Mitotic trafficking of silicon microparticles†
Rita E.
Serda‡
a,
Silvia
Ferrati‡
a,
Biana
Godin
a,
Ennio
Tasciotti
a,
XueWu
Liu
a and
Mauro
Ferrari
*abc
aUniversity of Texas Health Science Center, Department of Nanomedicine and Biomedical Engineering, 1825 Pressler, Suite 537, Houston, TX 77030. E-mail: Mauro.Ferrari@uth.tmc.edu; Tel: +713-500-4444
bUniversity of Texas MD Anderson Cancer Center, Department of Experimental Therapeutics, Unit 422, 1515 Holcombe Blvd., Houston, TX 77030
cRice University, Department of Bioengineering, Houston, TX 77005
First published on 5th October 2009
Abstract
Multistage carriers were recently introduced by our laboratory, with the concurrent objectives of co-localized delivery of multiple therapeutic agents, the “theranostic” integration of bioactive moieties with imaging contrast, and the selective, potentially personalized bypassing of the multiplicity of biological barriers that adversely impact biodistribution of vascularly injected particulates. Mesoporous (“nanoporous”) silicon microparticles were selected as primary carriers in multi-stage devices, with targets including vascular endothelia at pathological lesions. The objective of this study was to evaluate biocompatibility of mesoporous silicon microparticles with endothelial cells using in vitroassays with an emphasis on microparticle compatibility with mitotic events. We observed that vascular endothelial cells, following internalization of silicon microparticles, maintain cellular integrity, as demonstrated by cellular morphology, viability and intact mitotic trafficking of vesicles bearing silicon microparticles. The presence of gold or iron oxide nanoparticles within the porous matrix did not alter the cellular uptake of particles or the viability of endothelial cells subsequent to engulfment of microparticles. Endothelial cells maintained basal levels of IL-6 and IL-8 release in the presence of silicon microparticles.
This is the first study that demonstrates polarized, ordered partitioning of endosomes based on tracking microparticles. The finding that mitotic sorting of endosomes is unencumbered by the presence of nanoporous silicon microparticles advocates the use of silicon microparticles for biomedical applications.
Introduction
Silicon-based nanostructured materials are widely studied for use in bioapplications based on their readily modified physico-chemical and biophysical properties, including surface functionalization, size, shape and porosity.1–3 Initial reports on the biocompatibility of silicon were presented in 19954 when it was discovered that porosification of the silicon imparted biodegradation properties to the material. Current biomedical applications based on nanostructured porous silicon include BioSilicon™ (pSivida Corp.) for drug delivery, and two FDA-approved sustained-release devices for the treatment of chronic eye disease. While based on an entirely different approach to mesoporous (or “nanoporous”) silicon, our research employs carriers that comprise this innovative material within the first stage of multi-functional devices capable of releasing successive stages of carries and drugs intended to negotiate sequential biological barriers to reach the intended target. We have envisioned a multistage delivery system comprised of stage-one nanoporous silicon particles loaded with one or more types of second-stage nanoparticles that in turn either carry active agents or higher level stages of nanoparticles.5 Each level of complexity presents a solution for overcoming barriers, such as enzymatic degradation, crossing the vascular endothelium, and molecular efflux pumps. One strategy for the negotiation of biological barriers is targeting the Stage 1 MicroParticles (“S1MP”) to vascular endothelial cells expressing specific markers of pathology.6 S1MPs carry and release Stage 2 NanoParticles (“S2NP”), which in turn may also vector higher-stage payloads, in a nested-particle system that possesses the ability to provide both time-sequential release and concurrent modes of action.The porous nature of the first stage permits loading the microparticle with drugs, such as cancer therapy agents, and diagnostic agents, such as contrast agents. The tunable pore size introduces control over the rate of degradation of the silicon,7,8 which thereby imparts control over the release of therapeutic and contrast agents. Employing methods of photolithography,5,9,10 nanoporous silicon particles can be fabricated with precise control over particle geometry, such as size and shape, which govern vascular navigation, avoidance of biological barriers, and localization of the particles.11–13Oxidation of the silicon surface creates hydroxyl units that can be used to functionalize the microparticle with surface moieties for targeting specific cellular populations. Functionalization, as well as the scalability, precision and reproducibility of methods used to fabricate silicon particles, make silicon devices especially attractive for clinical applications.
In this study, we have examined the biological impact of nanostructured porous S1MPs on vascular endothelial cells using in vitroassays to assess cellular morphology, cell viability, impact on cell cycle, mitotic potential, and pro-inflammatory responses following cellular engulfment of S1MPs. The mitotic division of cells after internalization of S1MPs and mitotic sorting of vesicles carrying S1MPs were explored.
