Shedding light on surfaces—using photons to transform and pattern material surfaces
Ellane J.
Park
,
Gregory T.
Carroll
,
Nicholas J.
Turro
and
Jeffrey T.
Koberstein
*
Department of Chemistry and Chemical Engineering, Columbia University, 500 West 120th St, MC4721, New York, New York 10027. E-mail: jk1191@columbia.edu; Fax: +212 854 3054; Tel: +212 854 3120
First published on 9th October 2008
Abstract
The ultimate goal of surface modification is to quantitatively control surface properties by precise manipulation of surface chemical structure at the molecular level. Advances in the understanding of molecular design principles for soft matter surfaces can be combined with the available arsenal of interesting photochemical reactions to create an exciting paradigm for surface modification: the use of photons to both transform and pattern chemical functionality at soft matter surfaces. The success of the paradigm is predicated on the ability to design and synthesize “photochemical surface delivery vehicles”, complex photoactive molecules that form stable surface monolayers and subsequently deliver photoactive moieties to the surface. Shedding light onto these smart, modified surfaces brings about a wide variety of precise photochemical reactions that are preprogrammed within the surface delivery vehicle. Surface chemical patterns are formed by exposure through a mask. Some photochemical surface transformation can be considered as “green” chemistry since only photons are required as reagents. In this review, we provide a brief tutorial on photochemistry fundamentals to illustrate the nature of possible photochemical surface reactions and discuss the principles of design for photochemical surface delivery vehicles. Applications of the paradigm drawn from a variety of fields emphasize the tremendous potential for photochemical surface transformation and patterning on both hard and soft substrates.
 Ellane J. Park |
 Gregory T. Carroll |
 Nicholas J. Turro |
 Jeffrey T. Koberstein |
1 Introduction
It is not an exaggeration to say that material surfaces have a profound effect on everyday life. Every object has a boundary (i.e., surface) that defines its shape and size and allows us to distinguish that object from its surroundings. In fact, a number of material properties are defined exclusively to describe surfaces: adhesion, friction, gloss, wear and wetting to name but a few. Many interesting characteristics of surfaces have evolved naturally to benefit certain animals and organisms, while others are intentionally applied in some man-made fabrication technology. For example, water striders1 have developed an ability to use surface tension to glide smoothly across water; gecko feet2 have sophisticated surface structures that impart adaptable adhesive properties and allow them to walk up walls and across ceilings; and sharks3 and dolphins4 have evolved skins that naturally reduce drag and resist biofouling.Even cells are reliant on surface properties, as so-called anchorage dependent cells become activated only when cell wall receptors receive specific stimuli by interaction with an appropriate surface.5
The ubiquitous Teflon® coated frying pan provides sufficient testimony to illustrate the tremendous impact of man-made surface treatments.
Given the enormous importance of surfaces, it is not surprising that researchers have gone to great lengths to develop a capability for the design and control of surface properties, both on natural and synthetic materials. The purpose of this review is to describe the remarkable and unique potential associated with the use of light to transform and pattern surfaces. The ultimate goal of photochemical surface transformation is to develop surfaces that can be modified using only photons as reagents. While photochemical surface modification is a well-known technique, in terms of industrial and academic applications, we describe herein a new paradigm for photochemical surface modification that is based in part on the rapidly growing understanding of how to manipulate the chemistry of surfaces at the molecular level. Coupled with the rich and extensive base of knowledge of photochemistry, this molecular design capability offers exciting potential for quantitative modification of surfaces; that is, deliberate control of both the density and nature of surface chemical groups. The new paradigm for the transformation of surfaces involves the synthesis of “smart” molecules that might be termed “photochemical surface delivery vehicles”. These complex molecules are preprogrammed to spontaneously assemble onto hard and soft substrates in a fashion that both anchors the molecule at the surface and delivers a specific photochemical group. Appropriate photochemical groups are designed to transform into a desired surface functionality upon illumination with light.
An outline of this review is presented in Fig. 1. First we review relevant principles and concepts that form the basis of surface modification strategies and organic photochemistry. With this foundation, we discuss sequentially the advantages of using light to transform surfaces, the fundamental molecular design of “photochemical surface delivery vehicles”, and how these vehicles are used to construct preprogrammed photoactive surfaces on both hard and soft substrates.
 | ||
| Fig. 1 An outline of this introduction to photochemical methods of surface modification. | ||
1.1 Surface modification techniques
Basic surface modification strategies rely on covering an object with a coating that is usually organic in nature, or, in other words, a soft matter material. Such applications have become ubiquitous in modern society. Take the automobile for example: several layers of paint are applied to metal surfaces to protect against corrosion and to add color, while waxes are employed, causing water to bead up and drain off the exterior, a process that keeps the surface clean and adds an aesthetically pleasing luster. The challenge of surface modification is to change the surface properties of a material without deleteriously affecting the pre-optimized bulk material properties.There is a myriad of surface modification strategies, each with a particular set of advantages and disadvantages. The method of choice depends not only on the technical requirements of the application, but also on considerations such as cost and environmental impact. The interest in “green” technologies, for example, has led to the replacement of chromate primers for metals,6 the development of aqueous based coatings,7 and the removal of copper and lead from anti-fouling paints.8,9
Methods that have been used to modify surface properties can be roughly divided into the following three categories:
(1) Chemically-based methods modify soft matter surfaces by applying a broad spectrum of known chemical reactions. The reagents and reactions required depend on the chemical nature of the substrate undergoing modification and the desired surface transformation. In certain cases these methods can provide a unique ability to modify surfaces. One such case is the modification of fluorocarbon surfaces that are resistant to many chemical reagents but can be modified with sodium naphthalene etch.10 It may be difficult to control the depth of a chemical treatment, however, and the chemical reagents required can be hazardous and lead to undesirable environmental consequences.11
(2) Plasma and corona discharge processes11,12 offer another effective means to modify surfaces, particularly when an inexpensive and rapid process for increasing the surface tension is desired, for example, when printing onto plastic articles such as polyethylene bags. The disadvantage with plasma treatments is that they can be difficult to control and usually do not afford the possibility to specify the precise nature or surface density of functional groups created. In addition, they are generally not thermodynamically stable, and are prone to reorganization that can undo the desired modification.12,13
(3) A third method that might be referred to as “physical” modification, involves coating the surface with surfactant-like molecules that form monolayer structures at the surface. It is this third method, specifically with preprogrammed photochemical surface delivery vehicles, that forms the basis of the methods described in this review. Before we consider the advantages that this method offers, we must be aware of the possibility that light may cause degradation to a surface via radical formations and may yield byproducts, in the case of photodeprotection.
There are a number of advantages in using light to transform surfaces compared to chemical, plasma and mechanical (e.g., stamping14) based methods for surface modification. First, light directly produces the desired surface transformation in a single step. Secondly, because effort is placed on the molecular design of the preprogrammed photochemical surface delivery vehicle, the fabrication process is straightforward, usually requiring only photons as the reagents for the surface transformation reaction. Thirdly, surface patterning with light has the inherent resolution of photolithography techniques and requires only illumination through masking equipment (which is commercially available). Lastly, in many photochemical reactions, light acts as a clean reagent with little or no environmental impact, thereby advancing the cause of “green” soft matter chemistry and manufacturing.
With this background, we begin the review with a brief tutorial describing the fundamental basis of photochemical surface modification strategies.
