A novel isotachophoresis of cobalt and copper complexes by metal ion substitution reaction in a continuous moving chelation boundary†
Wei
Zhang
a,
Jian-Feng
Chen
a,
Liu-Yin
Fan
*a,
Cheng-Xi
Cao
*a,
Ji-Cun
Ren
b,
Si
Li
a and
Jing
Shao
a
aLaboratory of Analytical Biochemistry & Bio-separation, Key Laboratory of Microbiology of Educational Ministry, School of Life Science and Biotechnology, Shanghai Jiao Tong University, Shanghai, 200240, China. E-mail: cxcao@sjtu.edu.cn; Fax: +86 21-3420 5820; Tel: +86 21-3420 5682lyfan@sjtu.edu.cn; Tel: +86 21-3420 5682
bCollege of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai, 200240, China
First published on 9th November 2009
Abstract
A novel separation mode of isotachophoresis (ITP) was advanced for the study on the continuous moving chelation boundary (MCB) formed with EDTA and two metal ions of Co(II) and Cu(II). The experiments were performed systemically. The relevant results indicated that: (1) there were three boundaries in the whole system, viz., a sharp MCB, a wide moving substitution boundary (MSB) and a sharp complex boundary (CB); (2) within the MSB, an ion substitution reaction occurred between [Co-EDTA]2− and Cu(II), and the reaction resulted in the release of Co(II) and EDTA from [Co-EDTA]2− and the binding of Cu(II) with the released EDTA due to log KCu(II) (= 18.80) > log KCo(II) (= 16.31); (3) because of the novel ITP mode induced by the MSB as well as the merging of the MCB and CB, the original low concentration Co(II) and Cu(II) were chemically separated as two characteristic coloured zones of pink [Co-EDTA]2− and blue [Cu-EDTA]2−, and the sensitivities for detection of the two metal ions were greatly enhanced. The quantitative analyses of the zone composition by ICP-AES and UV-vis spectrophotometry supported the mechanism of the novel separation mode induced by the MSB. The further theoretical and experimental results indicated that the separation mode was a novel ITP relied on moving reaction boundary (MRB), rather than a classic ITP based on the moving boundary system developed about 60 years ago. These findings provide guidance for the development of the MRB and the MCB-based ITP separation of metal ions in environmental and biological matrices.
1. Introduction
Since 1970, three kinds of moving reaction boundaries (MRBs) have been investigated. The three MRBs were the moving precipitate boundary (MPB) advanced by Deman1–4et al. and Cao5et al., the moving neutralization boundary (MNB) developed by Cao5,6et al., and the moving chelation boundary (MCB) evolved by Fan7et al. and Jin8et al. from the important work of EDTA-based sweeping contributed by Quirino9et al. and Isoo and Terabe 10.In 1970, Deman1–4et al. advanced the important concepts of the ‘precipitate reaction front’ and ‘moving reaction front (MRF)’, actually the prototype of MPB mode1,2 and observed the separative phenomena of different metal ions, such as Cd(II) and Co(II), in accordance with the solubility products in different MPB systems.2–4 Although being unnoticed almost completely,5 the idea of MRF had great importance in the analytical and separative fields. At first, Deman3,4 developed the concept of elution boundary (EB) and performed the EB-induced separation of metal ions in a MRF. The EB-induced separation was actually the prototype of the novel isotachophoresis (ITP) developed herein. The novel ITP relied on the physico-chemical model of MRB system, as revealed by eqn (19) in Ref. 5 and demonstrated herein. Further more, the novel ITP was different from the classic ITP11,12 which was based on the traditional physico-chemical model of moving boundary system (MBS) developed about 60 years ago,11–24 as shown in Section 3.3 herein. Secondly, the concept of MPB provides an idea for solving a series of fundamental scientific problems,25–30 which have existed in isoelectric focusing (IEF) for about 40 years, as has been shown in detail in Section 3 and 4.4 of Ref. 5.
About ten years ago, Cao5,6et al. developed the concept of MNB from the important ideas of MPB1–4 and the stationary neutralization boundary created in capillary electrophoresis (CE).36 As has been revealed in Ref. 5, the concept of MNB could systemically solve a series of fundamental scientific problems existing in classic IEF (see Section 4.4 in Ref. 5), e.g., the dynamics of classic IEF,5,6 the quasi-stable pH gradient in IEF,5 the pH gradient drifting5,25–30 and plateau of pH gradient during IEF,5,29,30 and the Hjertén's mobilization of pH gradient after IEF.5,30–35 Furthermore, the relevant method37–39 and theory of MNB-based sample stacking40 were developed for improving the detection sensitivity in CE and for computer simulation.5,41,42
In 2008, Fan5,7et al. further advanced the theory of MCB from the previous studies,1–6,9,10 verified the existence of a transient MCB and a complex boundary (CB) in the EDTA-based metal ion sweeping,9,10 performed the theoretical investigations for boundary movements and controllable sweeping of metal ion. The theoretical studies manifested that the difference between the MCB and CB velocities played a key role in the stacking efficiency of one metal ion. This year, Jin8et al. developed the mathematic mode of MCB and relevant computer software, and used the software for the quantitative simulation of EDTA-based sweeping of metal ion in a continuous MCB. With the simulative and experimental results on MCB, Zhang43et al. demonstrated the theoretical relationship between the Kohlrausch regulating function (the oldest function in the electrophoretic field) and MRB equations.
However, if two metal ions (e.g., Co(II) and Cu(II)) were used to create a continuous MCB system, a series of interesting results were observed, including the separation of two characteristic zones of pink [Co-EDTA]2− and blue [Cu-EDTA]2−, the formation of MCB, moving substitution boundary (MSB) and CB, and the condensation of the two metal ions as well. As shown in Section 3 herein, the separation mechanism of two metal complexes is different from the present electrophoretic modes. Firstly, the relevant mechanism of separation could not be explained by the present concept of MCB,5,7,8 but could be illuminated by the combination of MCB and MSB concepts. Secondly, the simultaneous separation and condensation of two metal ion complexes could not be explained by the current separative mechanisms of inorganic ions, such as, a classic ITP11,12 based on the physico-chemical mode of MBS,11–24 capillary zone electrophoresis (CZE),44,45 capillary electro- chromatography (CEC), ionic exchange CEC, micellar electrokinetic chromatography (MEKC),46,47 electrostatic CE, and ion selection system etc.
