms. Together our data verifies that this represents a novel reproducible procedure for the analysis of BMAA and DAB in complex biological matrices. Our findings may also to some extent explain the published controversies in regards to the presence/absence of BMAA in different types of biological systems.Acknowledgements
Funded by The Foundation For Strategic Environmental Research, Swedish Research Council for Environmental, Agricultural Sciences and Spatial Planning, K., A. Wallenberg Foundation, The Royal Swedish Academy of Sciences (FOA08H-242), Stockholm University Proteomics Facility (SUPF) and GAUK (119308/B/2008) and the Czech Ministry of Education (Project MSM 0021620822).References
- A. Vega, A. Bell and P. Nunn, Phytochemistry, 1967, 6, 759–762 CrossRef CAS.
- S. A. Banack, S. J. Murch and P. A. Cox, J. Ethnopharmacol., 2006, 106, 97–104 CrossRef.
- P. A. Cox, S. A. Banack, S. J. Murch, U. Rasmussen, G. Tien, R. R. Bidigare, J. S. Metcalf, L. F. Morrison, G. A. Codd and Birgitta Bergman, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5074–5078 CrossRef CAS.
- K. D. Hill, Australian Systematic Botany, 1994, 7, 543–567 Search PubMed.
- S. D. Rao, S. A. Banack, P. A. Cox and J. H. Weiss, Exp. Neurol., 2006, 201, 244–252 CrossRef CAS.
- S. J. Murch, P. A. Cox, S. A. Banack, J. C. Steele and O. W. Sacks, Acta Neurol. Scand., 2004, 110, 267–269 CrossRef CAS.
- T. J. Montine, K. Li, D. P. Perl and D. Galasko, Neurology, 2005, 65, 768–769 CrossRef CAS.
- M. Esterhuizen and T. G. Downing, Ecotoxicol. Environ. Saf., 2008, 71, 309–313 CrossRef CAS.
- T. Kubo, N. Kato, K. Hosoya and K. Kaya, Toxicon, 2008, 51, 1264–1268 CrossRef CAS.
- S. Moura, M. D. Ultramari, D. M. L. de Paula, M. Yonamine and E. Pinto, Toxicon, 2009, 53, 578–583 CrossRef CAS.
- J. Rosen and K. E. Hellenäs, Analyst, 2008, 133, 1785–1789 RSC.
- M. M. Kushnir and J. Bergquist, Eur. J. Mass Spectrom., 2009, 15, 439–443 CrossRef CAS.
- J. Pablo, S. A. Banack, P. A. Cox, T. E. Johnson, S. Papapetropoulos, W. G. Bradley, A. Buck and D. C. Mash, Acta Neurol. Scand., 2009, 120, 216–225 CrossRef CAS.
- R. M. O'Neal, C. H. Chen, C. S. Reynolds, S. K. Meghal and R. E. Koeppe, Biochem. J, 1968, 106, 699–706 CAS.
- L. R. Snyder, R. Cruz-Aguado, M. Sadilek, D. Galasko, C. A. Shaw and T. J. Montine, Toxicol. Appl. Pharmacol., 2009, 240, 180–188 CrossRef CAS.
- P. M. Scott, B. Niedzwiadek, D. F. K. Rawn and B. P.-Y. Lau, J. Food Prot., 2009, 72, 1769–1773 CAS.
- M. Reverter, T. Lundh and J. E. Lindberg, J. Chromatogr., B: Biomed. Sci. Appl., 1997, 696, 1–8 CrossRef CAS.
- R. Y. Stanier, R. Kunisawa, M. Mandel and G. Cohen-Bazir, Bacteriol. Rev., 1971, 35, 171–205 CAS.
- J. Eriksson, S. Jonasson, D. Papaefthimiou, U. Rasmussen and B. Bergman, Amino Acids, 2009, 36, 43–48 CrossRef CAS.
- European Pharmacopoeia 6th edition (Ph. Eur. 6). Council of Europe, Strasbourg, 2007 Search PubMed.
- D. Uwe, J. Chromatogr., A, 2005, 1079, 153–161 CrossRef CAS.
- International Conference on Harmonization (ICH). Q2(R1): Text on Validation of Analytical Procedures. US FDA Federal Register. ( 2005) Search PubMed.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2010 |