1.2 Organic photochemistry
How photochemistry can be used to modify surfaces can be illustrated by addressing the question: what happens when light (i.e., a photon) interacts with a molecule on a surface? First, consider the photochemical paradigm that governs how a substrate, originally derivatized with some functionality (PA), can be photo-modified to introduce a new functionality (P) at the surface as shown in Scheme 1, where PA represents a photoactive moiety that becomes excited to *PA upon irradiation with light and forms an intermediate I, before yielding the final product, P.15 | ||
| Scheme 1 A global paradigm for organic photochemical reactions. | ||
The first crucial step of the global paradigm is the primary photochemical process, in which *PA proceeds to form a reactive intermediate possessing an open valence, e.g., a free radical. The next essential step in the paradigm is the secondary thermal process converting I to P, where intermediates yield photoproducts by elimination, addition or rearrangement reactions. Several classes of these photoreactions have been studied:
Elimination: a catalytic species, such as an acid or base, can be produced on an external photoactive molecule to bring about an elimination reaction such as a photodeprotection or photo-cleavage.
Addition: one example of an addition reaction is photografting via an excited carbonyl group of benzophenone (BP). Upon illumination, a reactive intermediate forms at the carbonyl oxygen on BP, by abstracting a hydrogen atom from a neighboring external compound, a process that also creates a radical on the external compound. The external species can covalently link to the BP by radical–radical combination.
Rearrangements : rearrangements include cis–transisomerization where excitation of the double bond in an initially trans-azo compound, for example, overcomes the rotational energy barrier of the double bond leading to formation of a less stable cis-azo conformer. Sigmatropic15 and electrocyclic15rearrangements are also possible, but are beyond this review’s coverage.
The global paradigm for photochemical reactions (Scheme 1) and examples of primary photochemical processes (Table 1) illustrate that appropriate selection of the photoactive “PA” reactant is essential for highly efficient phototransformation of surfaces. The success of covalent photoimmobilization of proteins to a PA-derivatized surface, for example, is determined by the electronic nature of *PA. In this review we elucidate how considerations regarding the identity of *PA and the nature of the surrounding medium can be used to develop various surface modification strategies that use photons as reagents.
| Pre-light-exposure | Post-light-exposure | References | |
|---|---|---|---|
| PAG + Tert-butyl ester |

|
Carboxylic acid | 16–18 |
| PAG + Tert-butoxycarbonyloxyalcohol (TBOC) |

|
Alcohol | 19 |
| PAG + Tert-butyloxy carbamate (BOC) |
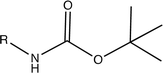
|
Amine | 20,21 |
| Ortho-nitrobenzyl |

|
Carboxylic acid or alcohol | 22,23 |
| Ortho-nitrobenzyl |
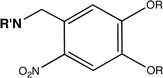
|
Amine | 24,25 |
| Phthalimide |

|
Free radical | 15,26 |
| Benzophenone |

|
Free radical | 15,27–30 |
| Diazirines |

|
Carbene | 28 |
| Aryl azides |

|
Free radical | 28 |
| Azo moiety |
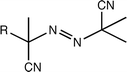
|
Free radical | 31 |
| Azobenzene | Ph—N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N—Ph N—Ph |
Trans–cisisomerization | 32,33 |
| Stilbene | Ph—CHCH—Ph |
Trans–cisisomerization | 34 |
Some examples of PA groups and the functionalities they produce upon illumination are listed in Table 1. In some cases (for example, photodeprotection of tert-butyl protected functional groups), the transformation requires the use of a small photocatalyst molecule, also known as a photoacid generator (PAG). Nitrobenzyl derivatives photo-cleave at the ortho position to produce a new functionality at the surface, varying from alcohols23 to amines.24,25 This photodeprotection feature has been used to fabricate bilayer lipid membranes.22 A few PA groups are capable of grafting macromolecules to a surface by abstracting hydrogen atoms from the C–H groups on organic molecules that come into contact with surface-bound PA groups. Hydrogen abstraction produces radicals that participate in a number of subsequent radical-based processes, some of which can lead to covalent surface grafting. Phthalimide26 and benzophenone27–30 derivatives, for example, graft macromolecules to a substrate by means of this hydrogen abstraction mechanism. Irradiation of diazo and azide compounds results in the loss of N2 and formation of reactive intermediates (e.g.carbenes and nitrenes) that undergo a variety of reactions leading to covalent bond formation, including insertion into σ and π bonds, addition of a nucleophile or electrophile and hydrogen abstraction.15,28Alkyl azo moieties31 can be cleaved in response to light or heat and form a free radical, serving as a catalyst to polymerization. Azobenzene32,33 and stilbene34 conformers can undergo reversible cis–transisomerization after exposure to light and/or heat.
2 Molecular design of photochemical surface delivery vehicles for the transformation of hard and soft material surfaces
The first step in developing a photoactive smart material surface is the design of an appropriate “photochemical surface delivery vehicle”, a molecule that self-assembles at a surface and is preprogrammed to transform into the desired surface functionality upon exposure to light. Preferred molecular designs13 provide precise, quantitative control of the surface density of specific chemical groups by forming surface monolayers. Monolayer coatings of this nature have found widespread usage both to passivate and functionalize hard substrates.35 Because the fundamental bases for organic modification of hard and soft substrates differ, the two subjects will be treated separately.2.1 Modification of hard substrates with photoactive SAMs
Conceptually, methods employed for the general surface functionalization of hard substrates require the synthesis of organic molecules that consist of three constituents, each with an important function: a sticky foot (SF), molecular spacer (SP), and functional group of interest, in this case, a photoactive group (PA). The most widely utilized architecture of organic molecules used to modify hard substrates is referred to as a self-assembled monolayer (SAM).35–37 SAMs are designed to spontaneously assemble onto a substrate surface as an ordered monolayer as depicted in Fig. 2. The organized structure is formed by a directed self-assembly process. The sticky foot provides anchoring to the substrate and orients the molecule to present the photoactive moiety at the surface, while attractive in-plane van der Waals interactions between the spacer groups drive molecular assembly. | ||
| Fig. 2 A schematic illustration of a photoactive self-assembled monolayer with a photoactive group (PA) at the air–polymer interface and sticky foot group (SF) at the polymer–substrate interface. | ||
The SF anchors the molecule to the surface via covalent bonding with or chemisorption onto the substrate. For example, thiol, disulfide and alkyne groups are known to have an affinity for gold substrates.37–39Inorganic phosphates, commonly found in commercial detergents, are well-known for their affinity for metal oxide substrates such as tin-doped indium oxide surfaces.40–43 Some of the more common examples of sticky feet employed in SAM designs are listed in Table 2, along with the types of substrates that they effectively modify.
| Sticky foot | Substrate | References |
|---|---|---|
| Thiol/thiolate/disulfide/thioacetate | Coinage metals (e.g.gold), gallium arsenide | 16,24,38,39,44–46 |
| Alkyne | Gold | 47 |
| Silane | Glass, silicon, indium-tin-oxide (ITO) coated glass | 48–50 |
| Phosphonates/esters | Metal oxides (e.g. ITO-coated glass) | 42,43,51 |
| Carboxylic acid | Ceramics, oxides (e.g.alumina) | 52,53 |
The spacer (SP) is typically a hydrocarbon chain, although spacers containing aromatic groups54 are not uncommon. Attractive in-plane van der Waals interactions among the SP portion of neighboring molecules drive monolayer self-assembly and can in some cases lead to two-dimensional crystallization of the SAM.