Therefore, the main purposes in this paper are to introduce the mode of novel separation of two metal ion complexes in a MCB system, describe the experimental procedures, report the relevant separative results and briefly discuss the relative mechanism of separation. To succinctly visualize the whole course of MCB system, we choose EDTA and two metal ions (Cu(II) and Co(II)) as the model ions of chelation in the studied MCB system due to their characteristic colours of complexes and non-absorption to the gel used herein. The relevant theoretical studies on the separation will be performed in another paper due to the much complexity of theoretical formation and computation of the novel ITP separation.
2. Experimental
2.1 Chemicals
Ethylenediaminetetraacetic acid disodium salt (Na2EDTA, Analytical Reagent Grade, AR), potassium chloride (Guaranteed Reagent grade, GR) and agarose (Biochemical Reagent grade, BR) were obtained from Shanghai Chemical Reagent Co. (Shanghai, China). Copper chloride (CuCl2·2H2O, AR) and cobalt chloride (CoCl2·6H2O, AR) were purchased from Sino-pharm Chemical Reagent Co., Ltd. (Shanghai, China). Sodium hydroxide (GR) was from Shanghai Zhungong Reagent Factory (Shanghai, China).2.2 Instruments
A home-made apparatus was used for the experiments on a MCB. The apparatus had been described in detail in Ref. 7. A glass tube (i.d. 3.60 mm × 170 mm) was used for the run of gel-filled tube experiment. Peristaltic pumps (HL-2, the Shanghai Hu-xi Instrumental Factor, Shanghai, China) were employed to pump the solutions into the tube. The electric field was yielded by a power supply (DYY-2C, the Beijing Liu-yi Scientific Instrument Factor, Beijing, China). A high resolution digital camera (6490, the Kodak Co., Rochester, NY, USA) was fixed above the tube to monitor the run of MCB and colour change. An UV-Vis spectrophotometer (UV-2550, Shimadzu Co., Kyoto, Japan) using 1.0 cm matched silica cells was employed for the detection of complexes in the gel. An inductive coupled plasma atomic emission spectrometry (ICP-AES, IRIS Advantage 1000, Thermo Jarrell Ash Co., USA) was employed for the determination of metal total concentrations. The experiments in the gel-filled tube were carried out at ambient temperature.An apparatus of CE (P/ACE MDQ, Beckman Coulter Co., Fullerton, CA, USA) was used for the run of continuous EDTA-based sweeping. The CE was equipped with a power supply (up to constant voltage 30 kV), a 32 Karat software and an UV-visible detector. A fused–silica capillary (35.1 cm total length, 25 cm effective length, I. D. 75 μm) was obtained from the Factory of Yongnian Optical Fiber (Hebei, China). The runs were carried out under 25 °C. Before runs, the new capillary was conditioned by rinsing with 1 M NaOH for 10 min, ultrapure water for 5 min, 1 M HCl for 10 min, and running buffer for 10 min, in order. Shorter wash of running buffer was used between runs.
Ultrapure water with conductivity down to 0.055 μS cm−1 was finely produced by a pure water system (SG Wasseraufbereitung und Regenerierstation GmbH, Fahrenberg, Barsbüttel, Germany). The pH values of buffers used were adjusted by a pH meter (320, Mettler-Toledo Instruments (Shanghai) Ltd., Shanghai, China).
2.3 Solution and procedure used in gel-filled tube run
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Then the mixtures were diluted to 10 mM as the stock solutions of [Cu-EDTA]2− and [Co-EDTA]2−. The pH value was adjusted to 5.0 with 1 M NaOH.
:1. Then the mixtures were diluted to 10 mM as the stock solutions of [Cu-EDTA]2− and [Co-EDTA]2−. The pH value was adjusted to 5.0 with 1 M NaOH.
| No. | CuCl2/mM | CoCl2/mM | KCl/mM |
|---|---|---|---|
| a The ionic strength of each solution above is 100 mM, adjusted by KCl. | |||
| 1 | 0.5 | 0.5 | 97 |
| 2 | 1.0 | 1.0 | 94 |
| 3 | 2.0 | 2.0 | 88 |
| 4 | 4.0 | 4.0 | 76 |
| 5 | 8.0 | 8.0 | 52 |
Two kinds of gels, viz., an EDTA gel and a metal gel, were prepared. The EDTA gel was prepared with the EDTA solution mentioned above, and its composition was 2.0% agarose, 30 mM Na2EDTA and 5.4 mM KCl (pH 5.0). The metal gel contained 2.0% agarose and the two metal ions with different concentration shown in Table 1.
![Mode of novel ITP in the MCB system formed with Cu(ii), Co(ii) and EDTA. (A) initial MCB just after the electric field; (B) formations of MCB, MSB, CB and two new phases (viz., phase α′ and β′) in the given MCB system; (C) mechanism of metal ion substitution within the MSB. The arrows indicate the movements of MCB, EDTA, Cu(ii), Co(ii), complex of [Cu-EDTA]2− and [Co-EDTA]2−. The symbols “+” and “–” represent the anode and cathode, respectively.](/image/article/2010/AN/b912799b/b912799b-f1.gif) | ||
| Fig. 1 Mode of novel ITP in the MCB system formed with Cu(II), Co(II) and EDTA. (A) initial MCB just after the electric field; (B) formations of MCB, MSB, CB and two new phases (viz., phase α′ and β′) in the given MCB system; (C) mechanism of metal ion substitution within the MSB. The arrows indicate the movements of MCB, EDTA, Cu(II), Co(II), complex of [Cu-EDTA]2− and [Co-EDTA]2−. The symbols “+” and “–” represent the anode and cathode, respectively. | ||
2.4 Solution and procedure used in CE
:1, and 50 mM [Cu-EDTA]2− or 50 mM [Co-EDTA]2− stock solutions were obtained. Then the stock solutions were diluted with 100 mM pH 5.0 acetate buffer to obtain 0.2, 1.0, 2.0, 5.0, 10, 15, 20 mM standard solutions. The compositions of metal ion solutions were 0.01/0.01, 0.03/0.03, 0.1/0.1, 0.5/0.5, 1.0/1.0, 2.0/2.0 mM of Cu(II)/Co(II) in 100 mM pH 5.0 acetate buffer.
