Chemical approach toward efficient DNA methylation analysis
Akimitsu
Okamoto
*
Advanced Science Institute, RIKEN (The Institute of Physical and Chemical Research), Wako, Saitama 351-0198, Japan. E-mail: aki-okamoto@riken.jp; Fax: 81 48 467 9205; Tel: 81 48 467 9238
First published on 6th November 2008
Abstract
The development of a reaction for the detection of the presence/absence of one methyl group in a very long DNA strand is a chemically and biologically challenging research subject. Several newly designed chemical assays for the typing of DNA methylation are reported and discussed in this paper. A new concept of sequence-specific short-term methylation analysis, supported by a chemical basis, is the starting point for a novel methylation-typing assay, which will supersede conventional methods.
 Akimitsu Okamoto Akimitsu Okamoto | Akimitsu Okamoto obtained his PhD in synthetic chemistry and biological chemistry from Kyoto University in 1998, where he was appointed Assistant Professor in 1999 after Postdoctoral Fellow in Massachusetts Institute of Technology for a year. He moved to RIKEN as Initiative Research Scientist (Unit Leader) in 2006. His research focuses on the design, synthesis, and physical properties of new, man-made biopolymers with various functionalities and the design of unique organic chemical systems for recognizing, transforming, and visualizing a single component or atom in biopolymers of interest. He is the winner of the OBC Lecture Award 2008. |
1 Introduction
Gene expression is regulated by the epigenetic modification of biopolymers independent of their primary sequences. In particular, cytosine methylation, in which the C5 position of the cytosine base is methylated enzymatically, plays a crucial role in the regulation of chromatin stability, gene regulation, parental imprinting, and X chromosome inactivation in females (Fig. 1).1–4 In addition, erroneous DNA methylation may contribute to the etiopathogenesis of tumorigenesis and aging.5,6Cytosine methylation is one of the most important epigenetic events, and its detection is very significant. Much effort has gone into developing a simple reaction for 5-methylcytosine detection. Analysis of the cytosine methylation status of a gene is very important for understanding the expression mechanism of genetic information. However, it is not easy to distinguish methylcytosine from cytosine, i.e., to detect the existence of only one methyl group in a long DNA strand. The development of a reaction for the detection of the presence/absence of one methyl group in a DNA strand is a chemically and biologically challenging research subject. | ||
| Fig. 1 Cytosine methylation and epigenetic control of gene expression. | ||
The establishment of methylation-typing techniques is necessary to elucidate the role of each methylation, in order to proceed to the next stage of the present research, which involves rough scanning and scoring of the methylcytosines in genes. The existence of a rapid and selective chemical reaction capable of distinguishing between methylated and nonmethylated cytosines would be very useful as a good method for efficiently analyzing the status of cytosine methylation at a specific site in a gene.
The five key points required for a new chemical assay for methylation detection are as follows.
(1) Sequence-selective assay: The focus of methylation studies is changing from the storage of information on the amount of methylation and the location of the methylation region to the elucidation of the status and role of each methylation. The development of a conceptually new chemical approach to site-specific detection is very important.
(2) Methylcytosine-positive assay: Reactions selective to methylation sites should be developed. Conventional methods are nonmethylated cytosine-positive and methylated cytosine-negative.
(3) Simple detection process: The most facile method for methylation detection is probably to directly label the signal-sending units, such as fluorescent dyes, to the methylation sites. To the best of our knowledge there is no fluorescent assay for cytosine methylation detection, except for a DNA microarray of sequence-converted DNAviabisulfite treatment.7,8
(4) Noncleavage assay: DNA samples are damaged by strand scission during bisulfite treatment, as described below.9 Nonspecific cleavage complicates the detection process and reduces quantification precision.
(5) Short-time assay: The bisulfite assay usually requires about half a day for complete modification to obtain reliable results. A shorter reaction time is desirable.
This paper introduces and discusses several newly designed chemical assays for DNA methylation typing. The chemistry-based assay includes many advantages that are quite different from conventional assays. The new concept of sequence-specific short-term methylation analysis supported by a chemical basis will be the starting point for a novel methylation-typing assay.