The SAMs present preprogrammed head groups at the surface that transform chemically upon exposure to light. The nature of the PA group (see Table 1 for examples) not only controls the surface chemical properties, but also affects surface physical properties such as wettability, adhesion, and friction.
2.2 Modification of hard substrates with photoactive polymers
The methods by which photoactive polymers have been used to produce smart organic surfaces on hard substrates12,55 generally fall into two categories: physisorption of thin polymer films, and the formation of photoactive polymer brushes. If a sticky foot is placed at the end of the polymer chain, the photoactive polymer is grafted to the substrate, forming what is referred to as a polymer brush, as depicted in Fig. 3a. If one or more SF and PA groups are located along the polymer backbone, a copolymer thin film resembling Fig. 3b is formed. The polymer is represented as a random copolymer, but may also be a block copolymer, with one copolymer sequence containing PA and a second copolymer sequence containing SF. The polymer backbone generally serves as the spacer, but may also function as a sticky foot. For example, poly(methyl methacrylate) is itself a Lewis base56 and can act as a sticky foot to substrates, such as glass,11 with Lewis acid character.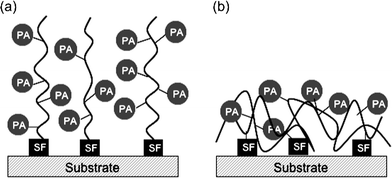 | ||
| Fig. 3 A schematic illustration: (a) photoreactive polymer brush with photoactive groups (PA) along each chain; (b) polyvalent polymer film. | ||
The surface structure manifest in thin copolymer films is determined by the location, type and number of functional groups along the polymer chain.57,58Copolymers containing more random distributions of the PA and SF groups form disordered surface structures, while block copolymers form individual surface layers comprising the two different copolymer sequences.59 The spatial distribution of the PA and SF groups depend on their relative preference to locate at the air interface, the substrate interface or within the polymer film. Preferential surface segregation depends on the architecture of the functional molecule and the relative surface tensions of the functional groups, the substrate and the polymer backbone.57,58,60Copolymer films are subject to reorganization phenomena, as lower energy species are preferentially adsorbed at interfaces with air, while higher surface tension species are favored at a surface in contact with a high energy fluid such as water.13
The stamping14 technique provides a means to pattern molecules onto surfaces. The technique involves first forming a topographical pattern on a silicone substrate, then contact-coating the patterned silicone substrate (i.e., the stamp) with the surface molecule of interest (i.e., the ink), and contacting the “inked” stamp with the substrate of interest, thereby transferring the patterned film of surface molecules onto the substrate to be modified. The stamping technique can create patterned photoactive surfaces by use of photoactive “inks”.
2.3 Modification of soft (i.e. polymeric) substrates
While light has been used extensively to modify the bulk properties of polymers, for example, in chemical amplification photoresist61 technology, there are relatively few examples of the use of photons to modify polymer surfaces.The simplest method for polymer surface modification by light involves direct photolysis of surfaces by exposure to UV in the presence of oxygen.62,63 The method, outlined in Fig. 4(a), involves exposure of oxygen to 185 nm radiation to form ozone, and simultaneous exposure to 254 nm radiation, which converts the ozone to molecular and atomic oxygen. Atomic oxygen reacts rapidly with organics to eventually produce water and carbon dioxide. The UV–ozone method is mild compared to other oxidative surface treatments such as oxygen plasmas and can be applied in ambient environments open to the atmosphere. The method is sufficiently mild that it is capable of producing a thin layer of oxygenated species at a polymer surface simply by brief exposure to appropriate UV radiation. When applied to silicone-based polymers, the method can be used to produce a SiOx (where x ≈ 2) surface layer on the polymer by exposure to UV at room temperature,64–67 something that cannot be achieved by other techniques. Since the technique is based upon exposure to light, the surface chemical transformations achieved can be spatially controlled by using a mask.68 The physical changes can be seen in the atomic force microscopy image shown in Fig. 4(b). The UV–ozone method, while simple and inexpensive to employ, is primarily used to hydrophilize polymer surfaces and generally cannot control the chemical nature of the species produced nor their surface density.
 | ||
| Fig. 4 (a) The UV–ozone conversion method used to modify polymer surfaces using oxygen and light. (b) An atomic force microscopy image of a spin-cast and selectively patterned PDMS–SiOx film.68 | ||
The molecular architectures for preprogrammed, photochemical surface delivery vehicles that are suitable for polymer surface modification have straightforward designs.13 The simplest design is a photoactive diblock copolymer that self-assembles at polymer surfaces as depicted in Fig. 5(a). The first block comprises a polymer that is miscible in the polymeric substrate (i.e., usually the same polymer as the substrate) while the second block must be immiscible with the substrate, surface-active and photochemical in nature. The layered structure results from self-assembly of the copolymer promoted by out-of-plane replusive interactions between two unlike copolymer blocks, and is directed by preferential surface segregation of the low surface tension, surface-active copolymer block to form a surface layer of that species.59 After illumination, the entire surface layer undergoes a phototransformation. The first literature example of surface modification of this nature was the use of a poly(styrene-b-tert-butyl acrylate) diblock copolymer to modify the surface of a polystyrene substrate.17 Upon exposure to UV light in the presence of a photoacid generator, the surface layer of poly(tert-butyl acrylate) was transformed into poly(acrylic acid).
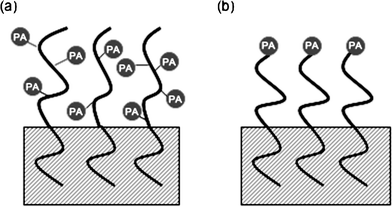 | ||
| Fig. 5 A schematic illustration of photoactive block copolymer brushes used to functionalize the surface of a polymeric substrate. (a) The surface-active copolymer sequence is also photoactive. (b) The surface-active copolymer sequence is terminated with a photoactive functional group. Photoactive functional groups are represented as (PA) in the figure. The anchor block fixes the structure at the surface by interpenetrating with the polymeric substrate. | ||
Quantitative control of the nature and areal density of functional groups at polymer surfaces can be achieved though the use of end-functional copolymers as depicted in Fig. 5(b). Formation of the surface layer directs the terminal functional groups toward the surface. The areal density of functional groups is identical to the areal density of block copolymer chains, a characteristic that can be externally controlled by adjusting the block copolymer molecular weight or the thickness of the block copolymer surface layer, the latter being accomplished by either adjusting the conditions for spin coating or controlling the copolymer's surface adsorption isotherm. The relationship between the surface areal density of functional groups, σ, the layer thickness, t, the molecular weight of the surface-active copolymer block, Mn, and the spin coating conditions (ω is the rotational speed, c is the concentration of the spin coating solution and η is its viscosity) is given by
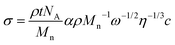
While end-functional block copolymers have been successfully applied to quantitatively functionalize surfaces69,70 and to immobilize DNA71 at controlled surface density, we are not aware of any examples where this method has been used to create preprogrammed surfaces that can be transformed with light.
