Synthesis of sulfone-based nucleotide isosteres: identification of CMP-sialic acid synthetase inhibitors†
Jessica H.
Wong‡
,
Urvashi
Sahni
,
Yanhong
Li
,
Xi
Chen
and
Jacquelyn
Gervay-Hague
*
Department of Chemistry, University of California, Davis, One Shields Avenue, Davis, CA, U.S.A.. E-mail: gervay@chem.ucdavis.edu.; Fax: +1 530 752-8995; Tel: +1 530 754 9577
First published on 17th November 2008
Abstract
A modular replacement approach to the synthesis of sulfo-nucleotide analogs prepared from condensation of nucleoside aldehydes with bis phosphonate Horner-Wadsworth-Emmons reagents is disclosed. These analogs were shown to be inhibitors of Neisseria meningitidis CSS (NmCSS), which is a key enzyme in the biosynthesis of the capsular polysaccharides required for bacterial infection.
Neisseria meningitidis B is a leading cause of bacterial meningitis for which there is no available vaccine. These bacteria express a linear homopolymer of α-2,8-linked sialic acid (1) as part of their capsular polysaccharides. In humans, polysialic acid (1) expression occurs early in development and is down regulated by about two years of age suggesting that its functional role in bacteria may be related to host mimicry.1–3 The bacteria may employ this polymer in cellular interactions that permit infection. Recently, we have focused our attention on developing methods for the synthesis of substrate-based inhibitors of enzymes involved in the biosynthesis of polysialic acids, which include CMP-sialic acid synthetases (CSS) and polysialyltransferases (PST). CSS is a bisubstrate enzyme that utilizes both sialic acid and cytidine triphosphate (CTP) as substrates in the production of CMP-sialic acid, which in turn is a substrate for PST, (Fig. 1). Activation of sialic acid to CMP-sialic acid is an essential step in the biosynthesis of sialoglyconjugates that is conserved from bacteria to humans: however, eukaryotic CSS are localized in the nuclear compartment whereas the bacterial enzymes are cytoplasmic. This fundamental difference offers the possibility of achieving selective inhibition as a function of nuclear transport. Presumably, inhibitors incapable of permeating the nuclear membrane would not inhibit mammalian enzymes. Development of substrate analogs of CSS enzymes as inhibitors and/or molecular probes represents an important area of investigation for potential drug development, as incorporation of sialic acid into capsular polysaccharides is required for the conversion of N. meningitidis to a serum resistant phenotype.4–6

 | ||
| Fig. 1 Modular replacement strategy for the synthesis of sulfo-nucleotide analogs. | ||
A number of CSS enzymes have been cloned from both bacterial and vertebrate sources. While several of these enzymes accept modified sialic acids as substrates,7–12 less is known about their tolerance to CTP modifications. Various nucleotides including CTP, CDP, UTP and UDP have been shown to be substrates of P. haemolytica A2 CSS,13 whereas vertebrate CSS appears to be highly specific for CTP.14 However, CDP and UTP serve as competitive inhibitors of these enzymes. Nucleotide analogs have been studied for other enzymes such as CTP synthetase,15Plasmodia orotidine monophosphate decarboxylase16 and the human P2Y6receptor.17 The promiscuity of N. meningitidis B CSS (NmCSS) toward CTP analogs has not been studied prior to this report.
In our general design of the nucleotide isosteres, phosphorus is substituted by sulfur, which is oxidized to the sulfone to avert potential stereochemical problems and the oxygen linkage of the phosphate is replaced with carbon. Our interest in this area is fuelled by the expectation that sulfones will serve as neutral, non-hydrolizable isosteres of phosphates. In support of this hypothesis, we recently reported crystal structure data comparing sulfones to phosphates. Geometrically, the sulfone functionality was found to be a good mimic for phosphate. The S-C-S and S-C-P backbones closely resemble the P-O-P backbone. However the S-C and P-C distances are slightly longer than the P-O distance and the S-C-S and S-C-P angles are more acute than the P-O-P angle.18
In targeting CSS, we envisioned implementing a modular replacement approach that would enable access to both nucleotide tri- and di-phosphate analogs employing the Horner-Wadsworth-Emmons reagents 219 and 3.20 Condensation of these activated phosphonates with appropriately protected nucleoside aldehydes would generate sulfo-nucleotide isosteres for biological evaluation. In this report, we demonstrate that 2 and 3 readily undergo condensation with nucleoside aldehydes derived from cytidine and uridine to produce sulfo-nucleotide analogs (e.g.4, 5) with potent inhibitory activity against N. meningitidis B CSS.