2.5 Mobility determination of metal ionic complexes in CE
To determinate the ionic mobilities of Cu(II)-EDTA and Co(II)-EDTA under the given conditions of novel ITP experiments, a running buffer and some complex samples were prepared. The running buffer contained pH 5.0 100 mM acetate buffer with 4.0 mM EDTA and the complex samples had 100 μM M-EDTA (M = Cu(II) or Co(II)) + 100 mM pH 5.0 acetate buffer + 4.0 mM EDTA + 2% acetone. The acetone was used as the EOF marker. After a run of CZE of complex samples, the complex mobility in the EDTA-BGS could be computed with the following equation,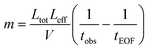 | (1) |
3. Results and discussion
3.1 Mode of novel continuous separation of metal ion complexes
Fig. 1 showed the experimental mode of the separation of two metal ion complexes by the ionic substitution in a MCB initially formed with EDTA and two metal ions of Cu(II) and Co(II). As shown in panel A, EDTA was in phase β, Cu(II) and Co(II) were in phase α. The anode and cathode were respectively set at the right and left sides. If voltage was applied, Cu(II) and Co(II) moved towards the cathode due to their positive charges, and EDTA electrically migrated towards the anode because of its negative charges. When they met each other, two chelation reactions took place between Cu(II), Co(II) and EDTA (see Panel A and Reaction (I) shown below).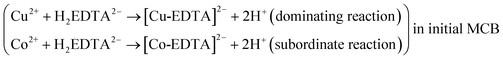 | (I) |
In the mode of Fig. 1, EDTA, Co(II) and Cu(II) were used as the model ions of the novel separation of two complexes in the MCB studied herein. This was because of the following reasons. At first, EDTA, a widely-used colourless chelator, could react with many metal ions in equivalent molar ratio and form a stable complex of [M-EDTA]n−4 (where M implied metal ion and n was the positive charge of the metal ion). Secondly, the complexes of [Cu-EDTA]2− and [Co-EDTA]2− had characteristic blue and pink colours, respectively, while the solutions of Co(II) and Cu(II) were almost colourless under the given low concentration. The characteristic colour contrast between the two complexes was very important for distinguishing the zones of [Cu-EDTA]2− and [Co-EDTA]2−. Thirdly, the reaction between EDTA and metal ions was rapid48–50 because of enough high reaction coefficient with EDTA.51–53 This kind of chelation reaction has been used for: (1) the probing for indirect photometric detection via the complexation prior to injection in the capillary zone electrophoresis (CZE),54,55 and (2) the online separation of metal ions via on-capillary complexation in CZE.56–60
In the chelation reaction system (I), the equilibrium reaction between Cu(II) and EDTA took dominant as compared with that between Co(II) and EDTA, because the stability constants of [Co-EDTA]2− (lgKCo = 16.31) was less than that of [Cu-EDTA]2− (lgKCu = 18.80).61 An initial MCB was formed because of the electromigration chelation reaction just after the voltage was applied in Panel A. The initial MCB in Panel B was expressed as
| EDTA (β,−) ‖ [→] Cu(II) + Co(II) (α,+) | (A) |
In the process of a run, complexes of [Co-EDTA]2− and [Cu-EDTA]2− were produced in the MCB system of Fig. 1. Because of the existence of ion substitution reaction within a MSB (see the following section), the two characteristic colour zones of complexes [Co-EDTA]2− and [Cu-EDTA]2− were separated with each other in accordance with their stability constants. The two characteristic colour zones were the pink [Co-EDTA]2− at the left side and the blue [Cu-EDTA]2− at the right side (see Panel B). In other words, two new phases, viz., phase β′ and phase α′, were simultaneously created, which mainly contain the [Co-EDTA]2− and [Cu-EDTA]2−, respectively. Clearly three boundaries were created in the whole system. The first boundary, from the left to the right, was the MCB created between the colourless EDTA and the characteristic pink [Co-EDTA]2−. The second was the MSB between the characteristic pink [Co-EDTA]2− and blue [Cu-EDTA]2−. The third was the CB between the blue [Cu-EDTA]2− and almost colourless metal ions in phase α. The three boundaries isolated the whole system into four zones, viz., phase β, β′, α′ and α from the left to the right of Panel B. As a result, the entire MCB system in Panel B could be expressed as
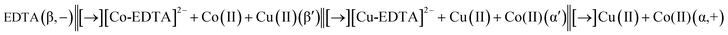 | (B) |
Evidently, a novel separation mode of metal ion complexes existed in the MCB system. As clearly shown in Fig. 1B, the newly-created MCB, MSB and CB separated the newly-produced two complexes as the blue [Cu-EDTA]2− and pink [Co-EDTA]2− zones. As revealed in the subsequent section, the principle of the novel separation mode was mainly based on the ionic substitution reaction in accordance with the complex stability constants within the MSB.