3 Types of surface photoreactions
Photochemical surface transformation reactions may be classified into several families: functional group elimination, addition, and rearrangements as illustrated in Fig. 6, where the surface to be modified is depicted as a SAM adsorbed onto a hard substrate for ease of presentation. | ||
| Fig. 6 Summary of photo-induced modifications of organic surfaces. (a) Photo-elimination: the most commonly known mechanism is of deprotection where irradiation cleaves the protective group to reveal a new functionality at the surface. (b) Photo-addition: irradiation grafts/couples the surface to an adsorbed overlayer that brings new functional groups to the surface. (c) Photo-rearrangements: irradiation oxidizes or induces rearrangements within the components of the surface to produce new chemical structures. Note: for the purpose of focusing on the photoactive groups that change upon UV irradiation, SF and SP were not specified in the figure because no light-induced change is assumed. | ||
Fig. 6(a) illustrates functional group elimination, which includes conventional deprotection, a common synthetic tool that has been used to photo-generate reactive groups at an interface. For example, upon irradiation with light in the presence of a PAG,72 a tert-butyl ester end-terminated self-assembled monolayer can be deprotected to yield carboxylic acids at the substrate–air interface.16 PAGs are compounds that can produce acids following UV exposure and are commonly used in the development of photoresists in the microelectronics industry.
Fig. 6(b) introduces the concept of photo-addition, more commonly known as surface grafting, where macromolecules are covalently bound to the substrate surface. Photografting can be approached in two ways: the photoactive group can be located on an external species that is immobilized onto a derivatized substrate (referred to as “grafting-to”) or the photoactive group can be located on the substrate (referred to as “grafting-from”73,74). “Grafting-to” includes the reaction of preformed polymers to the surface, while an example of “grafting-from” is surface-initiated polymerization wherein an initiator is immobilized on the substrate from which a polymer is grown.75 The grafting-to technique is hindered by some limitations in graft density and layer thickness as the polymer molecules face a strong kinetic hindrance in attaching to a thoroughly covered surface.76 With the grafting-from technique, polymers of high graft density and high molecular weight can be tethered to the substrate surface, surpassing the limitations of the former approach.77
Most photografting methods find their origins in a common photochemical mechanism, namely hydrogen abstraction via a free hydroxy radical. Ketones, ethylenes, conjugated enones, and azo compounds are some examples of functional groups that result in a photochemical primary process of hydrogen abstraction in the presence of hydrogen-atom-donating substrates (e.g. alcohols, amines, hydrocarbons, etc.), followed by product formation via secondary thermal processes.15
Lastly, Fig. 6(c) demonstrates a broad class of rearrangements, reactions without changes in the atomic composition, but with changes in atomic connectivity and stereochemistry. Rearrangements include a variety of reactions that modify an interface or component of a monolayer without coupling a second molecule to the surface, for example, photoisomerization about the double bond of surface-bound stilbene34 and azobenzene32,33 derivatives.
Surface phototransformation methods applied through a photomask78 (as shown in Fig. 7) have been used to achieve patterned deposition of a variety of biological and synthetic materials including proteins, DNA, cells, and nanoparticles. The feature size of the mask pattern and wavelength of incident light govern the resolution of such patterns. For example, poly(tert-butyl acrylate) block coppolymers were used to fabricate micropatterned polymer surfaces17 by selective photodeprotection. Unmasked regions of the surface were transformed into poly(acrylic acid) by ester hydrolysis catalyzed by use of a PAG.
 | ||
| Fig. 7 Photo-patterning scheme. All of the photo-induced surface modification reactions can be patterned onto self-assembled monolayers or films on hard and soft substrates by using a photomask. | ||
3.1 Photo-elimination for surface modification
While it is not uncommon to find internal photodeprotection reactions, this review will only focus on photodeprotection processes that use an external species, such as PAGs, to trigger the reaction. In the chemical amplification mechanism,61 one photochemical event generates an active species that catalyzes numerous chemical transformations. The active species can be either ionic or radical, but chemical amplification resist systems have commonly used PAGs. Fig. 8 illustrates how the PAG is excited and produces an acid that catalyzes formation of the final product. | ||
| Fig. 8 A schematic illustration of an external species, PAG, being excited and producing acid, thereby catalyzing a primary photochemical process that leads to a product via a secondary thermal process. (*PAG denotes a photo-excited PAG.) | ||
Of the available acid generators, triphenylsulfonium salts have been studied most comprehensively and are known to be the most thermally stable with decomposition starting at ca. 350 °C.61 These onium salts are typically sensitive to UV light and produce strong acids with excellent yields via the presumed mechanism, shown in Fig. 9(a). Upon illumination, the onium salt becomes a cation radical and undergoes an electronic rearrangement to produce H+, which is balanced in charge with the salt anion, X−, in solution.
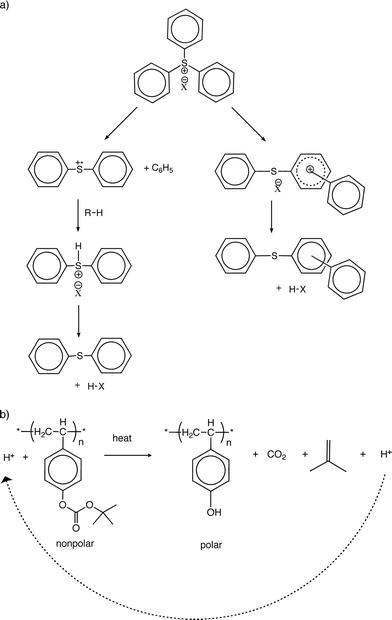 | ||
| Fig. 9 (a) In a deep-ultraviolet (DUV) chemically amplified resist, triphenylsulfonium salt, a photoacid generator (PAG), produces acid upon light-exposure.61 (b) Subsequent thermolysis of the polymer cleaves the tert-butoxycarbonyloxy (TBOC) groups to yield hydroxy groups.79 | ||
Wallraff and Hinsberg79 found that a subsequent thermolysis following irradiation with light in the presence of the onium salt leads to an acid-catalyzed deprotection of tert-butoxycarbonyloxy (TBOC) groups, yielding a hydroxy-derivatized substrate as shown in Fig. 9(b).
Established photoresist chemistry allowed for further advancements in surface patterning by adapting Wallraff and Hinsberg's technique to polymer films on substrates. Past attempts to pattern spin-cast films that are not covalently bound to the substrate often failed as the polymers were not stable in harsher conditions with good solvents.79 Subsequently, several groups have utilized photoactive polymer brushes to produce high-resolution micropatterns. Prucker and Rühe77,80 fabricated some of the first polymer brushes by surface-initiated polymerization, a “grafting-from” technique.
Hawker and coworkers18 developed an approach to control surface properties by photo-patterning stable polymer brushes. After preparing a thin polymer film on the polymer, the photoresist layer, which includes the PAG, was applied.