Results and discussion
Consistent with previous findings9,14 our present data demonstrates uptake of the silicon particles by Human Umbilical Vein Endothelial Cells (HUVECs), which internalize S1MPs by an active process that requires incubation at 37 °C.14Fig. 1 shows scanning electron micrographs of HUVECs incubated with either 1.6 or 3.2 µm S1MPs for 15 min at 37 °C. The S1MPs are pseudo-colored to emphasize their location. Following adhesion of S1MPs to the cell surface, the formation of lamellopodia and extension of surface ruffles initiates cellular engulfment of S1MPs. Based on the presented images, cellular uptake appears mechanistically similar for both sizes of S1MPs. We previously reported that this internalization process is an actin-dependent process inhibited by the presence of Cytochalasin B.9 | ||
Fig. 1
Phagocytosis of S1MPs by endothelial Cells. HUVECs were incubated with either 1.6 (A) or 3.2 (B) µm oxidized S1MPs (10 : 1 particles per cell) for 15 min at 37 °C in serum-free media. Pseudo-colored scanning electron micrographs show the formation of lamellopodia looping over the S1MPs, initiating internalization. Direct magnification 5000× and 30![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000×; bars 5 µm (left) and 1 µm (right). 000×; bars 5 µm (left) and 1 µm (right). | ||
The biological impact of nano- and microparticles is dependent on activation of signaling pathways, cellular uptake, and subcellular localization. In this study we examined the subcellular destination of S1MPs using scanning confocal fluorescence and transmission electron microscopies. Fig. 2 contains confocal micrographs of HUVECs 60 min after the introduction of S1MPs. Intact cytoskeleton filaments, stained with Alexa Fluor 555 conjugated Phalloidin, support cellular integrity following S1MP uptake. S1MPs are located in the perinuclear region of the cell, indicating motor-driven endosomal trafficking along the microtubules15 rather than random diffusion-based movement. A single HUVEC cell is shown in Fig. 2C as a cropped projection image of a z-stack which emphasizes the perinuclear location of multiple S1MPs within a single cell. These data indicate that phagosomes carrying S1MPs mature and migrate along microtubules similar to standard endosomal vesicles. Therefore early intracellular trafficking does not appear to be negatively impacted by the presence of S1MPs.
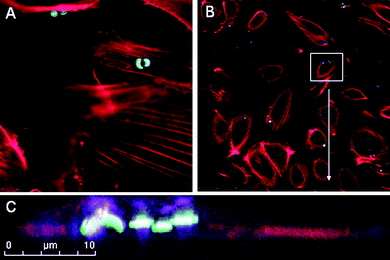 | ||
| Fig. 2 Perinuclear localization of S1MPs. HUVECs were incubated with 3.2 µm oxidized S1MPs (5 : 1) for 120 min at 37 °C, followed by staining with Alexa Fluor 555 Phalloidin. Subcellular location was visualized by scanning confocal fluorescence microscopy. Microparticles were visualized by autofluoresence following excitation at 633 nm. | ||
Transmission electron micrographs, shown in Fig. 3, display the cellular morphology of HUVECs 6 h after S1MP internalization. A single cell, incubated with 3.2 µm oxidized S1MPs (5 : 1; Fig. 3A) at 37 °C, is shown at increasing magnification to display both the overall cellular and the intricate subcellular morphology. The nucleus and subcellular organelles appear healthy, with dark chromatin staining and the presence of intact organelles, such as mitochondria and rough endoplasmic reticulum. Since the potential toxicity of particles is impacted by their surface charge, size, shape and chemical composition, we compared the cellular morphology and subcellular location of two sizes of S1MPs with either positive or negative surface charge. Fig. 3B contains transmission electron micrographs of internalized S1MPs showing tight vesicular membranes surrounding the S1MPs, accompanied by undisrupted organelles and cytoplasm surrounding each S1MP. Altering the surface charge and the size of the S1MP did not appear to alter the nature of the intracellular vesicle housing the S1MP.
 | ||
| Fig. 3
Transmission electron micrographs of HUVECs with internalized S1MPs. HUVECs were incubated with either oxidized (negative) or APTES-modified (positive) S1MPs for 6 h at 37 °C. A) Cell morphology at increasing magnification (2500×, 6000×, 25000×; bars 10, 2, and 0.5 µm) after internalization of 3.2 µm oxidized S1MPs. B) Internalized 1.6 and 3.2 µm S1MPs (direct magnification 50000×; bars 500 nm). | ||
To study the impact of S1MPs on normal cellular events, such as mitosis, we performed time-lapse experiments of cells subsequent to the introduction of S1MPs. Using phase contrast and confocal microscopy, four confocal planes were captured every 5 min for 24 h. A single Human Microvascular Vein Endothelial Cell (HMVEC) undergoing mitosis is shown in selected confocal micrographs in Fig. 4A (see movie 1 of the ESI† ). The time indicated in each image is time-lapsed since the introduction of S1MPs. Prior to mitosis, the endothelial cell contained 30 S1MPs. Following mitosis, each daughter cell had an equal number of S1MPs (15 per cell), providing support for a highly ordered process leading to equal partitioning of endosomes during mitosis. This highly ordered process maintains its integrity with the presence of S1MPs in the endosomes.
![Cellular proliferation of endothelial cells following engulfment of S1MPs. A) Real-time confocal imaging of a mitotic HMVEC cell following internalization of 1.6 µm S1MPs. B) MTT proliferation assays of HUVECs following internalization of either unloaded or loaded (gold [Au] or iron oxide IO S2NPs) S1MPs (particles to cell ratio: 5 : 1 [top] and 10 : 1 [bottom]). C) Scanning electron micrographs of HUVECs incubated with gold S2NP-loaded 3.2 µm S1MPs (10 : 1) for either 15 min (top) or 60 min (bottom) (direct magnification 20 000×; bar 1 µm).](/image/article/2009/NR/b9nr00138g/b9nr00138g-f4.gif) | ||
| Fig. 4 Cellular proliferation of endothelial cells following engulfment of S1MPs. A) Real-time confocal imaging of a mitotic HMVEC cell following internalization of 1.6 µm S1MPs. B) MTT proliferation assays of HUVECs following internalization of either unloaded or loaded (gold [Au] or iron oxide IO S2NPs) S1MPs (particles to cell ratio: 5 : 1 [top] and 10 : 1 [bottom]). C) Scanning electron micrographs of HUVECs incubated with gold S2NP-loaded 3.2 µm S1MPs (10 : 1) for either 15 min (top) or 60 min (bottom) (direct magnification 20000×; bar 1 µm). | ||
It has been reported previously that endosomes exist as distinct organelles and as an interconnected network during cell division.16 In a study of cells induced with high levels of invariant chain, enlarged endosomes were present during all mitotic phases.16 The large size of the endosomes in that study, as well as in our study, allows the fate of single endosomes to be followed with time. As demonstrated earlier with transmission electron microscopy, S1MPs (1.6 and 3.2 µm) are encased in tight vesicular membranes; therefore each S1MP potentially represents a single endosome. The perfect division of S1MPs in daughter cells suggests that this highly ordered endosomal sorting process is intact and supports the integrity of the mitotic event and cellular functioning in the presence of S1MPs. To our knowledge, this is the first study demonstrating mitotic trafficking of endosomes housing engineered microparticles.