2 Conventional bisulfite assay—disadvantages
Conventional techniques for methylation detection can be divided roughly into two types: DNA fragmentation by restriction enzymes10–16 and cytosine deamination by bisulfate salts.17–20 In the former method, the CpG methylation-sensitive restriction endonucleases were used to survey the extent of DNA methylation. The available target sequences are limited, and comparatively large amounts of genomic samples are required. The latter, bisulfite method has now been widely applied for epigenotyping at methylation hot spots and for the mapping of cytosine methylation in the promoter region. Sodium bisulfite causes the deamination of a cytosine residue in a single-strand DNA through formation of a 5,6-dihydrocytosine-6-sulfonate intermediate at acidic pH. The deaminated bisulfite adduct is converted into a uracil residue through elimination of bisulfite at alkaline pH (Fig. 2). Methylcytosine also yields thymine with sodium bisulfite, but the reaction rate for the bisulfite adduct formation is much slower.21,22 The difference in the rate of the adduct formation has been exploited in the discrimination between cytosine and methylcytosine. In this case, primer design and complete modification of DNA are very significant in order to avoid false positives for methylcytosine and thus to obtain reliable results. Thus, the bisulfite assay requires long reaction times (5–24 h, normally an overnight reaction). | ||
| Fig. 2 Bisulfite assay and decomposition of the sulfonic acid adduct. | ||
For precise mapping of DNA methylation patterns in CpG islands, methylation-specific PCR (MSP) was developed based on the modification of DNA by sodium bisulfite, which can rapidly assess the methylation status of cytosines within a CpG island.23,24 This assay entails initial treatment with sodium bisulfite, converting all unmethylated cytosines to uracil, and subsequent amplification with primers specific for methylated versus unmethylated DNA. MSP requires only small quantities of DNA, and is sensitive to small quantities of methylated alleles.
Recently, many further improved bisulfate assays have been reported by many assay kit suppliers. However, there are still serious problems. The biggest problem is DNA degradation during the long reaction times.9 This issue reduces the quantitativeness and reliability of the results of the methylation status analysis.
We investigated what percentage of the starting DNA degrades during bisulfite assay.25 The decrease in the percentage of the starting DNA strands during bisulfite treatment and the amount of recovered DNA strands after neutralization of the reaction mixture was analyzed using the intensities of HPLC peaks. The decomposition rate constants of pyrimidine bases were determined from the plot of the decrease in absorption versus the reaction time of bisulfite treatment. The degradation rate constants for the bisulfite adducts of uracil and thymine were calculated as kUD = 4.1 × 10−6 and kTD = 2.4 × 10−6 s−1, respectively, which indicate the decomposition rate constants of the adducts formed by bisulfate addition and subsequent deamination from cytosine and 5-methylcytosine, respectively (Fig. 2). Production of abasic structures at pyrimidine sites was observed by mass spectroscopy, suggesting that DNA degradation is caused via depyrimidination. The amount of target degradation during bisulfite incubation was quantified with quantitative PCR. Bisulfite treatment of a 100-mer DNA was executed for 16 h under typical bisulfate assay conditions. The sigmoid curves of quantitative PCR showed that the 1014 copies of the DNA before the bisulfate assay became only ca. 1011 copies after bisulfite treatment for 16 h. This is 0.1% of the original DNA amount, and suggests that a critical level of degradation was caused via depyrimidination during bisulfite treatment. The DNA sample should be carefully treated for methylation status quantification, and pleiotropic analysis including chemical or molecular biological techniques is essential to obtain more reliable methylation data.