Selected areas of the substrate are then exposed to UV light and heated to catalyze the photodeprotection of tert-butyl acrylate groups. With this surface-initiated photo-induced polymerization (SIPP) approach, patterned polymer films were generated, containing well-defined hydrophobic and hydrophilic regions as shown by the topographical changes in the optical micrograph of Fig. 10. Phototransformation of the poly(tert-butyl acrylate) (PtBA) brushes to poly(acrylic acid) (PAA) brushes decreased the advancing water contact angle from 92° to 15°. Atomic force microscopy (AFM) showed that the patterned surface was no longer smooth as the light-exposed regions decreased in film thickness from 130 to 80 nm.
 | ||
| Fig. 10 A schematic illustration of patterning continuous polymer films by combining photolithographic techniques with surface-initiated polymerization to convert poly(tert-butyl acrylate) (dark regions on the optical micrograph) to poly(acrylic acid) (light regions). Optical micrograph, without pretreatment, on the right (10 μm squares).18 | ||
Lee et al.16 demonstrated that the terminal tert-butyl groups on SAMs could be deprotected by exposure to 254 nm light in the presence of a PAG. The water contact angle changed from 89° to 28° as the hydrophobic tert-butyl ester groups were deprotected to expose hydrophilic carboxylic acid groups. When exposed to UV light through a mask, the photodeprotection reaction generated surface patterns of carboxylic acids (Fig. 11) that were subsequently used to template the deposition of a variety of different surface ligands. Amine-functionalized polystyrene colloidal particles (3 μm diameter), for example, selectively deposited onto patterns of surface carboxylic acids, mediated by acid–base interactions.
 | ||
| Fig. 11 The irradiation of an azobenzene self-assembled monolayer containing tert-butyl ester end-groups through a photomask results in a micropattern of carboxylic acids at the air–monolayer interface. Amine-coated PS microsphere particles selectively adsorb to the acid portions of the pattern as shown in the optical microscope image.16 | ||
As Fig. 12 illustrates, a poly(styrene-b-tert-butyl acrylate) diblock copolymer17 was used in similar fashion to modify and pattern the surface of a polystyrene substrate by acid-catalyzed photodeprotection of surface adsorbed tert-butyl acrylate groups. Unmasked regions of the copolymer monolayer on the polystyrene substrate undergo transformation to PAA, to form surface patterns of PtBA and PAA block copolymer brushes. These surface patterns17 were used to template the deposition of bovine serum albumin (BSA) on PtBA regions by hydrophobic interactions, polar dyes onto PAA regions by acid–base interactions, and fluorescent streptavidin by a combination of covalent bioconjugation and specific recognition. The steps of the bioconjugation process are shown in Fig. 13, along with a fluorescence micrograph of fluorescein-5 isothiocyanate tagged BSA that was templated onto a chemically micropatterned P(S-b-tBA)-modified PS substrate.
 | ||
| Fig. 12 PtBA in the block copolymer PS-PtBA undergoes a photo-elimination reaction as tert-butyl groups are cleaved off to introduce a hydrophilic functionality to the surface. The optical micrograph (right) shows physical changes in thickness of PtBA and pAA. | ||
 | ||
| Fig. 13 Biotin derivatization via bioconjugation at the carboxylic acid sites of the photo-patterned surface. | ||
3.2 Photo-addition for surface modification
The carbonyl functional group is generally reactive and plays a significant role in this particular class of photoreactions. Fig. 14 demonstrates how carbonyl-containing molecules can undergo hydrogen abstraction and subsequent radical recombination to form a variety of covalently linked species. When applied at surfaces, this mechanism can lead to covalent surface photografting between the substrate and molecules located at the surface. | ||
| Fig. 14 Carbonyls can participate in hydrogen abstraction reactions when irradiated with UV light. A photo-excited ketone can abstract a hydrogen atom from a donor, producing two radicals. Among the reactions the radicals can participate in are three potential recombination products.15 | ||
Phthalimides,26benzophenone81 and binaphthyl ketone82 have been used to prepare surfaces that photograft a wide variety of pre-existing polymersvia photo-addition reactions. Photografting reactions have been used to covalently immobilize numerous polymers in a host of applications including carbohydrate microarrays,83protein patterning,84lipid bilayers,85 and coatings for biomedical devices.86 The main advantage of the photografting technique is that the polymer can be immobilized onto the substrate without prior derivatization. However, intramolecular recombination can be a disadvantage as this process can limit reactivity to the surface.
These photochemical processes have also been applied in pharmaceutical research and development.28 Photoactive ligands enable researchers to recognize and locate drug targets, to gauge the level of interaction between the drug and target, and to locate the binding site on a target. Photoimmobilization of biopolymers (or photoactive ligands) provides information regarding the location and architecture of the ligand-binding site. Past research findings have demonstrated that tetrafluorophenyl azides, trifluoromethyl-phenyl diazirines, and benzophenone (BP) photophores are efficient and reliable in labeling target proteins.87 Of the photophores studied, BP delivers the best chemical function under varying conditions of light and solvent. BPs can be activated at a wavelength longer than 300 nm, thereby avoiding protein-damaging wavelengths; BPs are chemically more stable than aryl azides, diazirines, and diazo esters; and BPs selectively react with C–H bonds that are within a 3.1 Å radius of the carbonyl oxygen even in protic solvent conditions.27,28 These properties allow BPs to covalently bind to and modify macromolecules with enhanced control and efficiency.
Upon absorption of 365 nm UV light, the immobilized BP's carbonyl groups are excited to a reactive intermediate that is a very good hydrogen atom abstractor.15,88 Even hydrogen abstraction from relatively unreactive C–H bonds of a nearby polymer segment occurs followed by the formation of a stable carbon–carbon covalent bond. Although other photochemical processes such as disproportionation, recombination of two ketyl radicals or polymer radicals, and electron transfer are possible, they are not contributing factors to the photoimmobilization of the polymer to the surface.15Fig. 15 presents schematic illustrations of two different approaches to photoimmobilizing ligands to the desired substrate: “grafting-to”, (a), where the photoactive group is located on the molecule to be grafted to the surface, and “grafting-from”, (b), where the photoactive group is located on the substrate.
 | ||
| Fig. 15 Biopolymers are photoimmobilized via two different methods: (a) the protein-photophore is photografted to the surface; and (b) the photophore-derivatized surface selectively covalently binds to and thus immobilizes a macromolecule to the substrate.28 Each reaction is illustrated as a cartoon. | ||
Photografting-to and photografting-from techniques have both been studied for biomedical applications, but grafting-to is the lesser explored, due possibly to its lower binding selectivity and relative difficulty in chemically patterning surfaces. The photografting-from technique was applied using benzophenone as a hydrogen abstractor88 to create ultrathin patterned layers of partially-fluorinated polymers with well-defined features and film thickness.
Similar to benzophenone, the absorption of 300 nm UV light by phthalimide groups activates the carbonyl functionality to form a reactive intermediate that is conducive to a hydrogen abstraction, among other possible photochemical mechanisms [Fig. 16(a)].89 This mechanism was used to surface graft polysaccharides,26 which like nucleic acids and proteins play a significant role in biological processes. Photo-generated carbohydrate microarrays have been used to study viruses, such as HIV.90
 | ||
| Fig. 16 (a) Photografting-from with a phthalimide group: the mechanism for hydrogen abstraction occurring at the phthalimide's carbonyl group.89 (b) Irradiation of a (1,6)polysaccharide-coated phthalimide SAM on a silica substrate using a photomask results in a stabilized pattern of carbohydrates (MW of 2000 kDa) on the SAM. Water vapor preferentially condenses onto hydrophilic patterned polysaccharides as shown in the optical micrograph. The squares are approximately 280 × 280 μm2. | ||
The optical microscope image in Fig. 16(b) shows that water condenses preferentially onto hydrophilic (dark) regions of the substrate that are patterned with polysaccharides, and not onto hydrophobic (light) phthalimide regions that were not exposed to UV radiation. To enhance the interaction between carbohydrates and the phthalimide-modified substrate, mixed phthalimide–amine monolayers (PAM) were used to construct carbohydrate microarrays, which retain their immunological properties as defined by specific antigen–antibody interactions (Fig. 17). These carbohydrate microarrays were successfully applied to perform a high throughput characterization of antigen–antibody interactions, including various surface antigen receptors on Bacillus Anthracis.91 The phthalimide SAMs have been found to be useful for photografting virtually any hydrogen-containing polymers and are somewhat advantageous compared to the equivalent benzophenone SAMs, owing to the better wetting properties and higher surface tensions of phthalimide-based monolayers.