Based upon the knowledge that CTP and CDP are known substrates for P. haemolytica A2 CSS,13 we first determined if UTP, CDP and UDP could also serve as substrates for NmCSS. Reaction mixtures (20 μL) in Tris-HCl buffer (200 mM, pH 8.5) containing purified NmCSS recombinant enzyme (0.09 μg),8nucleotide at three different concentrations, N-acetylneuraminic acid (Neu5Ac, 1 mM) and MgCl2 (20 mM) were incubated at 37 °C for 30 min and the reactions were stopped by adding 10% SDS on ice to give a final concentration of 1% SDS. The samples were analyzed using a capillary electrophoresis system equipped with a UV detector. The formation of CMP-Neu5Ac and UMP-Neu5Ac was observed at 200 nm and confirmed by mass spectrometry indicating that CTP, UTP, CDP and UDP were all substrates for NmCSS. CTP was the preferred substrate with 78% CMP-Neu5Ac formation at 1 mM CTP. In contrast, only 5% UMP-Neu5Ac was formed at 1 mM UTP, and both CDP and UDP afforded only 1% C(U)MP-Neu5Ac under similar conditions. Moreover, the conversion of C(U)MP-Neu5Ac formation decreased as the concentration of substrates increased to 10 mM, which suggested that these substrates were also weak inhibitors. In subsequent studies (vide infra), we found that CDP weakly inhibits NmCSS (14%, Table 1).
| Entry | Compound | % Inhibition |
|---|---|---|
| 1 | Sulfo-CTP (4) | 39 ± 2 |
| 2 | Sulfo-CDP (5) | 47 ± 2 |
| 3 | Saturated sulfo-CDP (12) | 55 ± 4 |
| 4 | Sulfo-UTP (19) | 33 ± 5 |
| 5 | Sulfo-UDP (20) | 6 ± 3 |
| 6 | Saturated sulfo-UDP ester (21) | 24 ± 2 |
| 7 | Saturated sulfo-UDP (22) | 35 ± 2 |
| 8 | CDP | 14 ± 2 |
Since both CTP and CDP were shown to be substrates for NmCSS, we targeted the sulfo-CTP (4) and sulfo-CDP (5) substrate analogs for biological evaluation. Following the procedure of Wiemer et al.,21 the 5′-cytidine aldehyde (6) was prepared and coupled with 3 eq. of 2 or 3 providing the (E) alkenes 7 or 8 in 67–78% yields. We found this reaction to be sensitive to base exposure. When 9 eq. of DIEA was employed, the main product was the allyl sulfone where the double bond had migrated out of conjugation with the sulfone.22 This product was not observed in reactions using only 3 eq. of base. Deprotection of the vinyl sulfones 7 and 8 using TMSBr in CH2Cl2 gave the sulfone analogs 4 and 5 in 80–90% yields (Scheme 1). The stereochemistry at C-4′ was confirmed using two-dimensional nuclear Overhauser effect spectroscopy. The data show a cross peak between H4′ and H1′ of the ribofuranose indicating that H4′ is below the plane of the ring.
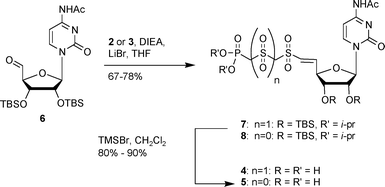 | ||
| Scheme 1 Synthesis of sulfo-CTP and CDP analogs. | ||
With the sulfo-analogs of CTP and CDP in hand, we turned our attention to testing these compounds as inhibitors of NmCSS. In the inhibition assays, reaction mixtures (20 μL) in Tris-HCl buffer (200 mM, pH 8.5) containing purified NmCSS recombinant enzyme (1.25 ng),8 1 mM CTP, 1 mM Neu5Ac and 20 mM MgCl2 were reacted in the presence of 1 mM inhibitor. The mixture was incubated at 37 °C for 15 min and the reaction was stopped by adding 10% SDS on ice to give a final concentration of 1% SDS. The samples were analyzed using capillary electrophoresis equipped with a UV detector. The percentage conversion for the formation of CMP-Neu5Ac was calculated based on a UV response standard curve of CTP and CMP-Neu5Ac. The UV response standard curve was obtained using mixtures of different known ratios of CTP and CMP-Neu5Ac. As shown in Table 1, to our delight, sulfo-CTP and sulfo-CDP showed inhibitory effects with 39% and 47% inhibition, respectively.