3 Osmium oxidation of methylcytosine
The existence of a more rapid, target-selective, DNA-friendly chemical reaction capable of distinguishing between methylated and unmethylated cytosines would be promising as a useful method for efficiently analyzing the status of cytosine methylation at a specific site in a gene. The reactions of the pyrimidine bases thymine and cytosine are well known, such as photodimerization,26,27Michael addition,28–30 and oxidation,31–34 but the number of reports on the chemistry of methylcytosine is still limited. The oxidation of pyrimidine bases may be applicable for the detection of the presence/absence of a methyl group at cytosine C5 because the number of the substituents of the C5–C6 double bond are different. The C5–C6 double bond of thymine and methylcytosine bases is known to be oxidized by osmium tetroxide and the bases are converted into their glycols.35,36 Thymines in single-stranded DNAs are also oxidized by osmium tetroxide and this reaction is sometimes used for thymine sequencing.37–41We have developed the sequence-selective oxidation of methylcytosines (Fig. 3).42 One example of the reaction conditions is as follows: 5 mM potassium osmate (an oxidant much less intractable than osmium tetroxide), 100 mM potassium hexacyanoferrate(III) (an activator), and 100 mM bipyridine (a reaction-accelerating ligand) in 100 mM Tris-HCl buffer (pH = 7.7), 1 mM EDTA, and 10% acetonitrile (for dissolution of bipyridine). The reaction mixture was incubated at 0 °C for 5 min. The oxidized strand was cleaved at a damaged pyrimidine base with hot piperidine, and then analyzed as a band for a shortened strand using polyacrylamide gel electrophoresis. Methylcytosines were oxidized efficiently by exposure to the reaction mixture. The strand cleavage at nonmethylated cytosine sites was negligible, whereas the methylcytosine-containing DNA strands were sequence-selectively cleaved at the methylcytosine sites. The MALDI-TOF MS data of methylated DNA treated with the reaction mixture suggested that a stable methylcytosine glycol–dioxidoosmium–bipyridine ternary complex was formed. In addition, methylcytosines in single-stranded DNA efficiently formed metal complexes, whereas the complexation of methylcytosines in a duplex was completely suppressed. This weak reactivity is probably attributed to inhibition of the attack of an osmium complex on the π-orbital of a C5–C6 double bond by the base stacking of the duplex structure.
 | ||
| Fig. 3 Osmium complexation of 5-methylcytosine. (a) A scheme of osmium complexation of 5-methylcytosine. (b) Sequence-selective osmium oxidation induced by a guide DNA. | ||
Osmium complexation occurs for thymines as well as methylcytosine in single-stranded DNA.42,43 We want to discriminate the target methylation site from thymines and other methylcytosines. The use of steric control, such as bulge structure formation, may be effective for sequence-specific osmium complexation. For example, efficient strand cleavage was observed at the methylcytosine site in the bulged structure, like the reaction in a single-stranded DNA. The calculated rate constants for methylcytosine-bulged and cytosine-bulged duplexes were 1.11 × 10−2 and 2.51 × 10−5 s−1, respectively. For easier analysis, we prepared a bulge-inducing DNA (guide DNA) fixed on polystyrene beads. After generation of the bulged structure by hybridization of the target DNA with the guide DNA, the duplex was incubated in a reaction mixture containing potassium osmate. After washing and treatment with hot piperidine, the DNA amplification was monitored with quantitative PCR. A curve with a retarded start and a small curvature was displayed for a methylated sequence. This is clearly distinguishable from the control curve, and may be a good method for the detection of the existence of only one methyl group in a long DNA strand.
The absolute structure of the resulting methylcytosine glycol–dioxidoosmium–bipyridine ternary complex was analyzed by X-ray crystallography.44 The ternary complex in DNA was digested to the nucleoside level using a mixture of snake venom phosphodiesterase, nuclease P1, and alkaline phosphatase, and then deaminated into the corresponding thymine derivative. The HPLC analysis showed two peaks with an area ratio of 81 : 19. The major product was crystallized in water and analyzed by X-ray crystallography. The complex had a slightly distorted octahedral geometry with coplanar glycol oxygens and bipyridine nitrogens. The two Os–O double bonds were trans, and tilted 3–6° to the bipyridine side. The structure of the glycol was the 5R,6S-configuration. The minor complex was a diastereomeric isomer containing 5S,6R-methylcytosine glycol. The predominant formation of 5R,6S-glycol is probably due to the larger contribution of the configuration of the nucleoside rather than that of the whole structure of the DNA strand.