Using Drosphila melanogaster embryos as a model for mitosis, it has been reported that cell membranes are targeted to invaginating furrows in mitotic cells via the formation of F-actin-associated vesicles in a process facilitated by proteins used in vesicular transport.17,18 Accompanying the transport of new F-actin-associated vesicles to the cleavage furrow along the central spindle are preexisting endosomal vesicles. Endosomes are trafficked to the central spindle where they form clusters, possibly by movement along the microtubule bundles, which could form tracks for vesicular transport towards the mid-body. In our study, the time-lapse images (movie 1 of the ESI† ) show S1MP-bearing endosomes clustering predominately as a single unit beginning at 13 h after S1MP introduction, and persisting for approximately 30 min (see insert in the top-right image of Fig. 4A). Bidirectional movement of endosomes towards this central cluster suggests that microtubules are involved in the process. It is reported that during cytokinesis, the final stage of mitosis, endocytic trafficking occurs within the intercellular bridge, and completion of cytokinesis is dependent on polarized delivery of endocytic recycling membranes.19 Polarized delivery of S1MP-bearing endosomes most likely occurs through the same process.
Continued proliferation of endothelial cells was monitored for 72 h using an MTTassay to measure mitochondrial enzyme activity (Fig. 4B). HUVECs were incubated with S1MPs at ratios of 5 (top graph) and 10 (bottom graph) S1MPs per cell. Based on the use of porous silicon particles as delivery vehicles for S2NPs, secondary gold or iron oxide nanoparticles were loaded into S1MPs by covalent attachment prior to their introduction to cells. Untreated cells were used as controls. Cellular proliferation was unaffected by the presence of either unloaded or loaded S1MPs. Transmission electron micrographs (Fig. 4C) show gold S2NP-loaded silicon particles adhering to the cell surface 15 min (upper image) after introduction of particles to the cell media. Within 1 h (lower image), gold S2NP-loaded silicon lies beneath the cell membrane.
To determine if polarized partitioning of S1MP-bearing endosomes persists with time, S1MP content per endothelial cell was monitored for 6 days. The doubling time of HMVEC endothelial cells is 48 h,20 supporting approximately three mitotic events during the course of the experiments. HMVECs were incubated with fluorescent-labeled 3.2 µm S1MPs and FACS-sorted to obtain a homogeneous population of cells containing similar levels of S1MPs. Phase contrast and fluorescent images were acquired each day and the number of particles per cell was quantified using bright-field images to define cell boundaries and fluorescent images to assess S1MP content. Representative images from days 2 and 6 are shown in Fig. 5A. A comparison across all days is illustrated in Fig. 5B as a Box and Whisker Chart. The mean particle content per cell for each day is marked with a small box and is also written as text above the bar. Significant changes in S1MP content over time are designated with an asterisk and are based on one-way ANOVA comparisons performed at the 0.05 level. The average number of S1MPs per cell decreased by half every 48 h, correlating with the doubling time of HMVECs, and supporting continued polarized partitioning of the S1MP-bearing endosomes over multiple mitotic events.
 | ||
| Fig. 5 Quantification of S1MPs per cell during multiple mitotic events. HMVECs were incubated with fluorescent S1MPs, then FACS-sorted to obtain a homogeneous population of cells with similar numbers of internalized S1MPs. A) Representative phase contrast and fluorescent microscope images of HMVECs at day 2 and day 6 following S1MP internalization. B) Statistical box charts displaying the number of S1MPs per cell with time. The graphs show the 25th, 75th (box range), and 50th (middle line) percentiles, as will as the average number of S1MPs per cell (small open square and number on top of box). Populations that differ significantly from the preceding significant time point are marked by a star. C) Dividing cell with internalized 3.2 µm silicon particles monitored by phase contrast and live confocal microscopy. | ||
To further support polarized partitioning and clustering of S1MP-bearing endosomes during mitosis, HMVEC cells were treated with 3.2 µm S1MPs. Live phase contrast confocal imaging of a single mitotic cell shows equal partitioning of S1MPs to daughter cells, with progeny receiving either 2 or 1 S1MP (see movie 2 of the ESI† ).