We next probed the importance of the vinyl sulfone functionality by preparing the saturated analog 12. Initial attempts at hydrogenation of 8 did not yield the desired compound; instead both the double bond and cytidine base were reduced with concomitant loss of the NH acetyl to afford 9. However, we were able to achieve reduction of the vinyl sulfone after removing the silyl protecting groups and then deprotecting the cytidine base to give compound 10, which readily converted to 11 upon hydrogenation. TMSBr mediated hydrolysis of the phosphonate esters delivered compound 12 (Scheme 2). Gratifyingly, analog 12 also showed moderate inhibition; slightly higher than 5 (55 vs 47%, respectively, Table 1).
 | ||
| Scheme 2 Synthesis of saturated sulfo-CDP. | ||
Encouraged by these results, we next turned our attention to the synthesis of sulfo-UTP (19) and sulfo-UDP (20) analogs (Scheme 3). Synthesis of the uridine analogs commenced with the preparation of alcohol 1323 and its subsequent oxidation to 14. Moffat oxidation of 13 had been previously reported, but the work-up for this reaction required aqueous conditions.23 Our experience with making nucleoside aldehydes indicates that they are susceptible to hydration and that it is best to avoid contact of the aldehyde with water. We find that the Horner-Wadsworth-Emmons reagents (2 and 3) are slow to react with acetals. Therefore, we elected to use Dess-Martin periodinane (DMP) as the oxidant. Subsequent reaction of 14 with 2 or 3 afforded the protected sulfo-uridine analogs 15 and 16, respectively. Deprotection of the p-methoxybenzyl groups with CAN also led to removal of the isopropylidenes to give 17 and 18, and the isopropyl esters were removed by the action of TMSBr to afford sulfo-UTP (19) and sulfo-UDP (20) analogs. Finally, hydrogenation of 20 afforded the saturated derivative 22.
 | ||
| Scheme 3 Synthesis of sulfo-uridine analogs. | ||
NmCSS inhibition assays indicate that the sulfo-uridine analogs are also potent inhibitors. There is little difference between sulfo-CTP and sulfo-UTP analogs (entries 1 and 4, Table 1). Although sulfo-UDP is essentially inactive (entry 5), saturation of the double bond restores activity (entry 7). Saturated sulfo-UDP is not quite as active as saturated sulfo-CDP (entry 3), which is consistent with the substrate specificity studies indicating that CDP is a better substrate than UDP. However, in both cases, saturation increases inhibition relative to the unsaturated analogs suggesting that increased flexibility enhances activity.
One motivation for replacing the phosphate with a sulfone functionality was to create neutral molecules that may be better suited for intracellular targets. We were curious as to whether or not the isopropylphosphonate esters would show biological activity. In preliminary investigations, the isopropyl protected form of 18, which is 21, was also tested (entry 6). While activity was diminished by about half, we still observed significant inhibition. These studies are encouraging from the perspective of developing pro-drug strategies.
As previously stated, there are notable differences between bacterial and vertebrate CSS. Bacterial enzymes are cytoplasmic and vertebrate CSS are active in the nucleus of cells. And although only a few enzymes have been assessed for substrate specificity, early indications suggest that there are distinct differences between mammalian and bacterial homologs with vertebrate enzymes being highly specific for CTP. The current study, which is the first to probe NmCSS activity as a function of unnatural nucleotide analogs, points to significant differences in inhibitor activity as a function of both the nucleotide base and degree of saturation. Kinetic studies are underway to determine the mechanism of sulfo-nucleotide inhibition, which we anticipate will provide a novel platform for discovery of selective CSS inhibitors.
Acknowledgements
This work was supported by TRDRP. NSF DBI-0722538 and NSF CHE-0443516 provided funding for the NMR spectrometers used on this project.References
- F. A. Troy, Glycobiology, 1992, 2, 5 CAS.