4 Methylation-selective fluorescence quenching
We used radio labels, piperidine treatment, and gel electrophoresis for methylation analysis, as described above. However, we preferred not to use these methods for methylation analysis because they are troublesome and time-consuming. We wished to use fluorescence or something easier. Therefore, a fluorescent probe was developed for methylation analysis, based on the control of the fluorescence emission from a fluorophore fixed on DNA, using the methylcytosine-selective addition of an osmium–bipyridine complex.45 We designed a DNA probe with a microenvironment-sensitive fluorophore, 6-dimethylamino-2-acylnaphthalene (DAN).46–52 The purine base modified with DAN (D) can form a wobble base pair with pyrimidines (Fig. 4). The treatment with potassium osmate and bipyridine drastically changed the fluorescence intensity of the D-containing duplex with high methylcytosine selectivity. The fluorescence of the duplex of osmium-complexed DNA and the D-labeled probe was significantly quenched compared to the fluorescence emission of the duplex of noncomplexed DNA and probe. Photoinduced electron transfer occurred from the excited D base to the osmium complex and this contributed to the quenching process. The fluorescence quenching of D was very sensitive only to complexation of methylcytosine opposite D.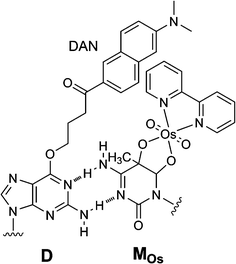 | ||
| Fig. 4 Methylcytosine-selective fluorescence quenching involving osmium complex formation at methylcytosine. | ||
5 Labeling of methylcytosine
A ligand bipyridine plays a very important role in acceleration of osmium oxidation. Functionalized bipyridines facilitate the incorporation of signal units into the methylcytosine in the DNA of interest through the coordination to the osmate. We designed tag-attachable bipyridine ligands for direct methylcytosine labeling and used them in fluorescent and electrochemical assays.53We first confirmed that substitution at C4 of bipyridine does not hinder the complexation with methylcytosine. Based on this knowledge, a new ligand, 4-(6-(4-aminobutylamino)-6-oxohexyl)-4′-methyl-2,2′-bipyridine, was synthesized. The formation of the methylcytosine-selective complex using a ligand with an amino linker made it possible to attach tags to the DNA methylation sites (Fig. 5). The labeling reaction for the triad proceeded quantitatively when using N-hydroxysuccinimidyl esters of the labels.
 | ||
| Fig. 5 Direct labeling to methylcytosine mediated by osmium complexation. R = fluorescent dyes and redox-active units. | ||
For example, a fluorescent dye was attached to the osmium complex. We applied this fluorescence label on methylcytosine to a fluorescence resonance energy transfer (FRET) system from the neighboring fluorophores fixed to the hybridizing DNA. We prepared a DNA strand labeled methylcytosine-selectively with hexachlorofluorescein (HEX), and investigated the FRET efficiency from the complementary probe containing a fluorescein unit. In the fluorescence spectrum of the hybrid on excitation at 495 nm, in which the fluorescein is excited selectively, a signal at 535 nm was observed from HEX, suggesting that FRET occurred from the complementary fluorescein probe to a DNA strand labeled methylcytosine-selectively with HEX. Cytosine methylation at the desired site was analyzed as having high sequence selectivity, although there are many labelable thymines and other methylcytosines in the sequence. FRET worked effectively only for the HEX label of the methylcytosine, which was located in the immediate neighborhood of the fluorescein of the complementary probe. Direct fluorescent labeling to methylcytosine enabled us to visually judge the presence/absence of methylation of a cytosine, and the sequence-selective analysis of cytosine methylation also became possible by the use of FRET.
6 Methylcytosine-selective interstrand crosslinking
We also designed a new “ligand”.54 An adenine base of a short DNA strand involved in a mismatched hybridization and the bipyridine ligand required for osmium-centered complex formation were connected (Fig. 6). The formation of a cytosine–adenine-mismatched pair causes partial disruption of π-stacking of the DNA duplex, and facilitates oxidation at the C5–C6 double bond of the cytosine forced out of the DNA major groove. The formation of a mismatched base pair and the location of bipyridine should result in complexation at a specific methylcytosine, regardless of other reactive bases in a long DNA strand. | ||
| Fig. 6 Structure of bipyridine–adenine conjugate B and sequence-selective interstrand crosslinking by osmium complexation. | ||
The ligand-tethered nucleoside, B, formed interstrand complexes at the target methylcytosine. The interstrand crosslinking of the osmium complex with B-containing nucleic acid (ICON) allowed sequence-selective labeling and the obstruction of PCR amplification at the target methylcytosine.