 | ||
| Fig. 17 Irradiation of carbohydrate-coated phthalimide–amine mixed monolayer results in grafted sugar moieties. Fluorescent tagged antibodies are spotted onto the carbohydrate microarray to indicate the presence of sugars.26 | ||
The hydrogen abstraction mechanism was also employed to prevent dewetting92,93 of thin polymer films upon heating to a temperature above their glass transition. Dewetting was prevented94 by crosslinking thin polymer films with a bifunctional photoactive molecule featuring two benzophenone chromophores (bis-BP) capable of hydrogen abstraction as shown in Fig. 18.94Bis-BP is capable of crosslinking hydrogen-containing polymers, in the glassy state, by forming a bridged crosslink between two polymer chains, when the chains do not have sufficient mobility to crosslink by radical termination with each other.
 | ||
| Fig. 18 Proposed mechanism for crosslinking a polystyrene film containing a bis-benzophenone additive by exposure to 300 nm radiation.94 | ||
A combination of a photopolymerization and thermal polymerization was used to create patterns of different polymer brushes, as shown in Fig. 19.31 An azo-initiator was employed due to its ability to cleave in response to both light and heat. The initiator-derivatized surface was first selectively irradiated with UV light (360 nm) through a contact mask to bring about a surface photo-initiated polymerization of poly(methyl methacrylate) and produce patterns of surface-bound PMMA. The reaction mixture was removed from unmodified regions of the substrate and the substrate was coated with a second monomer reaction mixture of hydroxyethyl methacrylate (HEMA) monomer. PHEMA brushes were then grown between areas of PMMA brushes by surface polymerization that was thermally initiated from still-intact azo intiators located in the previously shaded regions. During the second step, calcium ions were used as pseudo-protecting groups for the PMMA by making them temporarily insoluble with the reaction mixture containing HEMA. The end result was a substrate selectively patterned with both PHEMA and PMMA brushes.
 | ||
| Fig. 19 Surface-initiated photopolymerization of the binary-brush system poly(methacrylic acid) (PMAA)-poly(hydroxyethyl methacrylate) (PHEMA).31 | ||
3.3 Rearrangements for surface modification
Although there are several known photochemical rearrangement reactions, cis–transisomerization is by far the most useful. Both azobenzene32,33 and stilbene34 derivatives can be reversibly switched between trans and cis forms using light. When located at the surface, the cis isomer has a higher dipole moment and greater wettability by water.95,96 Irradiation of the derivatized surface at shorter wavelengths in the UV region (∼350 nm) switches the trans form to the cis form, while irradiation at longer visible wavelengths (∼455 nm) can be used to reverse the cis conformation to trans. In some instances, significant changes in contact angle measurements were achieved by incorporating azobenzene moieties along the backbone of polymers.97–99 The azobenzene-derivatized surfaces may undergo a cis–transisomerizationvia a zwitterionic or diradical intermediate.Möller et al.98 investigated a monolayer of polymer containing trifluoromethoxy terminated azobenzene side chains deposited on quartz and silicon substrates and demonstrated that they were able to manipulate the wetting behavior of the film using UV and visible light. Fig. 20 shows that only selected regions switched from trans to cis, as indicated by the illuminated areas of the optical micrograph. The difference in wettability of cis and trans states can be seen by the formation of water microdroplets. This cis–trans pattern was manipulated by selectively isomerizing regions with cis isomers back to the trans state, using visible-light-exposure.
 | ||
| Fig. 20 The azobenzene monolayer was irradiated with UV light in distinct regions using a mask to produce a cis–trans pattern. The cis and trans interfaces differ in wettability as seen in the optical microscope images, which were created by forming water microdroplets on the substrate. The width of the bars is 2 μm and the mesh size is 10 μm.98 | ||
Copolymers containing azobenzene side chains terminated with trifluoromethyl groups were formed on highly oriented pyrolytic graphite by Langmuir–Blodgett (LB) deposition.100 The NN double bond isomerized from trans to cis after illumination with UV light, and was accompanied by a water contact angle shift from 85° to 74° as shown in Fig. 21(a). Photoisomerization back to the trans state was almost completely reversible after exposure to visible light. The LB film in the trans state exposed fluorocarbon-groups leading to low surface energy, lower friction interaction, lower and hydrophobicity as suggested by Fig. 21 (b).
 | ||
| Fig. 21 (a) The contact angle changes for an 11-layer Langmuir–Blodgett film on glass with or without UV irradiation. After several cycles of irradiation, the contact angles gradually decreased. (b) Model of a single-layer Langmuir–Blodgett film of polymer undergoing a structural change upon exposure to UV light in addition to a reversible photoisomerization.100 | ||
Photoisomerization of azo dyes bound to surfaces has been used to alter the alignment of liquid crystal films between two photochromic command states.101 The azobenzene side chains undergo an isomerization from trans to cis, switching the liquid crystal alignment from homeotropic to parallel, respectively. The chirality of a surface can also be switched between left and right-handed forms of helical molecules with the presence of chiroptical photoisomerizable molecules.102,103 Such photoresponsive surface-bound molecules are expected to be critical components for the advancement of artificial molecular machines.104
4 Conclusions
Properly designed surface delivery vehicles can be used to functionalize the surface of both hard and soft substrates with preprogrammed photoactive groups that undergo a variety of photochemical transformation reactions upon exposure to light. Such surface phototransformation reactions, using only photons as reagents, advance the cause of “green” soft matter chemistry and manufacturing and have tremendous potential in surface modification and patterning applications. Photochemical surface transformation methods can be used to create specific surface functional groups, to change surface properties such as friction and surface tension, or to covalently bind a wide variety of organic molecules and biomacromolecules to a surface. Simple exposure to light though a mask allows for spatial patterning of surface functional groups and associated surface properties with pattern resolutions that match those of current photolithography techniques. Shedding light on soft matter surfaces has a bright future.Acknowledgements
This material is based upon work supported by, or in part by, U.S. Army Research Laboratory and the U.S. Army Research Office under contract/grant number DA W911NF-04-1-0282 and in part by the National Science Foundation under grant numbers DMR-0704054, CHE-04-15516 and CHE-07-17518. G.T. Carroll acknowledges an IGERT fellowship. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.References
- D. L. Hu, B. Chan and J. W. M. Bush, Nature, 2003, 424, 663–666 CrossRef CAS.
- K. Autumn, M. Sitti, Y. A. Liang, A. M. Peattie, W. R. Hansen, S. Sponberg, T. W. Kenny, R. Fearing, J. N. Israelachvili and R. J. Full, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12252–12256 CrossRef CAS.
- S. A. Wainwright, F. Vosburgh and J. H. Hebrank, Science, 1978, 202, 747–749 CrossRef.
- S. H. Ridgway and D. A. Carder, IEEE Eng. Med. Biol., 1993, 83–88 CrossRef.
- B. Alberts, D. Bray, J. Lewis, M. Raff, K. Roberts and J. D. Watson, Molecular Biology of Cell, Garland Publishing, Inc., New York and London, 1994 Search PubMed.
- S. M. Cohen, Corrosion, 1995, 51, 71–78 Search PubMed.
- D. L. Schmidt, C. E. Coburn, B. M. DeKoven, G. E. Potter, G. F. Meyers and D. A. Fischer, Nature, 1994, 368, 39–41 CrossRef CAS.