- F. A. Troy, Biology of Sialic Acids, 1995, Abraham Rosenberg, Ed. Plenum Press, New York, pp. 95 Search PubMed.
- F. A. Troy II, Encycl. Of Biol. Chem., 2004, 3, 407 Search PubMed.
- A. P. Moran, M. M. Prendergast and B. J. Appelmelk, FEMS Immunol. Med. Microbiol., 1996, 16, 105 CrossRef CAS.
- R. E. Mandrell and M. A. Apicella, Immunobiology, 1993, 187, 382 Search PubMed.
- A. Preston, R. E. Mandrell, B. W. Gibson and M. A. Apicella, Crit. Rev. Microbiol., 1996, 22, 139 CrossRef CAS.
- A. K. Münster-Kühnel, J. Tiralongo, S. Krapp, B. Weinhold, V. Ritz-Sedlacek, U. Jacob and R. Gerardy-Schahn, Glycobiology, 2004, 14, 43R CrossRef.
- H. Yu, H. Yu, R. Karpel and X. Chen, Bioorg. Med. Chem., 2004, 12, 6427 CrossRef CAS.
- M. Knorst and W.-D. Fessner, Adv. Synth. Catal., 2001, 343, 698 CrossRef CAS.
- D. Nakata, A.-K. Münster, R. Gerardy-Schahn, N. Aoki, T. Matsuda and K. Kitajima, Glycobiology, 2001, 11, 685 CrossRef CAS.
- E. Zbiral, E. Schreiner and R. Christian, Carbohydr. Res., 1989, 194, c15 CrossRef CAS.
- C. R. Petrie III, M. Sharma, O. D. Simmons and W. Korytnyk, Carbohydr. Res., 1989, 186, 326 CrossRef.
- I. G. Bravo, S. Barrallo, M. A. Ferrero, L. B. Rodriguez-Aparicio, H. Martinez-Blanco and A. Reglero, Biochem. J., 2001, 358, 585 CAS.
- L. B. Rodriguez-Aparicio, J. M. Lungo, C. Gonzalez-Clemente and A. Reglero, J. Biol. Chem., 1992, 267, 9257 CAS.
- K. H. Scheit and H.-J. Linke, Eur. J. Biochem., 1982, 126, 57–60 CrossRef CAS.
- A. M. Bello, E. Poduch, Y. Liu, L. Wei, I. Crandall, X. Wang, C. Dyanand, K. C. Kain, E. F. Pai and L. P. Kotra, J. Med. Chem., 2008, 51, 439–448 CrossRef CAS.
- P. Besada, D. H. Shin, S. Costanzi, H. Ko, C. Mathé, J. Gagneron, G. Gosselin, S. Maddileti, T. K. Harden and K. A. Jacobson, J. Med. Chem., 2006, 49, 5532–5543 CrossRef CAS.
- J. H. Wong, M. M. Olmstead, J. C. Fettinger and J. Gervay-Hague, Acta Cryst., 2008, C64, o132 CAS.
- M. J. Hadd, M. A. Smith, M. D. Carducci and J. Gervay-Hague, Tetrahedron Lett., 2001, 42, 5137 CrossRef CAS.
- J. H. Wong, M. M. Olmstead, J. C. Fettinger and J. Gervay-Hague, Acta Cryst. Sec. E., 2007, 63, 3046 Search PubMed.
- X. Chen, A. J. Wiemer, R. J. Hohl and D. F. Wiemer, J. Org. Chem., 2002, 67, 9331 CrossRef CAS.
- For related examples of double bond migration see: J.-F. Wang, X.-D. Yang, L.-R. Zhang, Z.-J. Yang and L.-H. Zhang, Tetrahedron, 2004, 60, 8535 Search PubMed.
- S. J. Danishefsky, S. L. DeNinno, S. H. Chen, L. Boisvert and M. Barbachyn, J. Am. Chem. Soc., 1989, 111, 5810 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details of compound syntheses and NmCSS substrate and inhibition assays; NMR, HRMS, HPLC and CE spectra. See DOI: 10.1039/b819155g |
| ‡ Recipient of the ACS Glaxo Smith-Kline Fellowship 2008. |
| This journal is © The Royal Society of Chemistry 2009 |