A model methylcytosine-typing experiment was carried out to construct a prototype for ICON-based methylation analysis. The mixture containing a RB1gene short fragment, potassium osmate and ICON probes was incubated at 55 °C for 1 h. Quantitative PCR assay after deionization showed sigmoid amplification curves (Fig. 7). The amplification starting point exhibited a linear dependence on the logarithm of the methylated duplex concentration. A linear relationship between the amplification starting point and the logarithm of the methylation proportion was also observed. This relationship allowed the calculation of the proportion of methylation at the target cytosine. An ICON with B-containing DNAs designed for each target cytosine was formed easily, depending on the amount of the DNA methylated at the target cytosine, and was completely independent of the amount of other methylated DNA. Blocking of PCR by ICON at the methylation site made possible the sequence-specific quantification of methylation.
 | ||
| Fig. 7 Sigmoid curves observed after quantitative PCR of ICON products. Different concentrations of the starting DNA were used for osmium oxidation. | ||
The sequence-specific ICON was used to quantify the methylation of the mouse genome, which was collected from different tissues. Two CpG sequences in chromosome 11 in a tissue-specific differentially methylated region were chosen as the targets, and four B-labeled DNAs were designed for these targets. A 60 min incubation and a 90 min PCR analysis were performed with 20 ng of the mouse genome. Samples from different tissues exhibited characteristic methylation levels for the two methylation sites, which is in close agreement with the degree of methylation as determined by mass spectroscopy analysis of fragmented and bisulfite-treated genome samples.
ICON probe resulted in the complexation at a specific methylcytosine regardless of other reactive methylcytosine and thymine bases in a long DNA strand. There still remain further aspects to be examined toward an easier-to-use methylation analysis, such as further improvement of the reaction yield for osmium complexation at methylcytosine and optimization of PCR conditions suitable for ICON. However, this new concept of sequence-specific short-term methylation analysis supported by the chemical basis will be the starting point for an epoch-making methylation-typing assay.
7 Other chemical assays
Other chemical reactions have also been studied for efficient methylation analysis. Tanabe and coworkers used photosensitized one-electron oxidation to detect methylcytosine in DNA.55 They used a sensitizing 2-methyl-1,4-naphthoquinone (NQ) chromophore tethered to the interior of DNA strands for oxidation (Fig. 8). Photoirradiation and subsequent hot piperidine treatment of the duplex consisting of a methylated DNA strand and an NQ-tethered complementary DNA strand cleaved the strand oxidized at the target methylcytosine site, because the NQ chromophore was arranged so as to be in close contact with the target methylcytosine. Furthermore, well designed incorporation of the NQ chromophore suppressed a competitive strand cleavage at consecutive guanines. The low sensitivity of the detection due to the low yield of methylcytosine-selective strand cleavage (ca. 5%) remains to be improved for establishment of a more general method with higher sensitivity.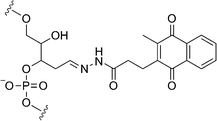 | ||
| Fig. 8 Photosensitizing unit 2-methyl-1,4-naphthoquinone tethered to DNA. | ||
This research group also developed a fluorometric detection system of DNA methylation based on a combination of a photooxidative DNA cleavage and an Invader assay.56 Enzymatic treatment of a mixture of a photochemically fragmented DNA strand at methylcytosine and a hairpin-shaped probe possessing a fluorophore-quencher pair resulted in an enhancement of fluorescence intensity. This system allows for the detection of methylcytosine in DNA in subfemtomole amounts by monitoring the fluorescence change.
Bareyt and Carell reported the oxidation of methylcytosine by vanadium species or sodium periodate.57 They used a combination of V2O5–lithium bromide or sodium periodate–lithium bromide (Fig. 9). The strand breaks in methylated cytosines were observed by denaturing polyacrylamide gel electrophoresis after treatment of the reaction product with hot piperidine. Although oxidation of guanine to 8-oxoguanine occurs with V2O5–lithium bromide, the sodium periodate–lithium bromide system suppressed the modification of all other nucleobases. They mentioned that the mechanism proceeds via the formation of a bromonium cation intermediate at the C5–C6 double bond of the pyrimidine rings. Although piperidine treatment is essential before analysis and we have to pay attention to reaction time and temperature to avoid cleavage at thymines and guanines, the methylcytosine-selective reaction with the sodium periodate–lithium bromide system would be one of useful chemical methods for a rapid and easy-to-use analysis of epigenetic information in genes.
 | ||
| Fig. 9 Methylcytosine-selective oxidationvia the formation of a bromonium cation intermediate. | ||
Conclusion
We have described a new method of chemical analysis of methylcytosine. New chemical reactions targeting 5-methylcytosine offer a promising prospect for a new methylation assay that is quite different from conventional ones. Further aspects still need to be examined, however, in order to arrive at simpler methylation analysis. Further improvements of the methylcytosine-selective reaction will afford a more reliable epigenotyping method that could replace conventional methylation analysis.References
- P. L. Jones and A. P. Wolffe, Semin. Cancer Biol., 1999, 9, 339–347 CrossRef CAS.