3.2 Demonstrations of novel continuous separation mode
Fig. 2 revealed the novel continuous separation of two metal ion complexes and the formation of MCB, MSB, CB and two new phases (viz., phase α′ and β′) in the given MCB system in a gel-filled tube. In Fig. 2, the initial chelation boundary between EDTA and metal ions was indicated by the arrow. In Panel A, two characteristic colour zones, viz., the pink in the left and the blue in the right, were clearly observed, as shown by the runs at 3, 6, 9, 12, 15 min. The pink zone was just phase β′ containing the newly produced [Co-EDTA]2− and the blue zone was phase α′ holding the newly-yielded [Cu-EDTA]2−. The long colourless zone at the left side of pink zone was phase β having EDTA. The colourless zone at the right side of blue zone was phase α containing Co(II) and Cu(II). The MCB was located between the colourless β phase and the pink β′ phase, the MSB was between the pink β′ phase and the blue α′ phase, the CB was created between the blue α′ phase and the colourless α phase. The experiments in Panel B demonstrated the similar results of Panel A, but the zones of two complexes [Co-EDTA]2− and [Cu-EDTA]2− became much sharper under the condition of low concentration metal ions used.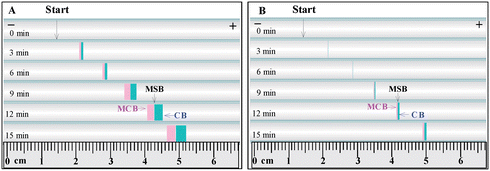 | ||
| Fig. 2 Formations of MCB, MSB, CB and new phases as well as boundary movements in the MCB systems initially formed by (A) 2.0 mM Cu(II) + 2.0 mM Co(II) + 88 mM KCl in phase α and 30 mM Na2EDTA + 5.4 mM KCl in phase β; (B) 0.5 mM Cu(II) + 0.5 mM Co(II) + 97 mM KCl in phase α and 30 mM Na2EDTA + 5.4 mM KCl in phase β. Experimental conditions: 4 mA electric current and 145 mV voltage; 2.0% agarose gel in the tube; I.D. of tube = 3.6 mm; length of tube = 170 mm; flow rate of the anolyte and catholyte = 2.0 mL min−1; “Start” means the initial boundary between metal ions and EDTA gels before electric field was applied; the symbol ‘+’ and ‘−’ indicate the anode and cathode respectively. | ||
Fig. 3 unveiled the experiments achieved in CE. As shown in Fig. 3, there was a long base plateau which indicated the existence of phase α containing two metal ions of Co(II) and Cu(II). After the plateau, an almost vertical line was present which represented a CB between phase α and α′. Then a high plateau peak was observed, the peak was just the newly-produced [Cu-EDTA]2− with strong absorbance at 254 nm. Behind the [Cu-EDTA]2− peak, an oblique line was observed. The oblique line indicated a wide MSB created between phase α′ and β′. A low plateau appeared behind the MSB. The low plateau was the newly-yielded complex of [Co-EDTA]2− with low absorbance at 254 nm, implying the existence of phase β′. After the plateau of [Co-EDTA]2−, a nearly vertical line was detected, which indicated the creation of MCB between EDTA and Co(II). Finally, one could observe the long plateau, which was lower than the frontal base plateau of phase α because of much low absorbance of EDTA.
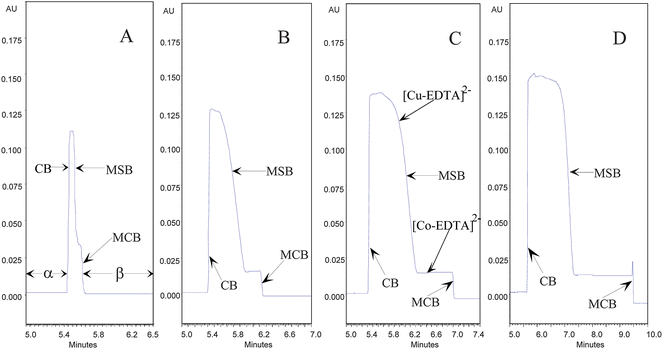 | ||
| Fig. 3 Electropherograms of MCB system in CE initially formed by: (A) 0.1 mM Cu(II) + 0.1 mM Co(II); (B) 0.5 mM Cu(II) + 0.5 mM Co(II); (C) 1.0 mM Cu(II) + 1.0 mM Co(II); (D) 2.0 mM Cu(II) + 2.0 mM Co(II) in 100 mM pH 5.0 HAc-NaAc buffer as phase α and 30 mM Na2EDTA in 40 mM pH 5.0 HAc-NaAc buffer as phase β. Conditions: UV-detector set at 254 nm, −15 kV, 35.1 cm (effective length 25 cm to the detector) and I.D. 75 μm capillary, 25 °C. The experimental FE values of Cu(II) in Fig. 3A, 3B, 3C and 3D were 96.8, 30.4, 14.1 and 7.60, respectively. That of Co(II) in Fig. 3A, 3B, 3C and 3D were 83.3, 22.8, 13.2 and 7.40, respectively. | ||
Evidently, the experiments both in Fig. 2 and Fig. 3 demonstrated that (1) the two metal ions Co(II) and Cu(II) were chemically separated as the pink [Co-EDTA]2− and blue [Cu-EDTA]2−, respectively, (2) the three boundaries, viz., MCB, MSB and CB, were formed; and (3) the four zones of EDTA, [Co-EDTA]2−, [Cu-EDTA]2− and metal ions were present in the whole system.
3.3 Mechanism of novel separation mode and its demonstration
Fig. 1C displayed the mechanism of this kind of continuous separation of metal ions Co(II) and Cu(II). As shown in Fig. 1C, there was a MSB created between the two characteristic color zones, viz., the pink zone [Co-EDTA]2− and the blue zone [Cu-EDTA]2−, and there was an ion substitution reaction within the MSB.62 When the two ions of Co(II) and Cu(II) moved from phase α into phase α′ and further towards the cathode under the electric field, the Co(II) could more freely pass through the MSB into phase β′ than Cu(II) because of the lower complexation constant with EDTA. As a result, there was an ion substitution reaction within the MSB,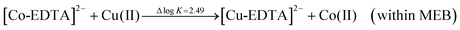 | (II) |
Due to the substitution reaction, Cu(II) was displaced as the blue complex [Cu-EDTA]2−. The copper complex electro-migrated towards the anode in the applied electric field owing to its divalent negative charges. The electro-migration of the complex resulted in the formation of copper CB and characteristic blue zone of [Cu-EDTA]2−. [Cu-EDTA]2− existed at much higher concentration than Co(II) complex because the stable constant of [Cu-EDTA]2− was evidently greater than that of [Co-EDTA]2−. Hence one could observe the concentrated [Cu-EDTA]2− zone in Fig. 2.
The substitution reaction also lead to the release of free Co(II) from its pink complex of [Co-EDTA]2−. The released Co(II), together with the free Co(II) migrating from phase α, migrated towards the cathode. When the Co(II) met the negatively-charged EDTA within the MCB, a chelation reaction took place between the Co(II) and EDTA. The reaction led to the existence of MCB between EDTA and Co(II), and the production of pink complex [Co-EDTA]2−. The pink complex moved towards the anode due to its negative charges. The movement of the complex created phase β′ characterized by its pink colour. When reaching the MSB, the pink complex joined a new circle of ion substitution reaction with the Cu(II) coming from phase α′.