In order to understand if the internalization of the S1MPs had a perturbing effect on the cell cycle, HUVEC cells were treated with S1MPs (cell:particle ratio 1 :1 0) and their DNA content analyzed through propidium iodide staining (PI). PI binds to DNA by intercalating between the bases with little or no sequence preference and with a defined and consistent stoichiometry.21 Upon binding to nucleic acids, PI fluorescence is significantly enhanced (20- to 30-fold) thus making it possible to quantify the amount of DNA through fluorescence-based techniques.22 Compared to an untreated, control population of HUVECs, the cells that internalized the S1MPs did not show any significant change in their relative cell cycle composition. At both the time points analyzed, the ability of the cells to transition from one phase of the cycle to the next one was not altered (Fig. 6A). When control HUVECs were compared to cells treated with a toxic dose of cisplatin (50 µg mL−1; positive control), several significant differences emerged in the analysis of their cell cycle. Cisplatin is a common drug used in many chemotherapeutic regimens for its ability to crosslink to nucleic acids and to prevent cell division by mitosis. Cisplatin damages DNA, elicits DNA repair mechanisms and eventually activates the apoptotic pathway if DNA repair proves impossible.23 As shown in Fig. 6B, cisplatin almost immediately induced the inhibition of cellular mitosis (reduction of the G2/M peak) and induced a massive induction of apoptosis (increase of the sub G0 peak). To further confirm these findings, HUVEC cells (control, treated with 50 µg mL−1 of cisplatin or incubated with S1MPs at the same ratio indicated before) were analyzed by FACS to study the change in their structural conformation. The distribution of the cells in a forward scatter vs. side scatter plot provides the overall size and shape of the cell population Fig. 6E. This distribution can be associated with cellular viability, since during apoptosis cells have reduced volume. This phenomenon of cell volume loss or cell shrinkage has been named apoptotic volume decrease and has been described as an ubiquitous aspect of apoptosis that facilitates the breakdown of the cell into smaller bodies that are eventually scavenged by macrophages.24 The two- and three-dimensional contour plots in Fig. 6C and D show the area in which the apoptotic bodies should appear as a result of induced cellular toxicity (see black arrows). While the HUVECs treated with cisplatin clearly display a major accumulation of cell fragments, both control HUVECs and HUVECs incubated with S1MPs did not show any major change in their distribution. The histograms in Fig. 6F summarize the data obtained 12 and 24 h after the treatment was started. Altogether these data confirm that the cell cycle was undisrupted and the cells did not enter a quiescent state as a consequence of exposure to S1MPs.
 | ||
| Fig. 6 Analysis of HUVEC cell cycle after exposure to S1MPs. HUVEC were exposed for 12 and 24 h to S1MPs, fixed stained with propidium iodide and analyzed by FACS to measure the DNA content and determine their distribution in the different phases of the cell cycle. A) Control cells grown in ideal conditions and cells cultured in the presence of S1MPs showed similar cell cycles and no sign of alteration of DNA content. B) Conversely, cells treated with the cytotoxic agent Cisplatin at the concentration of 50 µg mL−1 almost immediately underwent apoptosis and lost their ability to enter into mitosis. C, D) The Forward (FSC) and Side Scattering (SSC) of HUVECs treated as described before confirmed the structural integrity of the cells after 12 and 24 h (respectively) of incubation in the presence of S1MPs. Cells treated with Cisplatin were used as a positive control to verify the formation of apoptotic bodies. E) Exemplification of the 3D plots shown in C and D. F) Quantification of the percentage of cells accumulating in the areas pointed by the arrows (apoptotic bodies). | ||
It is well known that the administration of foreign material into the body can provoke acute or chronic inflammation, characterized by the release of inflammatory cytokines. For biomedical applications, drug delivery particles need to be biocompatible in terms of lack of immunogenic and inflammatory responses. Though silicon has been recognized as an essential trace element in the body which participates in the formation of connective tissue, especially cartilage and bone formation,25 some forms of crystalline silicon dioxide are known to induce toxicity in cells.26,27 Thus, it was important to assess the potential of S1MPs to induce a pro-inflammatory response in target populations.
HUVECs were treated with either positive APTES modified or near neutral PEGylated S1MPs and release of pro-inflammatory cytokinesIL-6 and IL-8 were monitored over a 24 h time period (Fig. 7). The presence of PEG on the S1MP surface inhibits cellular uptake,9 providing a comparison of S1MP internalization verses the presence of S1MPs in the cell culture media. Production of IL-6 and IL-8 was similar for cells incubated with S1MPs compared to controls. Conversely, when cells were incubated with a positive control, zymosan particles, a prominent increase in cytokine release was observed. Zymosan is a cell wall component of baker's yeast, Saccharomyces cerevisiae, which contains the immuno-modulating polysaccharidesmannan and β-1,3-glucan. These carbohydrates activate complement receptorsvia the alternative pathway, and subsequent binding of C3b/iC3b to zymosan particles enables internalization of the particles viacomplement receptors such as CR1 (CD35) and CR3 (CD11b/18) on phagocytes. The presence of complement receptors on endothelial cells mediates binding to zymosan, leading to particle engulfment and cytokines release.28
 | ||
| Fig. 7 Inflammatory response of HUVECs following engulfment of S1MPs. HUVECs were exposed to none, APTES- or PEG-5000 modified 3.2 µm S1MPs (5 : 1), or zymosan (25 µg mL−1) for 1, 4 or 24 h and the cell culture media was analyzed by ELISA for release of IL-6 (top graph) and IL-8 (bottom graph). | ||
Our results are in agreement with previously published studies, which demonstrated the biocompatibility of oxidized nanoporous silicon with primary hepatocytes.29 Choi et al30 reported a lack of cytotoxic and inflammatory responses of silicon nanoparticles compared with microparticles in RAW 264.7 murine macrophages at concentrations less than 20 µg mL−1. At elevated concentrations of silicon, microparticles were less toxic with respect to cell survival than nanoparticles, impacting cell survival only when elevated to 200 µg mL−1. Some studies have reported cellular toxicity related to cationic polymers and liposomes, for example DOTAP-containing liposomes, which has been suggested to be related to factors such as adsorption, aggregation, or to alterations in cell membrane properties by surfactant-type effects.31 However, for both silicon and silica particles, correlations with toxicity have been related to particle size and cell type, with no significant impact from surface charge.32 It has been reported that amine-derivatized mesoporous silica nanoparticles elicit no toxic effects on cells, based on LDH activity, WST-1 tests or flow cytometry.33 With respect to our porous silicon microparticles, once suspended in serum our cationic APTES silicon particles become highly negatively charged.9 We previously reported that the presence or absence of serum opsonins does not impact cellular association of APTES-modified microparticles with HUVEC endothelial cells, however, association of oxidized microparticles with cells was negatively impacted by serum opsonization.9
Conclusions
In the current study we showed that endothelial cell morphology, viability and mitosis are unaffected by internalization of porous S1MPs. Internalized S1MPs, which are encased in vesicles with tight membrane associations, irrespective of surface charge and across two sizes of S1MPs, exist predominately as single particles per endosomal unit. Mitosis, which is supported by endosomal trafficking proteins, is intact and results in even partitioning of endosomes bearing S1MPs in daughter cells. Cellular proliferation, across several days, was unaffected by vehicle- or S2NP-loaded silicon microparticles. Further supporting the safety of S1MPs is the lack of release of pro-inflammatory cytokines by endothelial cells in response to incubation with either cationic APTES-modified or PEGylated S1MPs, despite a strong response to the yeast cell wall component zymosan.The data presented here support the paradigm of endosomal clustering and polarized partitioning during mitotic events, emphasizing that biocompatible nanoporous silicon particles can function as tools for monitoring biological events. This is the first study to monitor mitotic trafficking of endosomes in the presence of microparticles. Nanoporosities render the silicon matrix degradable in biological environments, with tunable degradation rates. Biodegradability, combined with the inert nature of the material and the ability of the S1MPs to house secondary and tertiary S2NPs within the porous matrix, makes silicon an attractive option for the generation of drug delivery devices, as well as devices to sample or monitor biological processes.