- P. H. Tate and A. P. Bird, Curr. Opin. Genet. Dev., 1993, 3, 226–231 CrossRef CAS.
- V. Colot and J. L. Rossignol, BioEssays, 1999, 21, 402–411 CrossRef CAS.
- R. Feil and S. Khosla, Trend. Genet., 1999, 15, 431–435 Search PubMed.
- P. A. Jones and P. W. Laird, Nature Genet., 1999, 21, 163–167 CrossRef CAS.
- K. D. Robertson, Nat. Rev. Genet., 2005, 6, 597–610 CrossRef CAS.
- Y. Zhou, J. M. S. Lum, G.-H. Yeo, J. King, S. K. H. Tay and S. S. Chong, Clin. Chem., 2006, 52, 1492–1500 CrossRef CAS.
- V. L. Boyd, K. I. Moody, A. Z. Karger, K. J. Livak, G. Zon and J. W. Burns, Anal. Biochem., 2006, 354, 266–273 CrossRef CAS.
- M. Raizis, F. Schmitt and J.-P. Jost, Anal. Biochem., 1995, 226, 161–166 CrossRef CAS.
- M. F. Kane, M. Loda, G. M. Gaida, J. Lipman, R. Mishra, H. Goldman, J. M. Jessup and R. Kolodner, Cancer Res., 1997, 57, 808–811 CAS.
- A. Okamoto, K. Tanabe and I. Saito, J. Am. Chem. Soc., 2002, 124, 10262–10263 CrossRef CAS.
- A. Okamoto, Bull. Chem. Soc. Jpn., 2005, 78, 2083–2097 CrossRef CAS.
- P. S. Nelson, T. S. Papas and C. W. Schweinfest, Nucleic Acids Res., 1993, 21, 681–686 CrossRef CAS.
- J. G. Tasserondejong, J. Aker and M. Giphartgassler, Gene, 1988, 74, 147–149 CrossRef CAS.
- V. Butkus, L. Petrauskiene, Z. Maneliene, S. Klimasauskas, V. Laucys and A. Janulaitis, Nucleic Acids Res., 1987, 15, 7091–7102 CrossRef CAS.
- G. M. Church and W. Gilbert, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 1991–1995 CrossRef CAS.
- H. Hayatsu, Y. Wataya, K. Kai and S. Iida, Biochemistry, 1970, 9, 2858–2866 CrossRef.
- M. L. Gonzalgo and P. A. Jones, Nucleic Acids Res., 1997, 25, 2529–2531 CrossRef CAS.
- J. G. Herman, J. R. Graff, S. Myöhänen, B. D. Nelkin and S. B. Baylin, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9821–9826 CrossRef CAS.
- P. M. Warnecke, C. Stirzaker, J. Song, C. Grunau, J. R. Melki and S. J. Clark, Methods, 2002, 27, 101–107 CrossRef CAS.
- R. Shapiro, R. E. Servis and M. Welcher, J. Am. Chem. Soc., 1970, 90, 422–424 CrossRef.
- H. Hayatsu, Y. Wataya and K. Kai, J. Am. Chem. Soc., 1970, 90, 724–726 CrossRef.
- S. J. Clark, J. Harrison, C. L. Paul and M. Frommer, Nucleic Acids Res., 1994, 22, 2990–2997 CrossRef CAS.
- M. Frommer, L. E. McDonald, D. S. Millar, C. M. Collis, F. Watt, G. W. Grigg, P. L. Molloy and C. L. Paul, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 1827–1831 CAS.
- K. Tanaka and A. Okamoto, Bioorg. Med. Chem. Lett., 2007, 17, 1912–1915 CrossRef CAS.
- A. Wacker, H. Dellweg, L. Träger, A. Kornhauser, E. Lodemann, G. Türck, R. Selzer, P. Chandra and M. Ishimoto, Photochem. Photobiol., 1964, 3, 369–394 CrossRef CAS.
- D. Weinblum and H. Johns, Biochim. Biophys. Acta, 1966, 114, 450–459 CAS.
- H. Hayatsu, Prog. Nucleic Acids Res. Mol. Biol., 1976, 16, 75–124 Search PubMed.
- R. Shapiro, in Chromosome Damage and Repair, ed. E. Seeberg and K. Kleppe, Plenum Publishing Corp., New York, 1981, pp. 3–12 Search PubMed.