In accordance with the separation mode of two complexes induced by the MSB in Fig. 1, we have the following four predictions.
First, the total concentration of cobalt in phase α′ ought to be much lower than that of copper in the same phase; conversely, the total concentration of cobalt in phase β′ should be much higher than that of copper in phase β′.
Second, the concentration of the cobalt complex in phase α′ is much less than that in phase β′ while the concentration of the copper complex in phase α′ is much greater than that in phase β′.
Third, the concentration of free cobalt ion in phase α′ is evidently less than that in phase β′, because the concentration of free cobalt ion in phase β′ comprises the two parts, one is the free cobalt ion coming from that in phase α′ and the other is that released from the complex of [Co-EDTA]2− within the MSB.
Fourth, the concentration of free copper ion in phase α′ is significantly greater than that in phase β′, since the ion substitution reaction in the MSB greatly transforms the free copper ion coming from phase α and α′ as the complex of [Cu-EDTA]2−, and the substitution reaction prevents most of free copper ion from penetrating into phase β′.
To demonstrate the novel mechanism of separation of the two EDTA complexes in Fig. 1, and support the four predictions above, systemic quantitative investigations were carried out on the free and bound (viz., complex) metal ions in phase α, α′, β′ and β. The total metal ion concentrations (including the free and bound metal ions) in each of the four phases were detected by ICP-AES. As shown in Table 2, the total concentration of cobalt in phase β′ was 21.9 mM while that of copper in phase β′ was 1.87 mM. The concentrations of total cobalt and copper ions in phase α′ are 4.0 and 36.8 mM, respectively. Evidently, the results in Table 2 demonstrated the validity of the first prediction given above.
| Phase | Total coppera/mM | [Cu-EDTA]2− | Cu(II)/mM | Total cobalta/mM | [Co-EDTA]2− | Co(II)/mM | ||
|---|---|---|---|---|---|---|---|---|
| concentration/mM | fraction (%) | concentration/mM | fraction (%) | |||||
| a The concentrations deal with both the free metal ion and bound metal ion. b The experimental conditions were the same as those of Fig. 5E. | ||||||||
| α | 4.0 | — | — | 4.0 | 4.0 | — | — | 4.0 |
| α′ | 36.8 | 32.4 | 92.8 | 4.37 | 4.0 | 2.52 | 7.20 | 1.49 |
| β′ | 1.87 | 0.525 | 5.70 | 1.34 | 21.9 | 18.7 | 94.3 | 3.25 |
| β | 0 | — | — | — | 0 | — | — | — |
To detect the concentrations of bound metal ions in phase α′ and β′, we conducted the quantitative analyses of bound metal concentrations by UV-vis spectrophotometry at multiple wavelengths. The original amounts in the gel were obtained according to the procedure shown in the ESI†. The concentrations of bound metal ions in phase α′ and β′ as well as the molar concentration fractions were also shown in Table 2. It was clearly shown in Table 2 that (1) the concentrations of bound copper ion in phase α′ and β′ were respectively 32.4 and 0.525 mM, and the relevant molar concentration fractions were 92.8% and 5.70%, respectively; (2) the concentration of bound cobalt ion in phase α′ and β′ were respectively 2.52 and 18.7 mM and the relative fractions were respectively 7.20% and 94.3%. Obviously, the results in Table 2 validated the second prediction.
With the concentrations of total metal ions and the bound metal ions, we finally computed these concentrations of free copper and cobalt ions in phase α′ and β′. Table 2 further gave the computation results on the free metal ion concentrations. The results revealed that (1) the concentration of free cobalt ion in phase α′ (= 1.49 mM) was lower than that in phase β′ (= 3.25 mM) evidently; and (2) the concentration of free copper ion in phase α′ (= 4.37 mM) was highly greater than that in phase β′ (= 1.34 mM). The results verified the validity of the third and fourth predictions obviously.
Therefore, the quantitative detection in Table 2 demonstrated the validity of novel separation mode in Fig. 1, especially the four predictions based on the mode of Panel C of Fig. 1. Ideally, there should be no free and bound copper ions to be detected in phase β′ if a complete ion substitution reaction occurred in the MSB. However, a small amount of free and bound copper ions were found in phase β′. In addition, the concentration of bound cobalt ion in phase α′ was also higher than the expected.
The imperfect results were possibly caused by the low kinetic constants63 that slowed down the ion substitution reaction within the MSB. Normally, in the interval 3 < pH < 6, the substitution reactions of the metal ions of the complexes occurred predominantly by hydrogen ion-catalyzed dissociation of the complexes, while direct attack of the exchanging metal ions played a relatively minor role.64 Meanwhile, in the reactions of transition-metal EDTA complexes, such as [Cu-EDTA]2− and [Co-EDTA]2−, a substantially higher contribution was made to the substitution by direct attack of the exchanging metal ions.64 In general, the pseudo-first-order rate constants of substitution reaction increased with increasing hydrogen ion concentration and with the rise of the concentration of the exchanging metal. It was revealed that the pseudo-first-order rate constant of the relevant substitution reaction was in the range of 10−4∼10−2 s−1,63,64 not a much high value. Hence, it was theoretically analyzed that the MSB was wide, while the MCB was sharp. The theoretic analyses were verified by the experiments in Fig. 3C, in which the oblique MSB implied a wide boundary and the nearly vertical MCB, together with CB, showed a sharp boundary. Hence, little of [Co-EDTA]2− in phase β′ might deeply move into phase α′ due to slow ion substitution reaction. Consequently, little of bound cobalt ion was detected in phase α′. At the same time, little of free copper ion in phase α′ could penetrate into phase β′ through the MSB, and the free copper ion in phase β′ was partially transformed as the complex of [Cu-EDTA]2−. The partial transformation led to the co-existence of both free and bound copper ions in phase β′.
Due to the presence of more free cobalt ion in phase β′ compared with the free copper in the same phase, the dominating chelation reaction within the MCB in Fig. 1C took place between Co(II) and EDTA, and subordinate chelation reaction occurred between free copper ion and EDTA in the MCB. Namely, there was the following reaction within the MCB in Fig. 1C
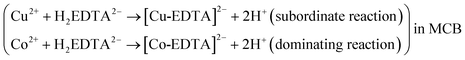 | (III) |
Reaction (III) in Fig. 1C was clearly different from Reaction (I) in Fig. 1A.