Experimental
Porous S1MP fabrication
Nanoporous quasi-hemispherical S1MPs were designed and fabricated in the Microelectronics Research Center at The University of Texas at Austin. The mean particle diameter was either 1.6 ± 0.2 or 3.2 ± 0.2 µm, with pore size of either 6.0 ± 2.1 or 26.3 ± 14.6 nm. Processing details were recently published by our laboratory.5,9,10 Briefly, heavily doped p++ type (100) silicon wafers with resistivity of 0.005 Ω cm (Silicon Quest, Inc, Santa Clara, CA) were used as the silicon source. Silicon nitride (Si3N4) was deposited on the wafer by low-pressure chemical vapor deposition and standard photolithography was used to pattern the S1MPs over the wafer using a contact aligner (K. Suss MA6 mask aligner) and AZ5209 photoresist. The porous silicon particles were then produced using our proprietary two-step electrochemical etching method in a hydrofluoric acid and ethanol (1 : 3 v/v) solution.5,9Surface modification of S1MPs
The isopropyl alcohol (IPA) suspension containing S1MPs was transferred to a glass Petri dish and IPA was evaporated using a hotplate set at 60 °C overnight. The hotplate temperature was then raised to 120 °C for 15 min to insure evaporation of residual IPA from the porous matrix. The dried S1MPs were then treated with piranha solution (1 volume H2O2 and 2 volumes of H2SO4) with heating to 110–120 °C for 2 h with intermittent sonication to disperse the S1MPs. The suspension was then washed in deionized (DI) water until the pH of the suspension was ∼5.5–6.Oxidized S1MPs were washed in IPA 3–4 times. They were then suspended in IPA containing 9% (v/v) 3-aminopropyltriethoxysilane (APTES) (Sigma-Aldrich, St. Louis, MO) for 2 h at room temperature. The APTES-modified S1MPs were washed in IPA and evaluated by zeta-potential analysis. Unless specified, all experiments were performed with APTES-modified S1MPs.
For attachment of poly(ethylene glycol) (PEG) to the S1MP surface, APTES modified S1MPs were reacted with 10 mM mPEG-SCM-5000 (methoxy PEG succinimidyl carboxymethyl; purchased from Laysan Bio Inc.) in acetonitrile for 1.5 h. The S1MPs were then washed in distilled water 4–6 times to remove any unreacted mPEG. Zeta-potential measurements were used to indicate adequate surface coating.
APTES-modified S1MPs were either unloaded or loaded with gold (6 nm; Ocean NanoTech, LLC, Springdale, AR) or Fe3O4 (10 nm; Ocean NanoTech, LLC) nanoparticles (S2NPs). Loading was achieved by covalent attachment of S2NPs to the S1MPs using sulfo-N-hydroxysulfosuccinimide (NHS) (Pierce Biotechnology, Rockford, IL) and 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide (EDC) according the “Procedure for EDC/NHS Crosslinking of Carboxylates with Primary Amines” protocol supplied by Pierce Biotechnology. Briefly, 1 × 107 S1MPs were loaded with a solution containing 100 µg mL−1 S2NPs in 0.5 mL 0.1 M 2-(morpholino)ethanesulfonic acid, (MES) buffer, 0.5 M NaCl, pH 6.0.