- H. Chen and B. R. Shaw, Biochemistry, 1993, 32, 3535–3539 CrossRef CAS.
- M. Beer, S. Stern, D. Carmalt and K. H. Mohlenrich, Biochemistry, 1966, 5, 2283–2288 CrossRef CAS.
- K. Frenkel, M. S. Goldstein, N. J. Duker and G. W. Teebor, Biochemistry, 1981, 20, 750–754 CrossRef CAS.
- B. Demple and S. Linn, Nucleic Acids Res., 1982, 10, 3781–3789 CrossRef CAS.
- S. V. Jovanovic and M. G. Simic, J. Am. Chem. Soc., 1986, 108, 5968–5972 CrossRef CAS.
- L. R. Subbaraman, J. Subbaraman and E. J. Behrman, Bioinorg. Chem., 1971, 1, 35–55 CrossRef CAS.
- C.-H. Chang, H. Ford and E. J. Behrman, Inorg. Chim. Acta, 1981, 55, 77–80 CrossRef CAS.
- H. Ford, C.-H. Chang and E. J. Behrman, J. Am. Chem. Soc., 1981, 103, 7773–7779 CrossRef CAS.
- H. Ide, Y. W. Kow and S. S. Wallace, Nucleic Acids Res., 1985, 13, 8035–8052 CrossRef CAS.
- E. Palecek, Methods Enzymol., 1992, 212, 139–155 CAS.
- M. Beer, S. Stern, D. Carmalt and K. H. Mohlhenrich, Biochemistry, 1966, 5, 2283–2288 CrossRef CAS.
- K. Nakatani, S. Hagihara, S. Sando, H. Miyazaki, K. Tanabe and I. Saito, J. Am. Chem. Soc., 2000, 122, 6309–6310 CrossRef CAS.
- A. Okamoto, K. Tainaka and T. Kamei, Org. Biomol. Chem., 2006, 4, 1638–1640 RSC.
- A. Nomura and A. Okamoto, Org. Biomol. Chem., 2008 10.1039/B813172D.
- T. Umemoto and A. Okamoto, Org. Biomol. Chem., 2008, 6, 269–271 RSC.
- K. Tanaka, K. Tainaka and A. Okamoto, Bioorg. Med. Chem., 2007, 15, 1615–1621 CrossRef CAS.
- A. Okamoto, K. Tainaka and I. Saito, Bioconjugate Chem., 2005, 16, 1105–1111 CrossRef CAS.
- E. Cohen, T. B. McAnaney, E. S. Park, Y. N. Jan, S. G. Boxer and L. Y. Jan, Science, 2002, 296, 1700–1703 CrossRef CAS.
- T. Kimura, K. Kawai and T. Majima, Org. Lett., 2005, 7, 5829–5832 CrossRef CAS.
- A. Okamoto, K. Tainaka and I. Saito, Photomed. Photobiol., 2006, 28, 31–32 Search PubMed.
- K. Tainaka, K. Tanaka, S. Ikeda, K.-i. Nishiza, T. Unzai, Y. Fujiwara, I. Saito and A. Okamoto, J. Am. Chem. Soc., 2007, 129, 4776–4784 CrossRef CAS.
- A. Okamoto, K. Tainaka, T. Unzai and I. Saito, Tetrahedron, 2007, 63, 3465–3470 CrossRef CAS.
- K. Tainaka, I. Saito and A. Okamoto, Photomed. Photobiol., 2007, 29, 12–13 Search PubMed.
- K. Tanaka, K. Tainaka, T. Kamei and A. Okamoto, J. Am. Chem. Soc., 2007, 129, 5612–5620 CrossRef CAS.
- K. Tanaka, K. Tainaka, T. Umemoto, A. Nomura and A. Okamoto, J. Am. Chem. Soc., 2007, 129, 14511–14517 CrossRef CAS.
- K. Tanabe, H. Yamada and S.-i. Nishimoto, J. Am. Chem. Soc., 2007, 129, 8034–8040 CrossRef CAS.
- H. Yamada, K. Tanabe and S.-i. Nishimoto, Bioconjugate Chem., 2008, 19, 20–23 CrossRef CAS.
- S. Bareyt and T. Carell, Angew. Chem., Int. Ed., 2007, 47, 181–184.
| This journal is © The Royal Society of Chemistry 2009 |