The separation of two complexes in Fig. 1C looked like a classic ITP.11,12 However, Fig. 2–4A in fact indicated a novel kind of ITP relied on the physico-chemical mode of MRB, but not the classic ITP that was based on the mode of MBS.11–24 To demonstrate the novel ITP, we further conducted the experiments on the novel ITP separation of two complexes. Fig. 4B showed the relevant experiments on the novel ITP. In Fig. 4B, the 1.5 cm Co(II)-gel zone was previously prepared between the EDTA-gel and the Cu(II)-gel, and the EDTA and Cu(II) were continuously feed from the cathodic and anodic vials, respectively. After the use of electric field, one could observe a unclear reaction zones of Co(II) and Cu(II) complexes initially. However, after nine minutes of electric field, two clear zones of pink and blue complexes were observed, and the two boundaries (viz., the MCB and MSB) beside the pink zone of Co(II) complex moved at the same velocities while the two of MSB and CB besides the blue zone of Cu(II) complex migrated with different velocities. The experiments of Fig. 4B vividly showed the novel ITP migration of MCB and MSB.
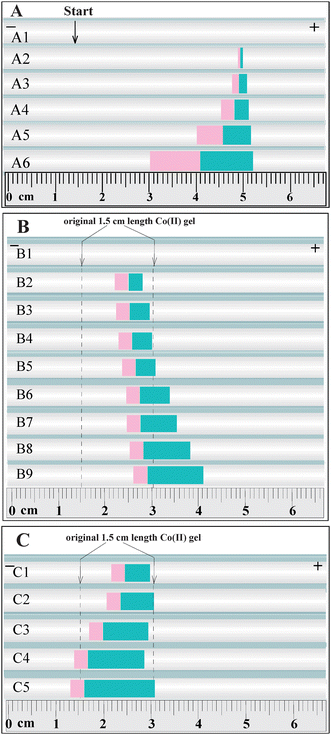 | ||
| Fig. 4 The movements of MCB, MSB and CB in the gel-filled tube experiments. (A) The boundary movements and controllable FE at the 15th minute in MCB system initially formed between phase β with 30 mM Na2EDTA + 5.4 mM KCl and phase α of (1) blank contrast, (2) 0.5 mM Cu(II) + 0.5 mM Co(II) + 97 mM KCl, (3) 1.0 mM Cu(II) + 1.0 mM Co(II) + 94 mM KCl, (4) 2.0 mM Cu(II) + 2.0 mM Co(II) + 88 mM KCl, (5) 4.0 mM Cu(II) + 4.0 mM Co(II) + 76 mM KCl, (6) 8.0 mM Cu(II) + 8.0 mM Co(II) + 52 mM KCl; (B) Novel ITP movement of MCB and MSB in the system initially formed with the right gel having 15 mM Cu(II), the 1.5 cm middle gel holding 7.0 mM Co(II) and the left gel containing 30 mM Na2EDTA at (1) 0, (2) 9, (3) 12, (4) 15, (5) 18, (6) 21, (7) 24, (8) 27 and (9) 30 min running times. The arrows indicate the original 1.5 cm Co(II) gel. Notice: the complex zones cannot be distinctly observed by the camera and naked eye during 0–9 min of using electric field; (C) The bidirectional ITP movements of MCB and MSB at the 18th minute in the system initially formed with the left gel having 30 mM Na2EDTA, the 1.5 cm middle gel holding 7.0 mM Co(II), and the right gel having (1) 15, (2) 20, (3) 25, (4) 30, and (5) 33.3 mM Cu(II). The conditions and the significances of symbols are the same as those in Fig. 2. | ||
However, the novel ITP in Fig. 4B was evidently different from the classic ITP. At first, some new complexes, which were not presented in the initial system of MCB, were produced due to the electromigration reaction between metal ions and EDTA during the separation of novel ITP; while no such complexes were yielded during a classic ITP.11–24 Secondly, the mode of novel ITP was based on the physico-chemical mechanism of MRB, while the one of classic ITP was relied on that of MBS (see the materials only used for the review). Thirdly, the concepts of leading and terminating ions developed from the classic MBS-based ITP could well explain the unidirectional boundary movement in a classic ITP,11–24 but could not illuminate the bidirectional movement of reaction boundary in a MCB system. The unidirectional movement of displacement boundary occurred in a classic ITP.11–24 For example, the boundary movement in the ITP system of LiCl (+, γ) || NaCl (β) || KCl (-,α) was always towards the cathode, the leading (K+) and terminating (Li+) ions could well illuminate the relevant boundary motion.11,12 However, the similar MCB system had bidirectional motion, it could move towards the anode (see Panel 1 and 2 in Fig. 4C), or the cathode (see Fig. 4C-4 and 4C-5), or almost stationary (see Panel 3 of Fig. 4C) under different reaction ion concentrations.
The separation mechanism in Fig. 1C was also different from other classic separative modes, such as CZE, IEF and MEKC etc. Because the former was based on the ion substitution reaction (II) according to metal ion stability constants with EDTA within a MSB. However, in a run of CZE analytes were separated in line with their mobilities. It was detected with CZE and eqn (1) that the mobility of Co(II) complex was 3.94 × 10−4 cm2 V−1 s−1, and the one of Cu(II) complex was 4.13 × 10−4 cm2 V−1 s−1. The two close mobilities indicated the difficulty to separate the two complexes by a zone electrophoresis. However, one could chemically separated Co(II) and Cu(II) as the two complexes with the manner of shoulder-to-shoulder. In an IEF, zwitterionic analytes were focused within a pH gradient in accordance with their pIs. And in a MEKC, neutral analytes were separated in line with their distribution coefficients between micellar and aqueous phases. Furthermore, the separation of metal ions in Fig. 1C was also different from that in precipitate boundary in which the metal ions were precipitated in different precipitate zones in accordance with their solubility products.