Cell culture
HUVECs, purchased from Lonza Walkersville, Inc. (Walkersville, Maryland), were cultured in EBM®-2 Endothelial Cell Growth Medium-2 (Clonetics®, CC-3162). Human microvascular endothelial cells (HMVECs), a kind gift from Rong Shao at the University of Massachusetts, were cultured in EGM® Endothelial Cell Growth medium (Clonetics®, CC-3124). Cells were maintained at 37 °C in a humidified 5% CO2 atmosphere, and detached using 0.25 mg mL−1trypsin/EDTA solution (Clonetics®) upon reaching 80–90% confluency. For serum-free experiments with HUVECs, the media used was EBM®-2 medium (Clonetics®) supplemented with only hydrocortisone and GA-1000, plus 0.2% BSA. HUVECs were discarded after 7–8 passages.MTT cell proliferation assay
HUVECs were seeded into 96-well plates at 5000 cells/well in a final volume of 200 µL/well in EBM®-2 medium. 24 h later, media containing 3.2 µm APTES modified S1MPs, either unloaded or loaded with gold or Fe3O4 S2NPs, at a ratio of either 1 : 5 or 1 : 10 cells to S1MPs was added. At 24, 48, and 72 h, the medium was removed and media containing 0.5 mg mL−13-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma) was added at 200 µL/well for 4 h at 37 °C to the appropriate plates. The medium was then removed and dimethyl sulfoxide (180 µL/well) was added to each well. After 30 min at room temperature, absorbance was read at 570 nm using a SPECTRA max M2 plate reader (Molecular Devices).Cell cycle analysis by flow cytometry
HUVECs were grown in 25 cm2 tissue culture flasks to subconfluence. S1MPs (3.2 µm) were added to the cell culture medium at different ratios (1 : 1; 1 : 5; 1 : 10; 1 : 20), and cells were incubated using standard conditions for 12, 24, 48 and 72 h. Dissociation of cells was performed with EDTA-trypsin. Cell suspensions were centrifuged at 1000 rpm at room temperature for 5 min. The supernatants were thoroughly removed by aspiration and the tubes tapped vigorously to disperse the pellets. Fixation was carried out at 4 °C in 70% ethanol and cells were stored at −20 °C for at least 30 min before RNAse treatment and propidium iodide staining. A sterile filtered solution of 50 µg mL propidium iodide in 10 mM Tris, pH 7.3, containing 5 mM MgCl2 was slowly added with vigorously manual tapping of the tube (vortexing was avoided). One mL of PI solution was added for each l06 cells, and 50 µL of a 1.5 mg mL−1 solution of RNAse I in distilled water (Sigma) was added immediately to each of the PI-stained sample at 4 °C to give a final concentration of 75 µg mL−1 RNAse. Cell suspensions were then incubated for 1 h at 37 °C and washed three times with ice cold PBS to remove the unbound reagents, the pellet resuspended in the appropriate volume of PBS for FACS analysis. Cells were analyzed using a Becton Dickinson FACSCalibur Flow Cytometer.Enzyme-linked immunosorbent assay (ELISA)
HUVECs were seeded into 24-well plates at 50000 cells/well in a final volume of 1 mL/well in EBM®-2 medium. After 24 h, 3.2 µm APTES or PEG-500 modified S1MPs at a ratio of 5 particles per cell, or 20 µg mL−1 zymosan were added to the appropriate wells using 1 mL complete media per well. At 1, 4 and 24 h, 100 µL was removed from each well and stored at −80 °C. The volume of media removed was replaced with fresh media. Interleukin-6 (IL-6) and interleukin-8 (IL-8) were quantitated in the cell culture media using the Human IL-6 and IL-8 ELISA Kits from Cell Sciences®, according to the manufacturer's protocol. The absorbance was read using the SPECTRA Max M2450 spectrophotometer set at 450 nm.
Fluorescence and scanning confocal microscopy
HUVECs were grown on No. 1.5 glass cover slips. When confluent, cells were incubated with 3.2 µm S1MPs (1 : 5; cell:S1MP) for 120 min in serum-free media at 37 °C. Cells were then washed with PBS, fixed and premeabilized with 0.1% Triton X-100. PBS containing 1% BSA was used as a blocking agent prior to incubation with 200 nM Alexa Fluor 555 conjugated Phalloidin (Invitrogen). Coverslips were then washed and mounted on glass slides using Vectashield mounting media (Vector Laboratories, Burlingame, CA). Detection of S1MPs was based on autofluorescence following excitation at 633 nm. Images were acquired using a Leica DM6000 upright confocal microscope equipped with a 63× oil immersion objective.For live cell imaging, HMVECs were cultured in glass bottom 24-well plates purchased from MatTek Corporation (Ashland, MA) at 25000 cells/well. APTES modified S1MPs (1.6 or 3.2 µm; 1 : 20; cell:S1MPs) were added to the culture and cells were visualized using a 1X81 Olympus Microscope equipped with a DSU Confocal Attachment and a 20× objective, housed in a humidified 37 °C incubator with 5% atmospheric CO2. Five focal planes were imaged at 5 min intervals for 19 h. Still shots represent the best focal plane while the movie is comprised of projection images using in-focus light from all planes.
For quantitative analysis of S1MP content per cell with repeated cell divisions, HMVECs were grown to near confluence in a 150 mm Petri dish, then incubated with 488 Dylight (Pierce)-conjugated 3.2 µm S1MPs (1 : 10). The cells were incubated overnight to achieve complete internalization of the S1MPs by the cells. Cells were then trypsinized and sorted using the Becton Dickinson FACSDiva Flow Cytometer and Cell Sorter (BD Bioscience, San Jose, CA), gating on the population of cells with the highest fluorescence intensity. Cells were then plated in 24-well glass bottom plates. Five wells were seeded with different amounts of cells, starting from 25000 cells, followed by a 1 : 2 serial dilution to a final concentration of approximately 800 cells per well. Images of the cells were taken every 24 h for six days with a Nikon Eclipse TS 200 equipped with a 20× objective. Bright-field and fluorescence images were acquired separately for each time point and the number of particles per cell was determined manually.