- E. G. Bellinger and B. R. Benham, Environ. Pollut., 1978, 17, 71–81.
- K. Schiff, D. Diehl and A. Valkirs, Mar. Pollut. Bull., 2004, 48, 371–377 CrossRef CAS.
- R. C. Snogren, Handbook of Surface Preparation, Palmerton, NY, 1974 Search PubMed.
- S. Wu, Polymer Interface and Adhesion, Marcel Dekker Inc., New York, 1982 Search PubMed.
- F. Garbassi, M. Morra and E. Occhiello, Polymer Surfaces: From Physics to Technology Updated Edition, John Wiley and Sons, Inc., New York, 1998 Search PubMed.
- J. T. Koberstein, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 2942–2956 CrossRef CAS.
- A. Kumar, H. A. Biebuyck and G. M. Whitesides, Langmuir, 1994, 10, 1498–1511 CrossRef CAS.
- N. J. Turro, Modern Molecular Photochemistry, University Science Books, Sausalito, CA, 1991 Search PubMed.
- K. Lee, F. Pan, G. T. Carroll, N. J. Turro and J. T. Koberstein, Langmuir, 2004, 20, 1812–1818 CrossRef CAS.
- F. Pan, P. Wang, K. Lee, A. Wu, N. J. Turro and J. T. Koberstein, Langmuir, 2005, 21, 3605–3612 CrossRef CAS.
- M. Husemann, M. Morrison, D. Benoit, J. Frommer, C. M. Mate, W. D. Hinsberg, J. L. Hedrick and C. J. Hawker, J. Am. Chem. Soc., 2000, 122, 1844–1845 CrossRef CAS.
- T. Shinoda, T. Nishiwaki and H. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2760 CrossRef CAS.
- J. P. Pellois, X. C. Zhou, O. Srivannavit, T. C. Zhou, E. Gulari and X. L. Gao, Nat. Biotechnol., 2002, 20, 922–926 CrossRef CAS.
- K. L. Christman, R. M. Broyer, Z. P. Tolstyka and H. D. Maynard, J. Mater. Chem., 2007, 17, 2021–2027 RSC.
- X. Han, K. Critchley, L. Zhang, S. N. D. Pradeep, R. J. Bushby and S. D. Evans, Langmuir, 2007, 23, 1354–1358 CrossRef CAS.
- T. Ito, A. Terao, Y. Inao, T. Yamaguchi and N. Mizutani, J. Photopolym. Sci. Technol., 2007, 20, 591–598 CrossRef CAS.
- D. Ryan, B. A. Parviz, V. Linder, V. Semetey, S. K. Sia, J. Su, M. Mrksich and G. M. Whitesides, Langmuir, 2004, 20, 9080–9088 CrossRef CAS.
- K. Critchley, J. P. Jeyadevan, H. Fukushima, M. Ishida, T. Shimoda, R. J. Bushby and S. D. Evans, Langmuir, 2005, 21, 4554–4561 CrossRef CAS.
- G. T. Carroll, D. Wang, N. J. Turro and J. T. Koberstein, Langmuir, 2006, 22, 2899–2905 CrossRef CAS.
- G. Dormán and G. D. Prestwich, Biochemistry, 1994, 33, 5661–5673 CrossRef CAS.
- G. Dormán and G. D. Prestwich, Trends Biotechnol., 2000, 18, 64–77 CrossRef CAS.
- E. Delamarche, G. Sundarababu, H. Biebuyck, B. Michel, C. Gerber, H. Sigrist, H. Wolf, H. Ringsdorf, N. Xanthopoulos and H. J. Mathieu, Langmuir, 1996, 12, 1997–2006 CrossRef CAS.
- O. Prucker, C. A. Naumann, J. Ruehe, W. Knoll and C. W. Frank, J. Am. Chem. Soc., 1999, 121, 8766–8770 CrossRef CAS.
- R. Konradi and J. Rühe, Langmuir, 2006, 22, 8571–8575 CrossRef CAS.
- S.-K. Oh, M. Nakagawa and K. Ichimura, J. Mater. Chem., 2002, 12, 2262–2269 RSC.
- O. M. Tanchak and C. J. Barrett, Macromolecules, 2005, 38, 10566–10570 CrossRef CAS.
- M. O. Wolf and M. A. Fox, Langmuir, 1996, 12, 955–962 CrossRef CAS.
- A. Ulman, An Introduction to Ultrathin Organic Films: From Langmuir-Blodgett to Self-Assembly, Academic Press, Boston, 1991 Search PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS.
- A. Ulman, Characterization of Organic Thin Films, Butterworth-Heinemann Press, Boston, 1995 Search PubMed.
- J. M. Tour, L. Jones II, D. L. Pearson, J. J. S. Lamba, T. P. Burgin, G. M. Whitesides, D. L. Allara, A. N. Parikh and S. V. Atre, J. Am. Chem. Soc., 1995, 117, 9529–9534 CrossRef CAS.
- R. Matsuno, K. Yamamoto, H. Otsuka and A. Takahara, Macromolecules, 2004, 37, 2203–2209 CrossRef CAS.
- M. A. White, J. A. Johnson, J. T. Koberstein and N. J. Turro, J. Am. Chem. Soc., 2006, 128, 11356–11357 CrossRef CAS.
- N. D. Popovich, B. K. Yen and S. Wong, Langmuir, 2003, 19, 1324–1329 CrossRef CAS.
- M. G. Nooney, A. Campbell, T. S. Murrell, X.-F. Lin, L. R. Hossner, C. C. Chusuei and D. W. Goodman, Langmuir, 1998, 14, 2750–2755 CrossRef CAS.
- C. D. Bain, E. B. Troughton, Y. T. Tao, J. Evall, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 1989, 111, 321–335 CrossRef CAS.
- J. S. Park, A. C. Smith and T. R. Lee, Langmuir, 2004, 20, 5829–5836 CrossRef CAS.
- J. S. Park, A. N. Vo, D. Barriet, Y.-S. Shon and T. R. Lee, Langmuir, 2005, 21, 2902–2911 CrossRef CAS.
- S. Zhang, K. L. Chandra and C. B. Gorman, J. Am. Chem. Soc., 2007, 129, 4876–4877 CrossRef CAS.
- K. Uosaki, M. E. Quayum, S. Nihonyanagi and T. Kondo, Langmuir, 2004, 20, 1207–1212 CrossRef CAS.
- J. Areephong, W. R. Browne, N. Katsonis and B. L. Feringa, Chem. Commun., 2006, 37, 3930–3932 Search PubMed.
- E. P. Plueddemann, J. Adhes., 1970, 2, 184–201 CrossRef CAS.
- B. P. Nelson, R. Candal, R. M. Corn and M. A. Anderson, Langmuir, 2000, 16, 6094–6101 CrossRef CAS.
- A. Raman and E. Gawalt, Langmuir, 2007, 23, 2284–2288 CrossRef CAS.
- P. E. Laibinis, J. J. Hickman, M. S. Wrighton and G. M. Whitesides, Science, 1989, 245, 845 CAS.
- E. Sabatani, J. Cohen-Boulakia, M. Breuning and I. Rubenstein, Langmuir, 1993, 9, 2974 CrossRef CAS.
- R. A. L. Jones and R. W. Richards, Polymers at Surfaces and Interfaces, Cambridge University Press, Cambridge, 1999 Search PubMed.