As a novel ITP, the limitation of the developed approach for the separation of metal ions in complex matrices was that the metal ions were chemically separated as shoulder-to-shoulder zones of complexes and could not be completely separated as a series of absolute zone peaks as obtained in CZE. The second limitation was that the concentrations of metal complexes could not be achieved from peak heights or areas, as done in CZE or chromatography.
3.4 Controllable focusing of metal ions
The condensation efficiency of a MCB has been described in Ref. 7 and 8. Interestingly, there was also a continuous controllable focusing to two metal ions of Cu(II) and Co(II) in the MCB investigated herein. Fig. 2 evidently unveiled that the widths of stacking zones in Panel B were much sharper than those in Panel A when the concentration of metal ions in phase α was decreased. Fig. 4A further manifested the observation in Fig. 2. In Fig. 4A, the running times were all set at 15 min, but the concentrations of metal ions in Panel B, C, D, E and F were set at 0.5, 1.0, 2.0, 4.0, 8.0 mM, respectively. The comparisons in Fig. 4A indicated that: (1) the widths of coloured zones became sharper as the concentrations of metal ions in phase α decreased gradually; and (2) when the concentration of metal ions was set at 0.5 mM, the pink and blue zones was turned as a sharp line-like zone.Fig. 3 manifests the similar results in Fig. 4A. In Fig. 3A–3D, the concentrations of Cu(II)/Co(II) were set at 0.1/0.1, 0.5/0.5, 1.0/1.0, 2.0/2.0 mM, respectively. As could be seen from Fig. 3, the left vertical line, which represented CB, appeared at almost the same time in all panels, while the migration times of MCBs in Fig. 3A, 3B,3C and 3D were 6.5, 7.0, 7.4 and 10 min, respectively. The data in Fig. 3 indicated that as the concentrations of metal ions decreased, the focusing became better.
To quantitatively study the focusing efficiency (FE), standard calibration curves of the complex concentration in CE were prepared to determine the concentrations of the complexes in CE during the runs of two metal ions systems in Fig. 3. The FE value was directly calculated with
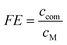 | (2) |
4. Conclusions
From above experimental results and discussions, we could achieve the following conclusions. At first, there were three boundaries, viz., MCB between EDTA and Co(II), MSB between [Co-EDTA]2− and Cu(II), and CB between [Cu-EDTA]2− and the two metal ions, and the three boundaries isolated the whole system into four zones or phases, (viz., EDTA in phase β, [Co-EDTA]2− in phase β′, [Cu-EDTA]2− in phase α′ and two metal ions in phase α). Secondly, a novel ITP separation mode of metal ions was induced by the ionic substitution in accordance with their stability constants within the MSB, rather than the classic ITP based on the physico-chemical mode of MBS. Lastly, a controllable focusing of metal ions could be simultaneously achieved in the given MCB system. These results obtained herein had evident significance for the development of the MCB-based ITP separation of metal ions in environmental and biological matrices, and the further studies on MRB.Acknowledgements
The project was supplied by the NSFC (Approved No. 20675051, 20805031 and 20821005), the national “863” Scientific-technological Key Program (Approved No. 2007AA10Z401), the National Basic Research Program of China (973 Program, Approved No. 2009CB118906), the Shanghai Leading Academic Discipline Project (Approved No. B203), the Instrumental Analysis Center of SJTU and the University.References
- J. Deman and W. Rigole, J. Phys. Chem., 1970, 74, 1122–1126 CrossRef CAS.
- J. Deman, Anal. Chem., 1970, 42, 321–324 CrossRef CAS.
- J. Deman, Anal. Chem., 1970, 42, 1699–1705 CrossRef CAS.
- J. Deman, Anal. Chem., 1970, 42, 983–989 CrossRef CAS.
- C. X. Cao, L. Y. Fan and W. Zhang, Analyst, 2008, 133, 1139–1157 RSC.
- C. X. Cao, J. Chromatogr., A, 1998, 813, 153–172 CrossRef CAS.
- L. Y. Fan, C. J. Li, W. Zhang, P. Zhou, C. X. Cao and Z. X. Deng, Electrophoresis, 2008, 29, 3989–3998 CrossRef CAS.
- J. Jin, J. Shao, S. Li, W. Zhang, L. Y. Fan and C. X. Cao, J. Chromatogr., A, 2009, 1216, 4913–4922 CrossRef CAS.
- J. P. Quirino, J. B. Kim and S. Terabe, J. Chromatogr., A, 2002, 965, 357–373 CrossRef CAS.
- K. Isoo and S. Terabe, Anal. Chem., 2003, 75, 6789–6798 CrossRef CAS.
- Electrophoresis: Theory, Methods and Applications, ed. M. Bier, Acadmic Press, New York, 1959, pp. 1–145 Search PubMed.
- Isotachophoresis: Theory, Instrumentation and Applications, ed. F. M. Everaerts, J. L. Beckers, T. P. E. M. Verheggen, Elsevier Scientific Publishing Co., Amsterdam, 1976, pp. 1–61 Search PubMed.
- D. A. MacInnes and L. G. Longsworth, Chem. Rev., 1932, 11, 171–230 CrossRef.
- V. P. Dole, J. Am. Chem. Soc., 1945, 67, 1119–1126 CrossRef CAS.
- L. G. Longsworth, J. Am. Chem. Soc., 1945, 67, 1109–1119 CrossRef CAS.
- H. Svensson, Acta Chem. Scand., 1948, 2, 841–855 CrossRef CAS.
- R. A. Alberty, J. Am. Chem. Soc., 1950, 72, 2361–2367 CrossRef CAS.
- J. C. Nichol, J. Am. Chem. Soc., 1950, 72, 2367–2374 CrossRef CAS.
- E. B. Dismukes and R. A. Alberty, J. Am. Chem. Soc., 1954, 76, 191–197 CrossRef CAS.
- J. C. Nichol, E. B. Dismukes and R. A. Alberty, J. Am. Chem. Soc., 1958, 80, 2610–2615 CrossRef CAS.
- T. M. Jovin, Biochemistry, 1973, 12, 871–879 CrossRef CAS.
- T. M. Jovin, Biochemistry, 1973, 12, 879–890 CrossRef CAS.
- T. M. Jovin, Biochemistry, 1973, 12, 890–898 CrossRef CAS.
- L. M. Hjelmeland and A. Chrambach, Electrophoresis, 1983, 4, 20–26 CAS.
- G. R. Finlayson and A. Chrambach, Anal. Biochem., 1971, 40, 292–311 CrossRef CAS.
- N. Y. Nguyen and A. Chrambach, Anal. Biochem., 1976, 74, 145–153.
- A. Murel, I. Kirjanen and O. Kirret, J. Chromatogr., A, 1979, 174, 1–11 CrossRef CAS.
- R. Hagedorn and G. Fuhr, Electrophoresis, 1990, 11, 281–289 CrossRef.
- G. Baumann, A. Chrambach, in Progress in isoelectric focusing and isotachophoresis, ed. P. G. Righetti, North-Holland Publishing Co., Amsterdam, 1975, pp. 13–23 Search PubMed.
- A. Chrambach, P. Doerr, G. D. Finlayson, L. E. M. Miles, R. Sherins and D. Rodbard, Ann. N.Y. Acad. Sci., 1974, 209, 44–64.
- S. Hjertén and M. D. Zhu, J. Chromatogr., 1985, 346, 265–270 CrossRef CAS.
- S. Hjertén, K. Elenbring, F. Kilar and J. L. Liao, J. Chromatogr., 1987, 403, 47–61 CrossRef CAS.
- F. Kilar and S. Hjertén, Electrophoresis, 1989, 10, 23–29 CAS.
- J. Wu and J. Pawliszyn, Electrophoresis, 1993, 14, 469–474 CAS.
- Y. J. Xu, S. Li, W. Zhang, L. Y. Fan, J. Shao and C. X. Cao, J. Sep. Sci., 2009, 32, 585–596 CrossRef CAS.
- J. Pospichal, M. Deml and P. Bocek, J. Chromatogr., 1993, 638, 179–186 CrossRef CAS.
- C. Guillo, J. M. Karlinsey and J. P. Landers, Lab on a Chip, 2007, 7, 112–118 RSC.
- X. Z. Wu, L. H. Zhang and K. Onoda, Electrophoresis, 2005, 26, 563–570 CrossRef CAS.
- C. A. Nesbitt, K. Jurcic and K. K. C. Yeung, Electrophoresis, 2007, 29, 466–474.
- C. X. Cao, W. Zhang, W. H. Qin, S. Li and W. Liu, Anal. Chem., 2005, 77, 955–963 CrossRef CAS.
- M. C. Breadmore, R. A. Mosher and W. Thormann, Anal. Chem., 2006, 78, 538–546 CrossRef CAS.
- M. C. Breadmore, Electrophoresis, 2007, 28, 254–281 CrossRef CAS.
- W. Zhang, J. Jin, L. Y. Fan, S. Li, J. Shao and C. X. Cao, J. Sep. Sci., 2009, 32, 2123–2131 CrossRef CAS.
- G. Rossella, P. Aldo, S. Daniela, P. P. Jennifer, B. Federica, L. Eloisa and T. Franco, Electrophoresis, 2008, 29, 4078–4087 CrossRef CAS.
- M. Pereira, E. P. C. Lai and B. Hollebone, Electrophoresis, 2007, 28, 2874–2881 CrossRef CAS.
- J. P. Quirino, S. Terabe and P. Bocek, Anal. Chem., 2000, 72, 1934–1940 CrossRef CAS.
- A. Macià, F. Vorrull, M. Calull and C. Aguilar, Electrophoresis, 2005, 26, 970–979 CrossRef CAS.
- Y. N. Ni and Z. H. Peng, Anal. Chim. Acta, 1995, 304, 217–222 CrossRef CAS.
- M. T. Vasconcelos and C. A. R. Gomes, J. Chromatogr., A, 1995, 696, 227–234 CrossRef CAS.
- D. Fangueiro, A. Bermond and E. Santos, Anal. Chim. Acta, 2002, 459, 245–256 CrossRef CAS.
- E. Norkus, A. Vaskelis and I. Zakaite, Talanta, 1996, 43, 465–470 CrossRef CAS.
- O. Gyliene, J. Aikaite and O. Nivinskiene, J. Hazard. Mater., 2004, B116, 119–124 CrossRef.
- S. Motomizu, M. Oshima, S. Matsuda, Y. Obata and H. Tanaka, Anal. Sci., 1992, 8, 619–625 CAS.
- A. R. Timerbaev, Electrophoresis, 2002, 23, 3884–3906 CrossRef CAS.
- C. Johns, M. Macka and P. R. Haddad, Electrophoresis, 2003, 24, 2150–2167 CrossRef CAS.
- H. Okamoto, A. R. Timerbaev and T. Hirokawa, J. Sep. Sci., 2005, 28, 522–528 CrossRef CAS.
- T. Takayanagi and S. Motomizo, Anal. Sci., 2006, 22, 1241–1244 CrossRef CAS.
- T. Hirokawa, H. Okamoto, Y. Gosyo, T. Tsuda and A. R. Timerbaev, Anal. Chim. Acta, 2007, 581, 83–88 CrossRef CAS.
- A. R. Timerbaev, J. Sep. Sci., 2008, 31, 2012–2021 CrossRef CAS.
- J. P. Hutchinson, C. J. Evenhuis, C. Johns, A. A. Kazarian, M. C. Breadmore, M. Macka, E. F. Hilder, R. M. Guijt, G. W. Dicinoski and P. R. Haddad, Anal. Chem., 2007, 79, 7005–7013 CrossRef CAS.
- Complexation in Analytical Chemistry: A Guide for the Critical Selection of Analytical Methods Based on Complexation Reactions, ed. A. Ringbom, Interscience, New York, 1963, pp. 1–43 Search PubMed.
- Coordination Chemistry: The Chemistry of Metal Complexes, ed. F. Basolo, R. C. Johnson, W.A. Benjamin, Inc., New York, 1964, pp. 54–62 Search PubMed.
- G. A. Lappin, M. C. M. Laranjeira and R. D. Peacock, Inorg. Chem., 1983, 22, 786–791 CrossRef CAS.
- E. Brucher and G. Laurenczy, Inorg. Chem., 1983, 22, 338–342 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details. See DOI: 10.1039/b912799b |
| This journal is © The Royal Society of Chemistry 2010 |