Scanning electron microscopy
S1MP association with HUVECs was observed by scanning electron microscopy. Cells were plated in 24-well plates containing 5 × 7 mm Silicon Chip Specimen Supports (Ted Pella, Inc., Redding, CA) at 5 × 104 cells per well. When cells were confluent, serum-free media containing either oxidized or gold S2NP-loaded S1MPs (1 : 10, cell:S1MPs, 0.5 mL/well) were introduced and cells were incubated at 37 °C for either 15 or 60 min. Samples were washed with PBS and fixed in 2.5% glutaraldehyde for 30 min (Sigma-Aldrich; St. Louis, MO). After washing in PBS, cells were dehydrated in ascending concentrations of ethanol (30%, 50%, 70%, 90%, 95%, and 100%) for 10 min each. HUVECs were then incubated in 50% alcohol–hexamethyldisilazane (Sigma) solution for 10 min followed by incubation in pure HMDS for 5 min to prepare for overnight incubation in a desiccator. Specimens were mounted on SEM stubs (Ted Pella, Inc.) using conductive adhesive tape (12 mm OD PELCO Tabs, Ted Pella, Inc.). Samples were sputter coated with a 10 nm layer of gold using a Plasma Sciences CrC-150 Sputtering System (Torr International, Inc.). SEM images were acquired under high vacuum, at 20 kV, spot size 5.0, using a FEI Quanta 400 FEG-SEM equipped with an ETD (SE) detector.Transmission electron microscopy
HUVECs were grown to 80% confluency in a 6-well plate. Using 1 mL of serum-free media per well, 1.6 or 3.2 µm oxidized or APTES-modified S1MPs were introduced at a cell:S1MP ratio of 1 : 10 at 37 °C for 6 h. HUVECs were then washed and fixed in a solution of 2% paraformaldehyde (Electron Microscopy Sciences; Hatfield, PA) and 3% glutaraldehyde (Sigma) in 0.01 M phosphate buffered saline pH 7.4 (2 mL/well; Sigma) for one hour. After fixation, the samples were washed and treated with 0.1% cacodylate buffered tannic acid, post-fixed with 1% buffered osmium tetroxide for 30 min, and stained en bloc with 1% uranyl acetate. The samples were dehydrated using increasing concentrations of ethanol, infiltrated, and embedded in Poly-bed 812 medium. The samples were polymerized in a 60 °C oven for 2 days. Ultrathin sections were cut in Leica Ultracut microtome (Leica, Deerfield, IL), stained with uranyl acetate and lead citrate in a Leica EM Stainer, and examined in a JEM 1010 transmission electron microscope (JOEL, USA, Inc., Peabody, MA) at an accelerating voltage of 80 kV. Digital images were obtained using the AMT Imaging System (Advanced Microscopy Techniques Cory, Danvers, MA).Acknowledgements
Special thanks to our silicon fabrication team, specifically Ciro Chiappini and Jean R. Fakhoury, and the Microelectronics Research Center at The University of Texas at Austin. We wish to thank the Rice Shared Resource Facility for training and use of the scanning electron microscope and Kenneth Dunner Jr. for sample processing and TEM analysis performed at the High Resolution Imaging Facility at the University of Texas MD Anderson Cancer Center (UT-MDACC), with support from the Institutional Core Grant #CA16672. We thank Jared K. Burks for live confocal imaging performed at the UT-MDACC Flow Cytometry and Imaging Facility (Core Grant #CA16672) and Matt Landry for help in assembling high resolution figures and pseudo-coloring of SEM images. This research was supported by the Department of Defense, grants DODW81XWH-07-1-0596, DODW81XWH-09-1-0212 and DODW81XWH-07-2-0101; NASA NNJ06HE06A; NIH RO1CA128797; and State of Texas, Emerging Technology Fund.References
- J. Salonen, A. M. Kaukonen, J. Hirvonen and V. P. Lehto, Mesoporous silicon in drug delivery applications, J. Pharm. Sci., 2008, 97(2), 632–653 CrossRef CAS.
- J. Salonen, L. Laitinen, A. M. Kaukonen, J. Tuura, M. Bjorkqvist, T. Heikkila, K. Vaha-Heikkila, J. Hirvonen and V. P. Lehto, Mesoporous silicon microparticles for oral drug delivery: loading and release of five model drugs, J. Controlled Release, 2005, 108(2–3), 362–374 CrossRef CAS.
- E. J. Anglin, L. Cheng, W. R. Freeman and M. J. Sailor, Porous silicon in drug delivery devices and materials, Adv. Drug Delivery Rev., 2008, 60(11), 1266–1277 CrossRef CAS.
- L. T. Canham, Bioactive silicon structure fabrication through nanoetching techniques, Adv. Mater., 1995, 7, 1033–1037 CAS.
- E. Tasciotti, X. Liu, R. Bhavane, K. Plant, A. D. Leonard, B. K. Price, M. M. Cheng, P. Decuzzi, J. M. Tour, F. Robertson and M. Ferrari, Mesoporous silicon particles as a multistage delivery system for imaging and therapeutic applications, Nat. Nanotechnol., 2008, 3(3), 151–157 CrossRef CAS.
- E. Ruoslahti, Vascular zip codes in angiogenesis and metastasis, Biochem. Soc. Trans., 2004, 32(3), 397–402 CrossRef CAS.
- S. H. C. Anderson, H. Elliot, D. J. Wallis, L. T. Canham and J. J. Powell, Dissolution of different forms of partially porous silicon wafers under simulated physiological conditions, Phys. Status Solidi A, 2003, 197, 331–335 CrossRef CAS.
- L. T. Canham, C. L. Reeves, J. P. Newey, M. R. Houlton, T. I. Cox, J. M. Buriak and M. P. Stewart, Derivatized mesoporous silicon with dramatically improved stability in simulated human blood plasma, Adv. Mater., 1999, 11, 1505–1507 CrossRef CAS.
- R. E. Serda, J. Gu, R. C. Bhavane, X. Liu, C. Chiappini, P. Decuzzi and M. Ferrari, The association of silicon microparticles with endothelial cells in drug delivery to the vasculature, Biomaterials, 2009, 30(13), 2440–2448 CrossRef CAS.
- E. T. C. Chiappini, J. R. Fakhoury, D. Fine, L. Pullan, Y. Wang, L. Fu and M. Ferrari, Tailored porous silicon microparticles for the delivery of nanoparticles, submitted.
- P. Decuzzi and M. Ferrari, Design maps for nanoparticles targeting the diseased microvasculature, Biomaterials, 2008, 29(3), 377–384 CrossRef CAS.
- P. Decuzzi, S. Lee, B. Bhushan and M. Ferrari, A theoretical model for the margination of particles within blood vessels, Ann. Biomed. Eng., 2005, 33(2), 179–190 CrossRef CAS.
- P. Decuzzi, R. Pasqualini, W. Arap and M. Ferrari, Intravascular delivery of particulate systems: does geometry really matter?, Pharm. Res., 2009, 26(1), 235–243 CrossRef CAS.
- R. E. Serda, J. Gu, J. K. Burks, C. Ferrari, K. Ferrari and M. Ferrari, Quantitative mechanics of endothelial phagocytosis of silicon microparticles, Cytometry Part A, 2009, 75A, 752–760 CrossRef CAS.
- S. Loubéry, C. Wilhelm, I. Hurbain, S. Neveu, D. Louvard and E. Coudrier, Different microtubule motors move early and late endocytic compartments, Traffic, 2008, 9(4), 492–509 CrossRef CAS.
- T. Bergeland, J. Widerberg, O. Bakke and T. W. Nordeng, Mitotic partitioning of endosomes and lysosomes, Curr. Biol., 2001, 11(9), 644–651 CrossRef CAS.
- R. Albertson, J. Cao, T. S. Hsieh and W. Sullivan, Vesicles and actin are targeted to the cleavage furrow via furrow microtubules and the central spindle, J. Cell Biol., 2008, 181(5), 777–790 CrossRef CAS.
- S. G. Zeitlin and K. F. Sullivan, Animal cytokinesis: breaking up is hard to do, Curr. Biol., 2001, 11(13), R514–516 CrossRef CAS.
- G. Montagnac and P. Chavrier, Endosome positioning during cytokinesis, Biochem. Soc. Trans., 2008, 36(3), 442–443 CrossRef CAS.
- R. Shao and X. Guo, Human microvascular endothelial cells immortalized with human telomerase catalytic protein: a model for the study of in vitro angiogenesis, Biochem. Biophys. Res. Commun., 2004, 321, 788–794 CrossRef CAS.
- A. Krishan, Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining, J. Cell Biol., 1975, 66(1), 188–193 CrossRef CAS.
- P. Pozarowski and Z. Darzynkiewicz, Analysis of cell cycle by flow cytometry, Methods Mol. Biol, .2004, 281, 301–311 Search PubMed.
- J. Z. Fuks, J. Aisner, D. A. Van Echo, H. Schipper, M. Levitt, S. Ostrow and P. H. Wiernik, Randomized study of cyclophosphamide, doxorubicin, and etoposide (VP16-213) with or without cisplatinum in non-small cell lung cancer, J. Clin. Oncol., 1983, 1(5), 295–301 CAS.
- C. D. Bortner and J. A. Cidlowski, Apoptotic volume decrease and the incredible shrinking cell, Cell Death Differ., 2002, 9(12), 1307–1310 CrossRef CAS.
- M. P. Absher, L. Trombley, D. R. Hemenway, R. M. Mickey and K. O. Leslie, Am. J. Pathol., 1987, 134, 1243–1251 Search PubMed.
- J. M. Buriak and M. J. Allen, J. Am. Chem. Soc., 1998, 120, 1339 CrossRef CAS.
- V. Kolb-Bachofen, J. Clin. Invest., 1992, 90, 1819–1824 CrossRef CAS.
- H. Langeggen, E. Namork, E. Johnson and G. Hetland, HUVEC take up opsonized zymosan particles and secrete cytokines IL-6 and IL-8 in vitro, FEMS Immunol. Med. Microbiol., 2003, 36(1–2), 55–61 CrossRef CAS.
- V. Chin, B. E. Collins, M. J. Sailor and S. N. Bhatia, Adv. Mater., 2001, 13, 1877–1880 CrossRef CAS.
- J. Choi, Q. Zhang, V. Reipa, N. S. Wang, M. E. Stratmeyer, V. M. Hitchins and P. L. Goering, Comparison of cytotoxic and inflammatory responses of photoluminescent silicon nanoparticles with silicon micron-sized particles in RAW 264.7 macrophages, J. Appl. Toxicol., 2009, 29(1), 52–60 CrossRef CAS.
- D. D. Lasic, Liposomes in gene delivery, CRC Press, Boca Raton, 1997, pp. 107–109 Search PubMed.
- L. C. Thomassen, A. Aerts, V. Rabolli, D. Lison, L. Gonzalez, M. Kirsch-Volders, D. Napierska, P. H. Hoet, C. E. Kirschhock and J. A. Martens, Synthesis and Characterization of Stable Monodisperse Silica Nanoparticle Sols for in vitro Cytotoxicity Testing, Langmuir, 2009 Search PubMed , in press.
- M. Fisichella, H. Dabboue, S. Bhattacharyya, M. L. Saboungi, J. P. Salvetat, T. Hevor and M. Guerin, Mesoporous silica nanoparticles enhance MTT formazan exocytosis in HeLa cells and astrocytes, Toxicol. in Vitro, 2009, 23(4), 697–703 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary movies 1 and 2. See DOI: 10.1039/b9nr00138g |
| ‡ Shared first authorship. |
| This journal is © The Royal Society of Chemistry 2009 |