- R. J. Good, M. K. Chaudhury and C. J. V. Oss, in Fundamentals of Adhesion, ed. L.-H. Lee, Plenum Press, New York, 1991 Search PubMed.
- D. N. Theodorou, Macromolecules, 1988, 21, 1411 CrossRef CAS.
- D. N. Theodorou, Macromolecules, 1988, 21, 1422 CrossRef CAS.
- F. S. Bates and G. H. Fredickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS.
- P. A. V. O'Rourke-Muisener, J. T. Koberstein and S. Kumar, Macromolecules, 2003, 36, 771–781 CrossRef CAS.
- H. Ito, Adv. Polym. Sci., 2005, 172, 37–245 CAS.
- B. Ranby and J. F. Rabek, Photodegradation, Photo-oxidation and Photostabilization of Polymers, Wiley, London, 1975 Search PubMed.
- J. F. Rabek, Intnl. Polym. Sci. Technol., 1985, 12, 91 Search PubMed.
- C. Mirley, Langmuir, 1995, 11, 1049–1052 CrossRef CAS.
- M. Ouyang, C. Yuan, R. Muisener, A. Boulares and J. T. Koberstein, Chem. Mater., 2000, 12, 1591–1596 CrossRef CAS.
- M. Ouyang, R. Muisener, A. Boulares and J. T. Koberstein, J. Membr. Sci., 2000, 177, 177–187 CrossRef CAS.
- M. Ouyang, P. P. Klemchuk and J. T. Koberstein, Polym. Degrad. Stab., 2000, 70, 217–228 CrossRef CAS.
- T. S. P. Bobin, R. J. Muisener, J. T. Koberstein and F. Papadamitrakopoulos, Adv. Mater., 2000, 12, 1257 CrossRef.
- J. T. Koberstein, D. E. Duch, W. Hu, T. J. Lenk, R. Bhatia, H. R. Brown, J.-P. Lingelser and Y. Gallot, J. Adhes., 1998, 66, 229–249 CrossRef CAS.
- W. Hu, H. R. Brown, J. T. Koberstein, R. Bhatia, J. P. Lingelser and Y. Gallot, C. R. Chim., 2006, 9, 45–59 CrossRef CAS.
- H. R. Rengifo, Langmuir, 2008 Search PubMed In Press.
- M. Shirai and M. Tsunooka, Prog. Polym. Sci., 1996, 21, 1–45 CrossRef CAS.
- G. J. Fleer, M. A. Cohen-Stuart, J. M. H. M. Scheutjens, T. Cosgrove and B. Vincent, Polymers at Interfaces, Chapman and Hall, London, 1993 Search PubMed.
- K. Bridger and B. Vincent, Eur. Polym. J., 1980, 16, 1017 CrossRef CAS.
- D. J. Dyer, J. Feng, C. Fivelson, R. Paul, R. Schmidt and T. Zhao, in Polymer Brushes: Synthesis, Characterization, Applications, ed. J. Rühe, Wiley, Weinheim, 2004, pp. 129–150 Search PubMed.
- R. Zajac and A. Chakrabarti, Phys. Rev. E, 1995, 52, 6536 Search PubMed.
- O. Prucker and J. Rühe, Macromolecules, 1998, 31, 602–613 CrossRef CAS.
- M. J. Madou, Fundamentals of Microfabrication, CRC Press, New York, 2002 Search PubMed.
- G. M. Wallraff and W. D. Hinsberg, Chem. Rev., 1999, 99, 1801 CrossRef CAS.
- O. Prucker and J. Rühe, Macromolecules, 1998, 31, 592–601 CrossRef.
- R. Toomey, D. Freidank and J. Ruehe, Macromolecules, 2004, 37, 882–887 CrossRef CAS.
- K. Dayananda, R. Dhamodharn, K. Vijayakumaran and T. Rajamannar, J. Polym. Sci. Part A: Polym. Chem., 2004, 42, 5413–5423 CrossRef CAS.
- S. Angeloni, J. L. Ridet, N. Kusy, H. Gao, F. Crevoisier, S. Guinchard, S. Kochhar, H. Sigrist and N. Sprenger, Glycobiology, 2005, 15, 31–41 CAS.
- L. F. Rozsnyai, S. P. A. Fodor, P. G. Schultz and D. R. Benson, Angew. Chem., Int. Ed. Engl., 1992, 104, 801–802 CrossRef CAS.
- W. W. Shen, S. G. Boxer, W. Knoll and C. W. Frank, Biomacromolecules, 2001, 2, 70–79 CrossRef CAS.
- H. Murata, B. J. Chang, O. Prucker, M. Dahm and J. Ruehe, Surf. Sci., 2004, 570, 111–118 CrossRef CAS.
- S. A. Fleming, Tetrahedron, 1995, 51, 12479–12520 CrossRef CAS.
- J. D. Jeyaprakash, S. Samuel and J. Rühe, Langmuir, 2004, 20, 10080–10085 CrossRef CAS.
- U. C. Yoon and P. S. Mariano, Acc. Chem. Res., 2001, 34, 523 CrossRef CAS.
- E. W. Adams, D. M. Ratner, H. R. Bokesch, J. B. McMahon, B. R. O'Keefe and P. H. Seeberger, Chem. Biol., 2004, 11, 875–881 CrossRef CAS.
- D. N. Wang, G. T. Carroll, N. J. Turro, J. T. Koberstein, P. Kovac, R. Saksena, R. Adamo, L. A. Herzenberg, L. A. Herzenberg and L. Steinman, Proteomics, 2007, 7, 180–184 CrossRef CAS.
- G. Reiter, Phys. Rev. Lett., 1992, 68, 75–78 CrossRef CAS.
- G. Reiter and P. G. D. Gennes, Eur. Phys. J. E, 2001, 6, 25–28 CrossRef CAS.
- G. T. Carroll, M. E. Sojka, X. Lei, N. J. Turro and J. T. Koberstein, Langmuir, 2006, 22, 7748–7754 CrossRef CAS.
- S. Wang, Y. Song and L. Jiang, J. Photochem. Photobiol. C: Photochem. Rev., 2007, 8, 18–29 CrossRef CAS.
- N. Delorme, J. F. Bardeau, A. Bulou and F. Poncin-Epaillard, Langmuir, 2005, 21, 12278–12282 CrossRef CAS.
- H. S. Lim, J. T. Han, D. Kwak, M. Jin and K. Cho, J. Am. Chem. Soc., 2006, 128, 14458–14459 CrossRef CAS.
- G. Möller, M. Harke, H. Motschmann and D. Prescher, Langmuir, 1998, 14, 4955–4957 CrossRef.
- T. Seki and K. Ichimura, Thin Solid Films, 1989, 179.
- C. L. Feng, Y. J. Zhang, J. Jin, Y. L. Song, L. Y. Xie, G. R. Qu, L. Jiang and D. B. Zhu, Langmuir, 2001, 17, 4593–4597 CrossRef CAS.
- H. Knobloch, H. Orendi, M. Buchel, T. Seki, S. Ito and W. Knoll, J. Appl. Phys., 1995, 77, 481–487 CrossRef CAS.
- R. A. van Delden, M. K. J. ter Wiel, M. M. Pollard, J. Vicario, N. Koumura and B. L. Feringa, Nature, 2005, 437, 1337–1340 CrossRef CAS.
- M. M. Pollard, M. Lubomska, P. Rudolf and B. L. Feringa, Angew. Chem., Int. Ed., 2007, 46, 1278–1280 CrossRef CAS.
- V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |