Naturally diverse: highlights in versatile synthetic methods enabling target- and diversity-oriented synthesis
Jared T. Shaw*
Department of Chemistry, University of California, One Shields Ave, Davis, CA, USA. E-mail: shaw@chem.ucdavis.edu; Tel: +011 (530) 752-9979
First published on 27th November 2008
Abstract
Covering: January 1993 to August 2008
This Highlight describes synthetic methods that have enabled new strategic approaches to the synthesis of natural product targets as well as libraries of compounds for unbiased screening experiments. The structural differences of the compounds produced by both efforts are illustrated.
 Jared Shaw | Jared Shaw received his B.S. from UC Berkeley (1993), and his Ph.D. from UC Irvine (1999) with Professor Keith Woerpel. He was an NIH postdoctoral fellow with David Evans at Harvard University (1999–2002) and an institute fellow at the Broad Institute of Harvard and MIT (2002–2007) before joining UC Davis. Prof. Shaw's research interests include multicomponent reactions, natural product synthesis, and chemical biology. |
1 Introduction
The synthesis of complex molecules has long been an important goal in organic chemistry. Targets often include molecules of medicinal, and biosynthetic, or physical/computational importance. The advent of combinatorial chemistry and nearly concurrent adoption of high-throughput screening in academia have opened up new vistas in the fields of chemical biology and systems biology.1 Many biologically active molecules discovered to date are derived directly or indirectly from natural sources2 and, as a result, many synthetic methods are developed to enable the construction of natural products. Diversity-oriented synthesis (DOS) emerged as a strategy to capitalize on the discovery potential of small molecules in unbiased screening experiments by enabling the rapid and parallel assembly of compound collections. Central to the goal of DOS is the assembly of organic molecules with diverse core structures, functional groups, and appendages.The construction of both complex natural products and structurally varied libraries depends on robust, flexible, and stereoselective synthetic methods. The conflux of natural product synthesis and DOS has resulted in the adaptation of many traditional methods to the preparation of “natural product-like” libraries. The strategy of making libraries of compounds that resemble natural products has been reviewed extensively.3,4 In a complementary fashion, DOS has tested the limits of many synthetic methods that produce complex unnatural products.5 This Highlight will review recent examples of synthetic methods that have enabled both natural product synthesis and DOS. In most cases, the end products of DOS bear little functional resemblance to the natural products made by the same strategies. The methods discussed have advanced both goals and underscore the fact that versatile synthetic methods are the key to success in the assembly of complex molecules, regardless of their origin or their intended application.
2 Assembly of complex heterocycles using the Schmidt reaction (Aubé)
The Aubé group has discovered fascinating and powerful intramolecular variants of the Schmidt reaction that enable the rapid assembly of complex lactams and oxazolines (Fig. 1). The initial discovery that hydroxyazides will temporarily tether to ketones and undergo azide addition and rearrangement via transient oxonium ion 2 has led to many applications.6 Equally powerful is the installation of an azide with a carbon tether to enable an intramolecular Schmidt reaction.7 Although it is outside the scope of this Highlight, this reaction was recently employed as the key transformation in the synthesis of 2-quinuclidone, a synthetic target that had eluded preparation for over 60 years.8 In several recent examples, this reaction is conducted in tandem with a Diels-Alder reaction between ketone- and azide-containing fragments to induce the required proximity.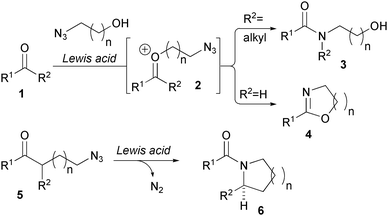 | ||
| Fig. 1 Intramolecular Schmidt reactions (Aubé). | ||
The intramolecular Schmidt reaction has enabled the efficient assembly of several complex alkaloid natural products isolated from Dendrobatidae or “poison dart” frogs in Central and South America (Fig. 2). In one of the first applications of the intramolecular Schmidt reaction, the Aubé group completed an enantioselective synthesis of dendrobatid alkaloid 209B.9 Cyclopentanone 7, prepared from pulegone, is converted to keto-azide 8, which is smoothly converted to the desired lactam 9 in high yield and with high diastereoselectivity. The remaining n-pentyl side chain is introduced in two steps to complete the synthesis of 209B (10). Nearly a decade later, the Aubé group disclosed an impressively efficient and selective route to alkaloid 251F (15, Fig. 3).10 Tandem ring-opening and ring-closing metathesis (ROM/RCM) of Diels-Alder product 11 produced bicyclo-octenone 12, which contains three of the requisite stereogenic centers for 15. This material was converted to keto-azide 13 and employed in an intramolecular Schmidt rearrangement to yield 14 in high yield as a single diastereomer. Reduction of the lactam provided alkaloid 251F in an overall sequence of 13 steps.
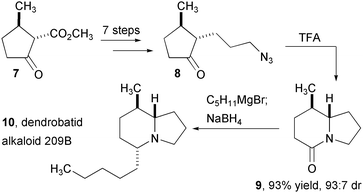 | ||
| Fig. 2 Synthesis of dendrobatid alkaloid 209B using the intramolecular Schmidt reaction. | ||
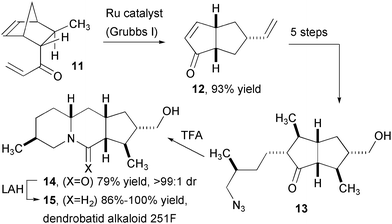 | ||
| Fig. 3 Synthesis of dendrobatid alkaloid 251F. | ||
The intramolecular Schmidt reaction also provides rapid access to polycyclic alkaloids of plant origin (Fig. 4). (+)-Aspidospermidine was efficiently assembled by intercepting the tricyclic ketone previously used by Stork in a Fischer indole synthesis.11 Bicyclic azido-ketone 18 was prepared in a straightforward sequence from an asymmetric Robinson annulation. Schmidt reaction of 18 using TiCl4 provided the desired ketone with a high level of both stereo- and regio-selectivity for the more distant ketone carbonyl. Stork's ketone 19b was produced in three more steps and the final product is accessed three steps thereafter. The use of a Diels-Alder reaction to assemble a Schmidt precursor from azide- and ketone-appended fragments provided a stunningly efficient assembly of the Stemona alkaloids stenine (25) and neostenine (27, Fig. 5). Enolsilane 21 and cyclohexanone (22) were combined with SnCl4, which effected both the cycloaddition and the subsequent rearrangement to provide tricyclic lactam 24 in high yield, with a 75:25 preference for exo approach of the dienophile. This one-pot assembly of the core of stenine installed three out of four rings and four stereogenic centers! A short sequence completed the most efficient preparation of this complex alkaloid reported. In an analogous fashion, the diastereoselectivity of the Diels-Alder reaction was switched to favor the endo product 26, which was converted to neostenine 27 in 9 steps.
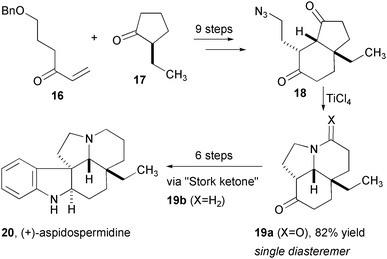 | ||
| Fig. 4 Synthesis of aspidospermine. | ||
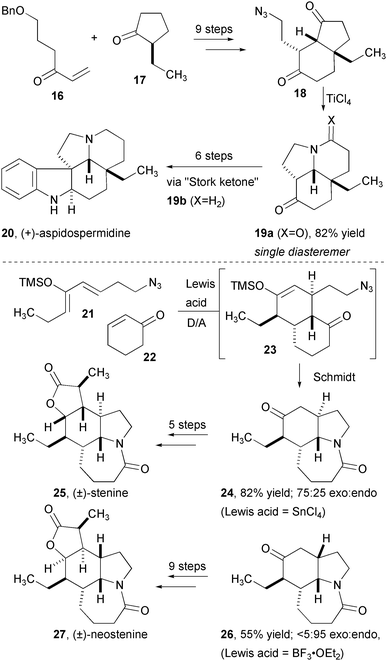 | ||
| Fig. 5 Synthesis stenine and neostenine using tandem Diels-Alder/Schmidt reactions. | ||
The intramolecular reactions of alkyl azides with carbonyl compounds has proven equally powerful for the preparation of diverse collections of small molecules (Fig. 6). The use of heterocyclic ketones and amino acid-derived hydroxyalkyl azides initially provided access to heterocyclic compounds aimed at mimicking peptide γ-turns.12 In this case, the Schmidt reaction is arrested at the iminium ether stage (32) and subsequent hydrolysis reveals the requisite N-hydroxyethyl 1,4-diazepin-5-one 33. After demonstrating the synthesis of a series of peptide mimics compounds, which could be used to modulate a variety of individual biological targets, Aubé and co-workers examined the breadth of reactivity associated with the iminium ether intermediates (Fig. 7).13 Their studies revealed that nucleophiles could attack in two different modes, namely Path A or Path B, to result in alkoxy group or iminium ion cleavage. Sodium borohydride and sulfide result in replacement of the carbonyl with methylene or thiocarbonyl groups, respectively, by way of Path A. Similarly, substituted amidines and dimethyl hydrazone were cleanly produced from corresponding amines and dimethyl hydrazine. Nucleophiles such as azide, cyanide, phenoxide and thiophenoxide reacted by Path B, as did soluble sources of fluoride and iodide. Active methylene compounds exhibit divergent reactivity wherein malononitrile produce the exo-enamine compound (Path A) and dimethylmalonate and bis(phenylsulfonyl)-methane produce N-alkylated amides (Path B).
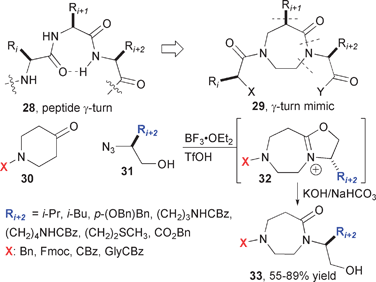 | ||
| Fig. 6 Synthesis of γ-turn mimics and diverse 7-membered ring heterocycles. | ||
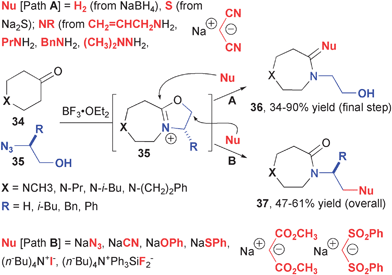 | ||
| Fig. 7 Regioselective nucleophilic opening of cyclic iminium ether intermediates of Schmidt reactions. | ||
Tricyclic amides based on 26 also offer an excellent launch point for DOS through regio- and diastereo-selective reactions of the ketone carbonyl (Fig. 8).14 Reductive amination with secondary amines, or with aniline followed by an aldehyde, provided access to amino-amides 38 with little control of selectivity in the formation of the new stereogenic center. In the step-wise method, the anilines are separable, providing divergent access to either diastereomer. In contrast, the carbonyl was selectively reduced to either the 10,11-syn or 10,11-anti alcohol, which is then acylated with a variety of isocyanates. In a complementary fashion, 26 was prepared with the epimeric configuration at the carbon appended with R1 and carried on to the corresponding diastereomers of 39 and 40 with opposite configuration at C10 and C11. Ketone 26a (R1 = H) was also used in regioselective syntheses of indoles 41 and quinolines 42 using Fischer and Friedländer protocols, respectively. Although the core structure of these compounds is based on the diversification of a synthetic intermediate from the stenine synthesis, it is unlikely that any would exhibit similar biology given that most lack a basic nitrogen and have different three-dimensional shapes. Stenine and the related alkaloids are related to compounds 38–42 more by the method from which they emerge than any biologically relevant structural similarity.
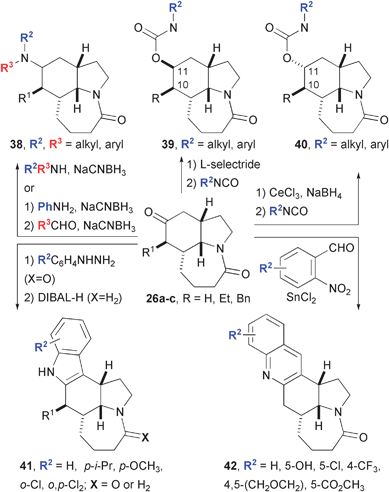 | ||
| Fig. 8 Diversification of intramolecular Schmidt reaction product 26. | ||
3 Multicomponent reaction (MCR) and hetero-Diels-Alder strategies (Batey)
The use of hetero-Diels-Alder reactions related to an initial discovery by Povarov provides the foundation for selective routes to tetrahdroquinolines and related structures (Fig. 9).15 Typically “aza-dienes” are formed from a condensation reaction between aldehydes 43 and anilines 44. A subsequent cycloaddition with an electron-rich alkene followed by re-aromatization produces the heterocyclic product. Batey has disclosed several variants of this reaction that are useful for the synthesis of natural products and diversely-substituted heterocycles.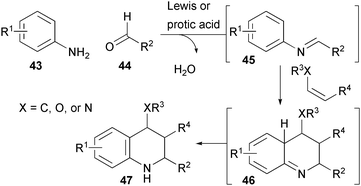 | ||
| Fig. 9 Tetrahydroisoquinoline synthesis from anilines, aldehydes, and alkenes (Batey). | ||
Batey and co-workers reported one of the earliest and most efficient syntheses of the alkaloids martinelline and martinellic acid (Fig. 10). These compounds were first isolated at Merck from the roots of Martinella iquitosensis based on the use of extracts from this vine by the indigenous inhabitants of the Amazon basin to treat eye infections.16 The heterocyclic core of these alkaloids, with just three fused rings and three stereogenic centers, have been described as “deceptively simple” and the diverse array of synthetic strategies aimed at their synthesis supports this assessment.17 Batey and co-workers recognized a tacit symmetry of the heterocyclic core that enabled rapid synthesis from a Povarov-type reaction wherein a pyrroline serves as both aldehyde synthon and dienophile.18 The diastereoselectivity could be tuned to favor either the endo or exo product.
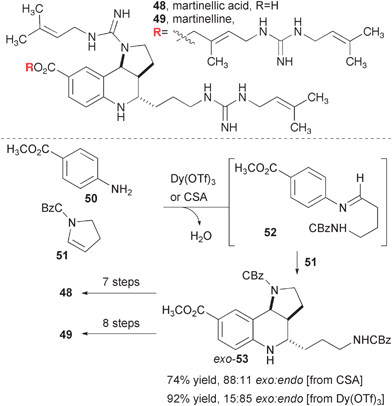 | ||
| Fig. 10 Synthesis of martinellic acid and martinelline. | ||
The hetero-Diels-Alder reactions of aniline imines has also proven effective for the assembly of the quinoline rings of luotonin A and camptothecin (Fig. 11).19 Aldehyde 54, assembled in 8 steps from isatoic anhydride, is converted to lutonin A upon treatment with aniline and Dy(OTf)3, which has proven optimal for this reaction in comparison to other Lewis acids. A similar reaction between aniline and pyridone 56, prepared from a multicomponent condensation in three steps, leads to nitrile 57, which is reported to be a precursor for the antitumor alkaloid camptothecin. In each case the intramolecular Povarov reaction enabled a one-step installation of three rings.
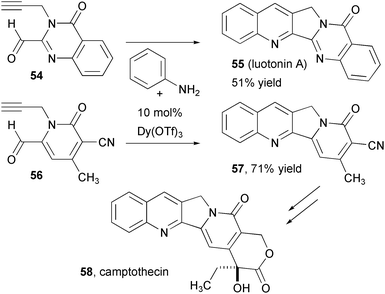 | ||
| Fig. 11 Synthesis of lutonin and formal synthesis of camptothecin using intramolecular variants of the Povarov reaction. | ||
Batey has also developed a series of MCRs that lead to martinelline analogs20 and related polycyclic structures (Fig. 12).21 In preliminary work that eventually led to the martinelline synthesis, a 2:1 adduct (62) formed from the reaction of dihydrofuran (60) and p-substituted anilines 43 was isolated in variable yield and diastereoselectivity. Electron-rich dienophiles are usually required for the Povarov reaction of aliphatic aldehydes in order to suppress enamine formation and the corresponding Doebner-von Miller side reaction (not shown). The Dy(OTf)3 catalyst in combination with slow addition of the aldehyde enables high yields and diastereoselectivities of a 3CR between anilines, aldehydes, and cyclopentadiene. Tricyclic tetrahydroquinoline 63 could prove useful for the synthesis of a variety of more complex structures that could emerge from reactions of the side chain and/or the cyclopentene double bond.
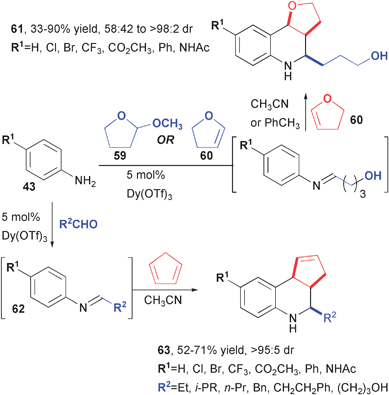 | ||
| Fig. 12 Synthesis of diverse heterocycles using the dysprosium-catalyzed Povarov reaction. | ||
4 Divergent transition metal-catalyzed reactions of alkynes and allenes (Brummond)
Cyclizations between allenes and alkenes or alkynes provide a wealth of routes to heterocyclic and carbacyclic compounds (Fig. 13). The Brummond group initially explored an allene variant of the Pauson-Khand (PK) reaction and found that the allene substitution dictated the formation of two possible regio-isomers (65 and 66) of alkylidene cyclopentenone formation.22 In a later study, Rh(I) catalysis was investigated and found to generally favor reaction at the distal π-bond of the allene (67 to 68).23 Moreover, when CO was excluded from the reaction mixture, a complementary allenic Alder-ene reaction (67 to 69) was observed from the same substrate and catalyst combination.24 Replacement of the alkyne appendage in the substrate leads to the formation of medium ring heterocycles 71.25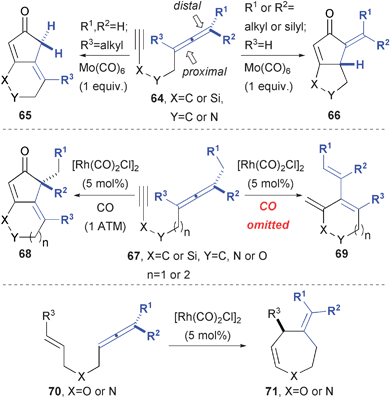 | ||
| Fig. 13 Transition metal-catalyzed and mediated reactions of alkyne- and alkene-tethered allenes. | ||
Natural products targeting cancer have provided a rich area for discovery in recent years, and the illudins are recent examples of secondary metabolites that have led to the discovery of a cancer drug, namely hydroxymethylacylfulvene (HMAF/Irofulven™).26 This compact and challenging structure has been the subject of several synthetic efforts27 and the Brummond approach using the allenic PK reaction was one of the earliest (Fig. 14).28 Allene-yne 75, available in five steps from 1,1-diacetyl cyclopropane 72, was converted to the anticipated cyclopentenone wherein the external alkene reacts, based on the disubstitution of the internal allene terminus. The bicyclic product was converted in three more steps to racemic HMAF. An enantioselective formal synthesis of either enantiomer using a Sharpless asymmetric dihydroxylation to access a single enantiomer and diastereomer of 75 was later reported.29
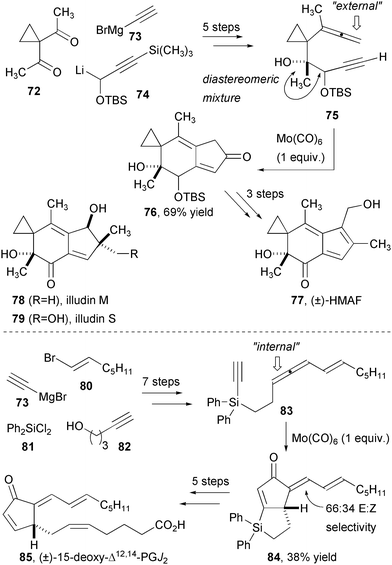 | ||
| Fig. 14 Synthesis of HMAF and a cyclopentenone prostaglandin using complementary PK reactions. | ||
The complementary PK reaction was employed in the synthesis of the 15-deoxy-Δ12,14-prostaglandin J2 (Fig. 14). The cyclopentenone subclass of natural products has attracted recent interest based on their atypical biological profile relative to other prostaglandins.30 Silicon-tethered PK substrate 83 was convergently assembled in seven steps. Monosubstitution at the internal carbon of the allene enforced reactivity at that same carbon in the molybdenum-mediated PK reaction. Although the yield and selectivity for E-alkene 84 are modest, the product contains almost all of the necessary functionality for the synthesis of the target. Moreover, the undesired Z isomer can be equilibrated photolytically to a 1:1 E:Z mixture to increase the yield of the desired compound. The silicon tether was proto-desilylated at the vinyl carbon and then oxidized α to the alkyl carbon to produce an alcohol that was oxidized for installation of the Z-alkene side chain.
The ability of the rhodium-catalyzed allenyl PK reaction to assemble 7–5-fused ring systems has enabled an efficient synthesis of the diterpenoid skeleton common to the guanacastepenes (Fig. 15). These antimicrobial diterpenoids were recently isolated from and endophytic fungus inhabiting the D. americana tree in the Guanacaste region of Costa Rica31 and have been the subject of many recent synthetic efforts.32 Unlike 6–5 and 5–5 ring systems, the 7–5 has been difficult to construct with the PK reaction.33 Allene-yne 89 was assembled from Smith's enone in ten steps with undetermined relative configuration between the two stereogenic centers of the six-membered ring. Standard rhodium-catalyzed PK conditions produced tricyclic enone 90 containing most of the oxygenation and all but one of the carbons of guanacastepene A. Although subsequent attempts to install the final angular methyl group at the A–B ring junction were unsuccessful, the efficiency with which the skeleton was fashioned from simple precursors is notable. A subsequent report details the rapid synthesis of a variety of related tricyclic products.34
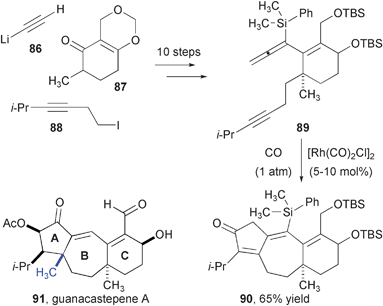 | ||
| Fig. 15 Synthesis of the guanacastepene framework. | ||
The high level of versatility achieved by obtaining structurally different products from the same starting material by choice of catalyst has allowed Brummond's metal-catalyzed and mediated reactions to be extremely useful for DOS (Fig. 16).75 Indeed, the author has drawn an apt analogy between the divergent structures obtained from a common allene/alkyne precursor to the diverse sesquiterpenoids that are formed biosynthetically from farnesyl pyrophosphate (FPP) when acted on by various enzymes. The common starting materials are derived from α-amino esters (93) and propargyl bromides. Allene installation was achieved by Claisen-type reactions of the amino esters. For R3 = Bz, the diastereoselectivity between the stereogenic center and the allene configuration was low (75:25), but a range of R2 substituents was tolerated. For R3 = CBz or Boc, diastereoselectivity was high, but only R2 substitution was limited to TMS. The resultant allenyl esters could be N-propargylated in 64–94% yield to provide substrates 94 in four steps total.
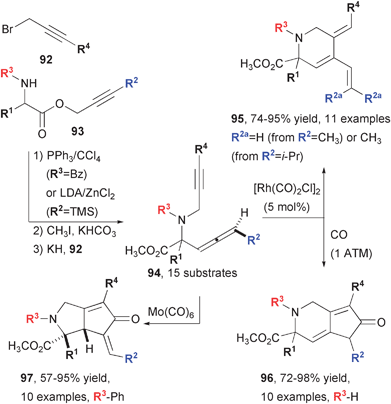 | ||
| Fig. 16 Synthesis of diverse heterocycles from a common allene-yne precursor. | ||
Allene-yne substrates were then subjected to three different cyclization reactions. The rhodium-catalyzed Alder-ene reaction was the most functional group-compatible of the three reactions and delivered the cross-conjugated trienes 95 in high yields (74–95%). In the case of the PK reactions, the diastereoselectivity between the two stereogenic centers, appended with R1 and R2, becomes an issue. Although there is a high level of stereochemical translation from 94 to 96 and 97, the low selectivity in the formation of the Bz-substituted starting materials resulted in complex product mixtures of these products. In the case of the rhodium-catalyzed reactions, terminal alkyne substitution is required (R4 ≠ H) in the formation of 96. For the molybdenum-mediated synthesis of the bicyclo-octenones 97, the E/Z alkene mixtures were problematic, and substrates with no terminal allene substitution (R2 = H) were best. Despite these shortcomings, the demonstration of a high level of control over diverse reaction products from a common precursor by choice of catalyst establishes a powerful strategy for DOS. A related approach was subsequently reported by Schreiber.35
The products of the PK and ene-type reactions provide excellent starting points for further diversification in a library synthesis setting (Fig. 17).76 The 5–5 bicyclic product of the molybdenum reaction (98) is an excellent substrate for a Stetter reaction to form 1,4-diketones 99. After reduction of the cyclopentenone double bond, the 1,4-diketone feeds directly into a Paal-Knorr pyrrole synthesis using a primary amine. This strategy was employed in the synthesis of a 178-membered library of tricyclic products with a diverse array of appendages. A subset of these compound were found to inhibit MAP kinase phosphatase (MKP-1) selectively over related phosphatases with IC50 values of 8–20 µM.36 A second library exploited the use of common allenyl alcohol 101 that was appended with either an alkyne or alkene for use in Alder ene-type reactions to produce dihydropyrans and oxepanes, respectively.77
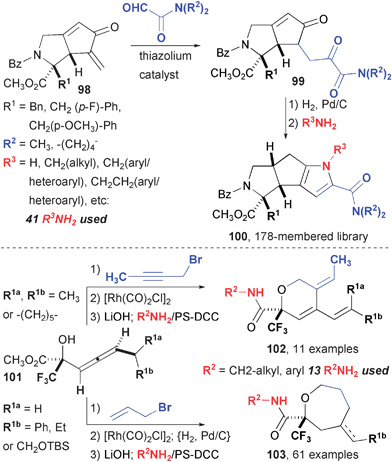 | ||
| Fig. 17 DOS pathways using common substrates and molybdenum-mediated and rhodium-catalyzed reactions. | ||
Finally, the development of an Alder-ene reaction using propiolamides37 to produce alkylidene dihydropyridones enabled the use of these cross-conjugated dienes in Diels-Alder cycloadditions (Fig. 18).78 Schreiber had previously demonstrated the ability for related substrates to undergo single or tandem cycloaddition reactions with either one or two equivalents of an electron-deficient dienophile.38 Treatment of substrates related to 104 with phosgene produced mixtures of the expected imidate products as well as imidates that also underwent an unexpected ene-carbamate formation. This mixture was treated with a variety of maleimides to form polycyclic imides 106 and 107, which were subsequently hydrogenated and further functionalized (not shown). In the event that the maleimide was in excess, double Diels-Alder adduct 107b was also observed in up to 35% yield.
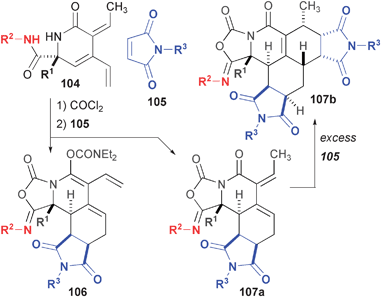 | ||
| Fig. 18 DOS pathways based on Diels-Alder reaction of Alder-ene products 104. | ||
5 Allylsilane addition and annulation reactions (Panek)
The stereoselective reactions of allylsilanes with various electrophiles has emerged as a powerful strategy for the assembly of complex molecules (Fig. 19). Research in the Panek group has focused on the use of enantiomerically pure allylsilanes related to 108 that are produced through Claisen rearrangement of appropriately silyl-substituted allylic alcohols. These nucleophiles undergo diastereoselective addition to aldehydes and other C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X π-bonds. Although the resultant double bond was oxidatively cleaved for the synthesis of polypropionates and related structures in early synthetic applications, recent examples make full use of the carbons installed using this reaction. An interesting variant of this reaction was recently reported that produces enantiomerically pure, trisubstituted 6-membered ring heterocycles. If the allylsilane is appended with either OTMS or NH2, condensation followed by intramolecular addition forms either the dihydropyran (DHP) or tetrahydropyridine, respectively. A high level of diastereoselectivity is observed in the translation of the two adjacent stereogenic centers in the starting material and the newly formed stereogenic centers of the product. In addition, nearly perfect transmission of absolute configuration is observed.
X π-bonds. Although the resultant double bond was oxidatively cleaved for the synthesis of polypropionates and related structures in early synthetic applications, recent examples make full use of the carbons installed using this reaction. An interesting variant of this reaction was recently reported that produces enantiomerically pure, trisubstituted 6-membered ring heterocycles. If the allylsilane is appended with either OTMS or NH2, condensation followed by intramolecular addition forms either the dihydropyran (DHP) or tetrahydropyridine, respectively. A high level of diastereoselectivity is observed in the translation of the two adjacent stereogenic centers in the starting material and the newly formed stereogenic centers of the product. In addition, nearly perfect transmission of absolute configuration is observed.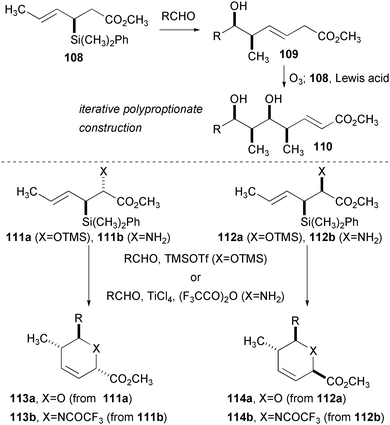 | ||
| Fig. 19 Diastereoselective aldehyde addition and annulation reactions of chiral allylsilanes. | ||
The allylsilane annulation reaction has been useful in several recent syntheses of complex tetrahydropyran (THP)-containing natural products,39 including leucascandrolide (121, Fig. 20). The interesting biological activity of this compound paired with the fact that it can no longer be isolated from its original source40 have made it the target of intense synthetic effort.41 Panek's approach used an allylsilane annulation to assemble each of the two THP rings. The first ring was prepared from a syn-selective annulation using silane 116. After four more steps, the second ring is introduced with high anti-selectivity using allylsilane 119. At this point the final ring is closed after five more steps. The high efficiency of this route (17 steps from 115 and 116) is notable.
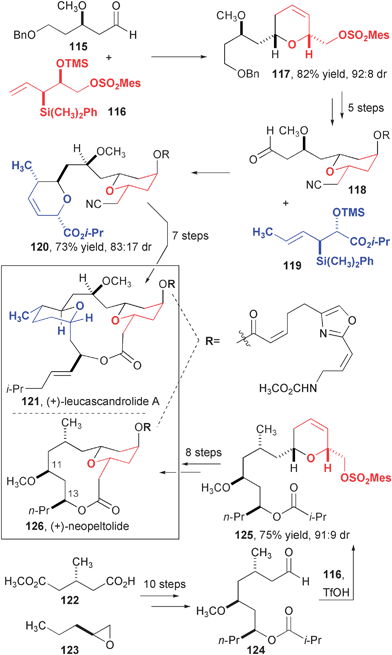 | ||
| Fig. 20 Synthesis of leucascandrolide and neopeltolide. | ||
Neopeltolide is an interesting THP-containing natural product recently isolated by Wright,42 which shares one THP ring and the oxazole-ester side chain with leucascandrolide. Panek targeted this compound based on its potent cytotoxicity, which was similar to that of leucascandrolide. Unpublished initial studies focused on the proposed structure of neopeltolide, which exhibited marked differences in the 1H- and 13C-NMR spectra. Upon close inspection of the structure, the authors inferred that the configuration of carbons 11 and 13 were misassigned. Complex aldehyde 124, which contained many of the stereogenic centers of the final product, was prepared and carried into an annulation reaction with 116, the same allylsilane used to prepare leucascandrolide. This key step occurred with high diastereoselectivity and a short sequence completed the synthesis and confirmed the structure of (+)-neopeltolide. The same conclusion regarding the structure of this compound was independently reached by Scheidt.43
Although the majority of natural product syntheses using allylsilane addition and annulation reactions has been directed at polyoxygenated compounds, the amino-allylsilane annulation enables the synthesis of polycyclic alkaloids (Fig. 21). Condensation of 127 with 5-(acyloxy)pentanal provided tetrahydropyridine 128 in high yield as a single diastereomer. This intermediate, which features all of the required stereogenic centers, was rapidly converted to alkaloid 217A.
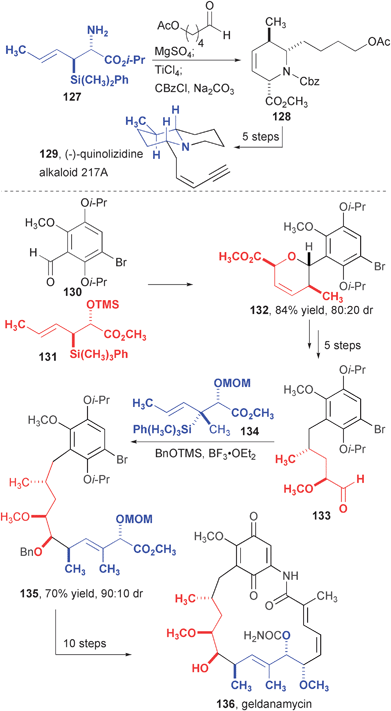 | ||
| Fig. 21 Synthesis quinolizidine alkaloid 217A and geldanamycin. | ||
Geldanamycin is produced in an efficient synthesis using allylsilane addition and annulation reactions as key carbon-carbon bond constructions. Geldanamycin is an antitumor compound that derives its activity from inhibition of Hsp90, and an analog of this compound is currently under clinical development.44 The structure is common to the ansamycin class of natural products that includes macbecin I45 and reblastatin.46 In the case of geldanamycin, it was recognized that two successive allylsilane reactions could assemble nine contiguous carbons of the backbone. Allylsilane annulation to aromatic aldehyde 130 in modest diastereoselectivity provided dihydropyran 132. This compound was opened by a reductive C–O bond cleavage and carried on to aldehyde 133, which was used in an allylsilane addition reaction to install the next 5 carbons in 70% yield and high (90:10) diastereoselection. Under the reaction conditions, which included the addition of BnOTMS, three stereogenic centers, a tri-substituted alkene, and the benzyl protecting group were installed in a single operation! Geldanamycin was formed in ten more steps, including a copper-mediated aryl amidation to close the large ring.
Allylsilane addition and annulation both enable highly efficient DOS strategies for creating libraries of complex compounds for screening (Fig. 22). Allylsilane addition to either a formaldehyde or glycoaldehyde equivalent yields hydroxy-terminated ester chains 137 and 139 that are suitable for macrodiolide formation using a fluorinated distannoxane transesterification catalyst.47 Shorter chain ester 137 provided 14-membered ring product 138 in 80% yield and with a >90:10 preference for dimer versus trimer. Similarly, 139 provided the 16-membered ring macrodiolide 140 in high yield. While these two products are highly reminiscent of closely related macrodiolide natural products, the method is applicable to a range of compounds unlike any known secondary metabolites. For example, an amino acid residue can be inserted for the synthesis of larger symmetrical compounds (141 to 142). Also, there were many cases in which unsymmetrical compounds could be made in high yield over the two potential symmetrical compounds. For instance, proline-derived ester 143 was successfully coupled to a series of monomers to produce a range of ring sizes (15–21 atoms) in high yields, considering the possibility for oligomerization.
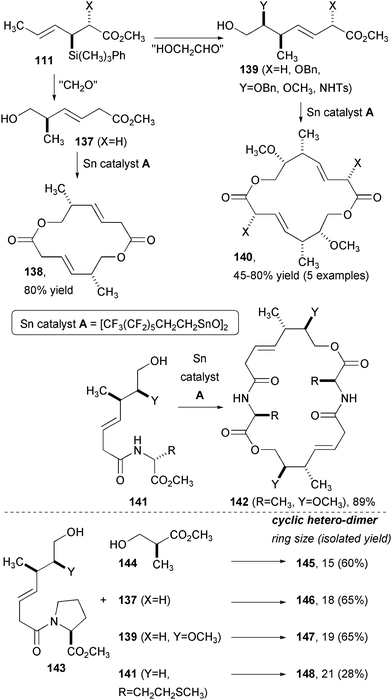 | ||
| Fig. 22 Synthesis of homo- and hetero-dimeric macrodiolides from allylsilane addition products. | ||
The tetrahydropyridines derived from allylsilane annulation have seen several DOS applications (Fig. 23). In a library reminiscent of the macrodiolides, diketopiperazines were synthesized from amino esters in a one-step protocol.48 Either 2,6-cis- or 2,6-trans-pipieridines (149 and 150, shown) were suitable for this reaction, as were pipecolic acids derived from other sources. Although a preference for homo- or hetero-dimer formation was observed in some cases, unselective reactions were generally clean and the three products could be separated easily on HPLC. Given that these compounds were destined for unbiased screening experiments, the lack of selectivity was, on some level, an asset that enhanced the diversity of the collection. In a subsequent report, Panek and coworkers developed a high-yielding annulation reaction between pipecolate esters and epoxides mediated by Mg(OTf)2.49 In most of the cases studied, this reaction proceeds in >95% yield, especially for the 2,6-cis-pipecolates. In the event that the epoxide was azido-substituted, a second diversification step involving rearrangement to a 6–7-fused bicyclic system could be effected using the Staudinger reaction followed by reduction of the resulting imine.
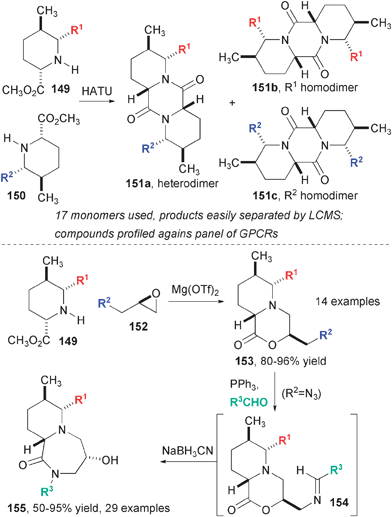 | ||
| Fig. 23 DOS pathways starting with allylsilane-derived pipecolate esters. | ||
In a fourth DOS strategy, the tetrahydropyridine products were engineered to contain at least one aryl halogen to serve as a starting point for radical cascade reactions (Fig. 24). The aryl halide was introduced either as the aldehyde starting material (159) or through diastereoselective alkylation (157), which also served to introduce other appendages. Stereoselective formation of fused or bridged bicyclic alkaloid structures proceeded in high yield and diastereoselectivity using a tin-free protocol with (TMS)3SiH. In addition, a tetracyclization could be induced if the alkylation substrate was either an allyl (162 to 163, shown) or propargyl group. The high level of complexity enabled by this strategy and the fact that these products do not resemble natural compounds made with this same method underscores the impact of robust synthetic methods on DOS and natural products synthesis.
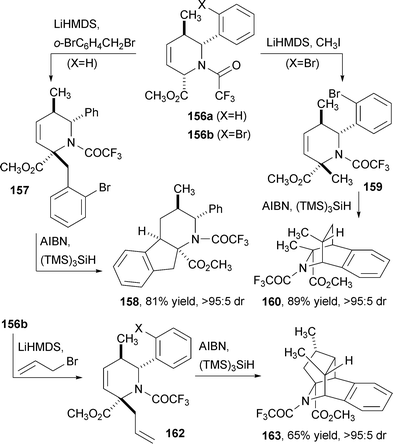 | ||
| Fig. 24 Radical cyclization reactions of allylsilane-derived pipecolate esters. | ||
6 Epoxyquinol cycloadditions for biomimietic natural product synthesis and DOS (Porco)
Exploration of biomimetic cycloaddition reactions have yielded interesting insights into the reactivity of polyfunctional expoxyquinols (Fig. 25). Initial investigations of these compounds were aimed at the assembly of natural products related to 164 that arise from Diels-Alder cycloaddition reactions of precursors 163 and 165. Porco's studies have revealed subtle structural effects that control the selectivity of these reactions in the formation of complex natural products. In addition, these substrates are primed for a variety of potential reactions that produce structurally diverse compounds that bear little resemblance to the dimeric natural products. Biological studies of both the natural compounds and the DOS molecules have yielded new information regarding the origin of their activity as well as novel medicinal leads.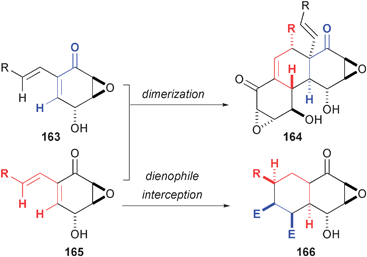 | ||
| Fig. 25 Diels-Alder reactions of epoxyquinols. | ||
Epoxyquinol and epoxyquinone natural products occur as monomers that can undergo dimerization through several different cycloaddition pathways. In the case of torreyanic acid (174), a Diels-Alder reaction between two diastereomers of oxidized ambuic acid (170) produced this complex and cytotoxic natural product (Fig. 26).50 Porco and co-workers completed a synthesis of t-butyl-protected ambuic acid (169) from dialkylhydroquinone 167. Upon oxidation with Dess-Martin periodinane (DMP), two products are observed, each of which arises by minimization of the interaction of the pentyl side chains during the Diels-Alder reaction. The combination of two diastereomeric pyrans, arising from reversible cyclization of the aromatic quinone aldehyde, produced (±)-torreyanic acid after removal of the t-butyl group with TFA. The analogous cycloaddition of two enantiomers of diastereomer 173 produced an equal amount of a dimer that yields a compound the authors term “iso-torreyanic acid.” The third possible product, arising from cycloaddition of 172 with its enantiomer, is not observed, which can be attributed to the aforementioned unfavorable steric clashes between pentyl side chains.
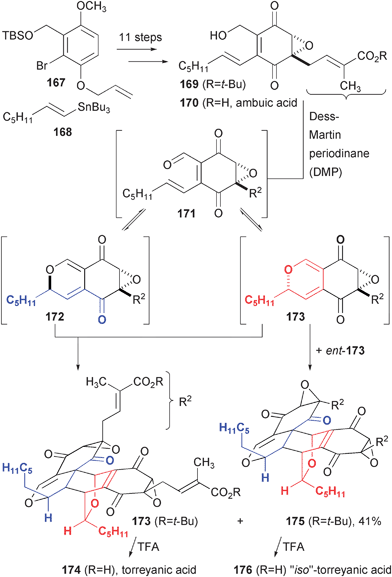 | ||
| Fig. 26 Biomimetic synthesis of torreyanic acid. | ||
A subsequent study of jesterone, an epoxyquinol natural product, yielded interesting chemical and biological results (Fig. 27). Jesterone (178a) was prepared as a single enantiomer using an auxiliary-controlled nucleophilic epoxidation of a quinone monoketal to produce 177 (not shown), followed by diastereoselective reduction with DIBAL-H. Upon treatment of quinone epoxide 177 with DMP, the hydroxyl group is oxidized and cycloaddition upon silica gel chromatography yields 178b, a compound the authors have named “jesterone dimer” in 84% yield as a single diastereomer! This compound has the same core configuration as torreyanic acid, although it is unclear why the smaller methyl group seems to have a greater directing effect when compared to the analogous pentyl group of ambuic acid. This “unnatural” (as of yet) product exhibits cytotoxicity toward cancer cells that is up to 100-fold greater than jesterone.51 A related synthesis of epoxyquinol A (not shown) allowed the authors to probe the propensity of this compound to undergo thermal isomerization.52 The importance of silica gel for catalyzing the cycloaddition was also probed in a [4 + 4] cycloaddition approach to RKB-3564D (epoxytwinol A53), an isomer of epoxyquinol.54
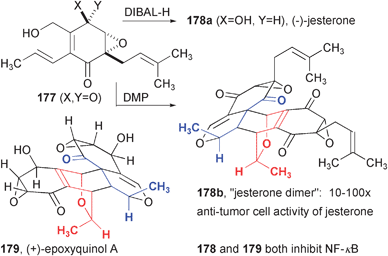 | ||
| Fig. 27 Synthesis of jesterone dimer. | ||
The biomimetic dimerization of epoxyquinols was later studied in detail for the synthesis of two fused tricyclic dimeric compounds (Fig. 28). In the synthesis of panepophenanthrin, Porco and co-workers examined the role of the formation of a ketal ring in the final product.55 The authors hypothesized that ketal formation might be either a pre-organization event for the cycloaddition or a means for preventing the reverse reaction. The latter hypothesis was confirmed with the successful and selective cycloaddition of a compound lacking the ability to form a ketal and the subsequent demonstration that this compound was, unlike the natural compound, prone to thermolytic scission back to the monomer.
A related synthetic route was used to settle the controversy regarding the structure of hexacyclinol that began with the reported synthesis of this compound by La Clair.56 The wholly unprecedented structure reported by Gräfe57 paired with a total synthesis from La Clair that lacked detailed supporting information58 caught the attention of several “bloggers” and set off a lively volley of discussion on many websites.59 Scott Rychnovsky published an insightful revision of the structure of hexacyclinol claiming that it was related to panepophenanthrin,60 which Porco and co-workers quickly demonstrated by synthesis.61 Dimerization of a suitably substituted epoxyquinol formed a compound that the authors termed “pre-hexacyclinol,” which cyclizes to the natural product at room temperature when exposed to K10 clay. As with panepophenanthrin, hexacyclinol is resistant to retrocycloaddition and “pre-hexacyclinol” readily thermolyses back to the monomeric starting material. An analogous synthesis featuring a different monomer synthesis was later reported by Mehta.62
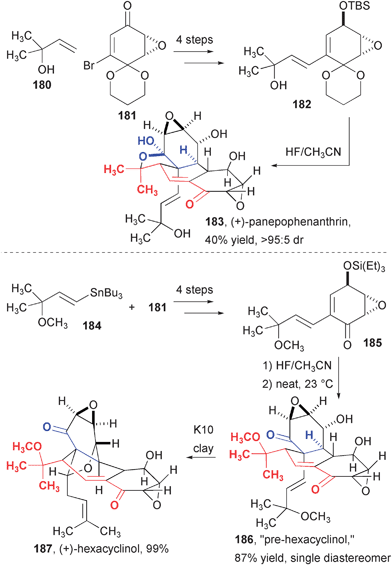 | ||
| Fig. 28 Synthesis of panepophenanthrin and hexacyclinol. | ||
The epoxyquinol nucleus can be employed in a variety of interesting transformation for DOS in which the diene undergoes cycloaddition to reactive dienophiles under conditions wherein dimerization does not occur (Fig. 29).63 Diene 188, in which dienophile reactivity is attenuated by ketal protection of the quinol carbonyl, undergoes smooth and diastereoselective addition to N-alkyl maleimides. The major diastereomer 193 arises from endo approach opposite to the TBS ether. A similar sense of facial induction is seen in the addition of a triazoline dione to produce 192. Removal of the TBS ether yields hydroxydiene 189 capable of undergoing Mg-directed cycloaddition to provide endo product 194 arising form approach syn to the hydroxy group. This reversal of stereochemistry provides a valuable branch point in the synthetic scheme that provides core structure or “skeletal” diversity. Additional skeletal diversity could be realized from the triazoline dione cycloaddition products by reductive cleavage and elimination of the epoxide (195a) or by N–N bond cleavage of the ketal-protected product with dissolving metal (Na/NH3; 197b). Finally, the cis bicyclic products could be diastereoselectively reduced with Adam's catalyst (PtO2) to yield 198 in which the epoxide has also been hydrogenolysed. This compound could be epimerized to the more stable trans isomer 200 in anhydrous HCl. Although this changes only a single stereogenic center, the effect on the overall three-dimensional shape is significant.
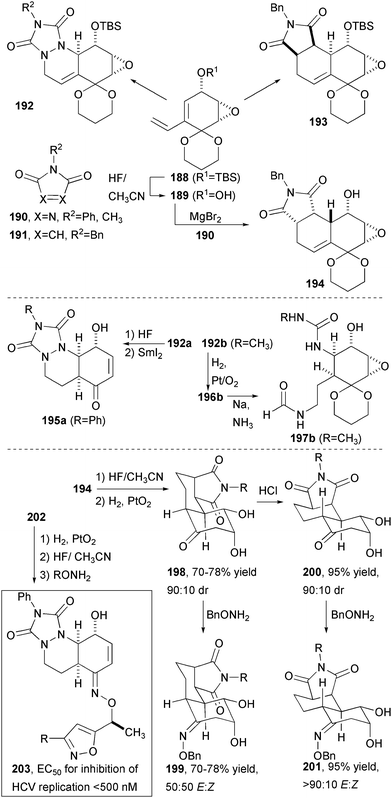 | ||
| Fig. 29 DOS pathways using stereoselective expoxy-quinone Diels-Alder reactions. | ||
The final library synthesis employed a diverse array of ketone core structures that were further diversified by oxime formation. In all, five ketones related to 199, 201, and 203 were treated with 14 different O-alkyl hydroxylamines to produce a library of 244 final compounds, which also included synthetic intermediates. As seen by oxime formation from 198 and 200, the selectivity of the CN bond geometrical isomers varied significantly. These compounds, along with many others from the Boston University CMLD have been used in a variety of unbiased screening experiments, many of which were conducted at the NCI-funded Initiative for Chemical Genetics screening center of the Broad Institute of Harvard and MIT. In one recent report, one of the epoxide oximes, along with several other epoxides, was found to inhibit the replication of hepatitis C virus at a concentration <500 nM.64 Although this was an interesting lead result, cytotoxicity at similar concetrations prevented further exploration of this series of HCV replication inhibitors. Porco and co-workers also noted inhibition of HSP72 (IC50 = 0.75 to 3.0 µM) by a series of epoxyketones (not shown) related to 194 (after ketal deprotection) differing only in the N-substituent.63
7 Imine anhydride and four-component heterocycle synthesis (Shaw)
Unlike many related DOS efforts, including the preceding ones, research in our group was initiated with the goal of increasing skeletal diversity in DOS. In the course of these studies, we discovered new methods that will be relevant to the targeted synthesis of natural products. We initially explored the reactions of imines with anhydrides, which had previously been demonstrated by Catagnoli,65 Cushman,66 and others,67 in order to prepare highly varied polycyclic lactams. During the course of these studies, we discovered a new substrate for this reaction that was highly selective and enabled further functionalization of the γ-lactam core (Fig. 30).68 Based on this observation, we later discovered a new four-component reaction (4CR) between amines, aldehydes, thiols and maleic anhydrides.69 This new reaction forms the same basic lactam product as the imine anhydride reaction with expanded substrate scope and the ability to install up to three stereogenic centers in one pot! Although the 4CR is a highly efficient, one-pot synthesis of densely-functionalized γ-lactams, this reaction is complemented by the reaction of imines with thio-succinic anhydride which protects sensitive substrates from aberrant reactions of free thiols and amines.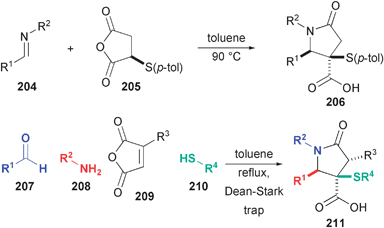 | ||
| Fig. 30 Diastereoselective synthesis of g-lactams from the formal cycloaddition of imines with anhydrides and by a new 4CR (Shaw). | ||
The reactions of imines with cyclic anhydrides has been employed in two DOS efforts. We initially explored the ability of common substrates to undergo multiple modes of cyclization as a way to realize a high level of skeletal diversity (Fig. 31).70 This was to be highly advantageous when using pool-and-split techniques wherein different substrates, bound to solid-phase synthesis (SPS) resin in Irori Kans, could be exposed to the same reaction conditions in one reaction vessel. The product of phenylsuccinic anhydride addition and subsequent amide formation was cyclized smoothly using stoichiometric CuI, a reaction developed by Tohkuyama71 that complements the related catalytic transformations of Buchwald,72 which were ineffective in this case. An alternate cyclization to produce spirocyclic oxindoles was envisioned from the product of o-iodophenylsuccinic anhdyride. Unfortunately, the formal cycloaddition process was unsuccessful, perhaps due to steric interference from the iodide preventing a co-planar arrangement of the phenyl ring and the presumed enolate intermediate. A related process was demonstrated with 2-fluoro-5-nitrophenylsuccinic anhydride, albeit with reduced (69:31) diastereoselection at the cycloaddition stage. Although this short and divergent sequence produced interesting polycyclic compounds, the differential conditions for cyclization prohibited further development for SPS.
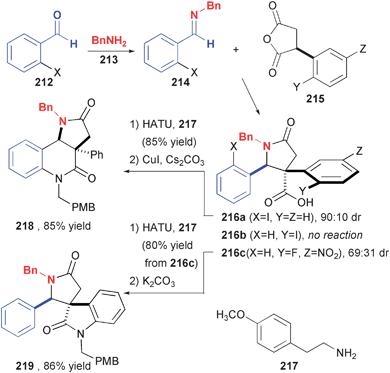 | ||
| Fig. 31 Divergent cyclization reactions of a lactam core derived from imine-anhydride cycloaddition. | ||
A subsequent library focused on more subtle changes that could result in diverse three-dimensional shape, and thus a wider variety of responses in unbiased biological screens (Fig. 32).73 A solid-phase library of 400 compounds was prepared using Irori Kans that contained polystyrene resin functionalized with a silicon tether.74 The first step of imine formation employed 12 different amines. The resulting imines were pooled and split into reactions with one of four different anhydrides to produce acids that are coupled to one of nine different primary amines. The two different succinic anhydrides offer significant shape complementarity and the two homophthalic anhydrides offer stereochemical complementarity. The kinetic product of homophthalic anhydride (220) addition is syn, which epimerizes to trans during amide bond formation. The product of 3-methylhomophthalic (221) anhydride exhibits similar kinetic selectivity and cannot epimerize due to the quaternary center. These compounds, along with the rest of the screening set at the ICG, had been employed in over 150 high-throughput screens at the time of publication. One compound was found to be an inhibitor of the binding between a transcription factor (HOXA13) and its DNA target oligonucleotide (IC50 = 6.7 µM). A subsequent reporter gene assay indicated that this compound indeed regulates transcription of the gene in cells.
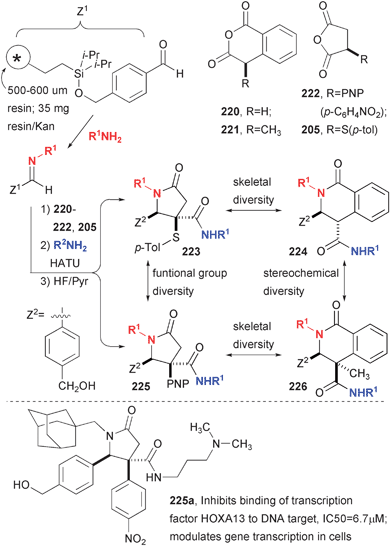 | ||
| Fig. 32 Efficient synthesis of lactam carboxamides with a high level of skeletal diversity. | ||
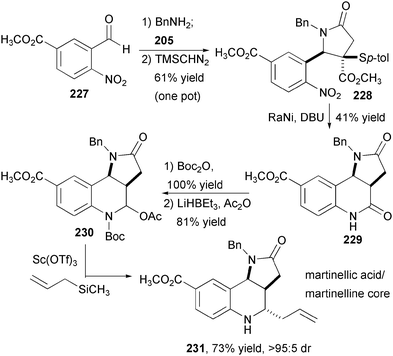 | ||
| Fig. 33 Diastereoselective synthesis of the tricyclic core of martinellic acid and martinelline. | ||
The formal cycloaddition of thio-phenylsuccinic anhydride has been employed in a synthesis of the tricyclic core of martinellic acid and martinelline (Fig. 33).68 Commercially available aldehyde 227 is employed in a one-pot imine formation, anhydride cycloaddition with 205, and methylation in 61% overall yield. This material is then treated with Raney nickel and DBU, which effects four processes: nitro group reduction, reductive desulfurization, epimerization of the ester α-carbon, and ring closure. This tricyclic bis-lactam is protected with a Boc group and then converted to an N-acyliminium ion precursor. The Lewis acid-mediated displacement with allyltrimethylsilane proceeds with concomitant Boc removal in 73% yield and with high (>95:5) diastereoselectivity. This compound contains all of the rings and stereogenic centers of the Martinella alkaloids. Establishing this route paves the way for a very efficient enantioselective synthesis once the imine-anhydride reaction can be made asymmetric, perhaps by using a chiral thiol-based variant of anhydride 205.
8 Conclusions
It is clear from the examples above that recent developments in synthetic methodology have had a dual and concurrent impact on both DOS and the assembly of complex natural product targets. As the importance of small molecule screening continues to develop as a tool for probing complex biological systems, so too will the need for diverse compounds that can be made rapidly and selectively. Although the synthesis of “natural product-inspired” libraries will likely continue to be an important strategy, the use of new and versatile synthetic methods for DOS will continue push the boundaries of structural diversity.9 References
- (a) D. W. Young, A. Bender, J. Hoyt, E. McWhinnie, G.-W. Chirn, C. Y. Tao, J. A. Tallarico, M. Labow, J. L. Jenkins, T. J. Mitchison and Y. Feng, Nat. Chem. Biol., 2008, 4, 59 CrossRef CAS; (b) D. M. Huryn and N. D. P. Cosford, Annu. Rep. Med. Chem., 2007, 42, 401 Search PubMed; (c) J. O. Jones and M. I. Diamond, ACS Chemical Biology, 2007, 2, 718 CrossRef CAS; (d) N. Tolliday, P. A. Clemons, P. Ferraiolo, A. N. Koehler, T. A. Lewis, X. Li, S. L. Schreiber, D. S. Gerhard and S. Eliasof, Cancer Research, 2006, 66, 8935 CrossRef CAS; (e) S. L. Schreiber, Nat. Chem. Biol., 2005, 1, 64 CrossRef CAS; (f) Z. E. Perlman, M. D. Slack, Y. Feng, T. J. Mitchison, L. F. Wu and S. J. Altschuler, Science, 2004, 306, 1194 CrossRef CAS; (g) J. R. Peterson and T. J. Mitchison, Chem. Biol., 2002, 9, 1275 CrossRef CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461 CrossRef CAS.
- A recent book covers many topics in this area: P. Eckard, U. Abel, H.-F. Rasser, W. Simon, B. Sontag and F. G. Hansske, Natural product-based, chemically and functionally diverse libraries, 2006 Search PubMed.
- Reviews from the last five years include: (a) C. Cordier, D. Morton, S. Murrison, A. Nelson and C. O'Leary-Steele, Nat. Prod. Rep., 2008, 25, 719 RSC; (b) L. Arve, T. Voigt and H. Waldmann, QSAR & Combinatorial Science, 2006, 25, 449 Search PubMed; (c) S. Shang and D. S. Tan, Curr. Opin. Chem. Biol., 2005, 9, 248 CrossRef CAS; (d) R. Messer, C. A. Fuhrer and R. Haener, Curr. Opin. Chem. Biol., 2005, 9, 259 CrossRef CAS; (e) J.-Y. Ortholand and A. Ganesan, Curr. Opin. Chem. Biol., 2004, 8, 271 CrossRef CAS; (f) D. S. Tan, Comb. Chem. High Throughput Screening, 2004, 7, 631 CAS; (g) A. M. Boldi, Curr. Opin. Chem. Biol., 2004, 8, 281 CrossRef CAS.
- M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46 CrossRef.
- V. Gracias, G. L. Milligan and J. Aubé, J. Am. Chem. Soc., 1995, 117, 8047 CrossRef CAS.
- E. Nyfeler and P. Renaud, Chimia, 2006, 60, 276 CrossRef CAS.
- K. Tani and B. M. Stoltz, Nature (London, U.K.), 2006, 441, 731 Search PubMed.
- J. Aubé, P. S. Rafferty and G. L. Milligan, Heterocycles, 1993, 35, 1141 CrossRef CAS.
- (a) A. Wrobleski, K. Sahasrabudhe and J. Aubé, J. Am. Chem. Soc., 2004, 126, 5475 CrossRef CAS; (b) A. Wrobleski, K. Sahasrabudhe and J. Aubé, J. Am. Chem. Soc., 2002, 124, 9974 CrossRef CAS.
- (a) R. Iyengar, K. Schildknegt, M. Morton and J. Aubé, J. Org. Chem., 2005, 70, 10645 CrossRef CAS; (b) R. Iyengar, K. Schildknegt and J. Aubé, Org. Lett., 2000, 2, 1625 CrossRef CAS.
- (a) D. S. Reddy, D. Vander Velde and J. Aubé, J. Org. Chem., 2004, 69, 1716 CrossRef CAS; (b) M. MacDonald and J. Aubé, Curr. Org. Chem., 2001, 5, 417 CrossRef CAS; (c) M. MacDonald, D. Vander Velde and J. Aubé, Org. Lett., 2000, 2, 1653 CrossRef CAS; (d) O. Kitagawa, D. Vander Velde, D. Dutta, M. Morton, F. Takusagawa and J. Aubé, J. Am. Chem. Soc., 1995, 117, 5169 CrossRef CAS.
- (a) E. Fenster, D. K. Rayabarapu, M. Zhang, S. Mukherjee, D. Hill, B. Neuenswander, F. Schoenen, P. R. Hanson and J. Aubé, J. Comb. Chem., 2008, 10, 230 CrossRef CAS; (b) E. Fenster, B. T. Smith, V. Gracias, G. L. Milligan and J. Aubé, J. Org. Chem., 2008, 73, 201 CrossRef CAS.
- K. Frankowski, J., B. Neuenswander and J. Aubé, J. Comb. Chem., 2008, 10, 721–725 CrossRef CAS.
- L. S. Pavarov, V. I. Grigos, R. A. Karakhanov and B. M. Mikhailov, Izv. Akad. Nauk SSSR, Ser. Khim., 1964, 179 CAS . This spelling of “Pavarov” is as it appears in this reference. Most subsequent publications refer to the “Povarov” reaction, so that spelling is used in the text of this Highlight.
- K. M. Witherup, R. W. Ransom, A. C. Graham, A. M. Bernard, M. J. Salvatore, W. C. Lumma, P. S. Anderson, S. M. Pitzenberger and S. L. Varga, J. Am. Chem. Soc., 1995, 117, 6682 CrossRef CAS.
- (a) See, for example: A. Shirai, O. Miyata, N. Tohnai, M. Miyata, D. J. Procter, D. Sucunza and T. Naito, J. Org. Chem., 2008, 73, 4464 Search PubMed; (b) S. Ikeda, M. Shibuya and Y. Iwabuchi, Chem. Commun., 2007, 504 RSC; (c) V. Badarinarayana and C. J. Lovely, Tetrahedron Lett., 2007, 48, 2607 CrossRef CAS; (d) C. Xia, L. Heng and D. Ma, Tetrahedron Lett., 2002, 43, 9405 CrossRef CAS; (e) B. B. Snider, Y. Ahn and S. M. O'Hare, Org. Lett., 2001, 3, 4217 CrossRef CAS; (f) D. Ma, C. Xia, J. Jiang and J. Zhang, Org. Lett., 2001, 3, 2189 CrossRef CAS.
- D. A. Powell and R. A. Batey, Org. Lett., 2002, 4, 2913 CrossRef CAS.
- H. Twin and R. A. Batey, Org. Lett., 2004, 6, 4913 CrossRef CAS.
- R. A. Batey and D. A. Powell, Chem. Commun., 2001, 2362 RSC.
- (a) D. A. Powell and R. A. Batey, Tetrahedron Lett., 2003, 44, 7569 CrossRef CAS; (b) R. A. Batey, D. A. Powell, A. Acton and A. J. Lough, Tetrahedron Lett., 2001, 42, 7935 CrossRef CAS; (c) R. A. Batey, P. D. Simoncic, D. Lin, R. P. Smyj and A. J. Lough, Chem. Commun., 1999, 651 RSC.
- (a) J. L. Kent, H. Wan and K. M. Brummond, Tetrahedron Lett., 1995, 36, 2407 CrossRef CAS; (b) K. M. Brummond and H. Wan, Tetrahedron Lett., 1998, 39, 931 CrossRef CAS.
- (a) K. M. Brummond, H. Chen, K. D. Fisher, A. D. Kerekes, B. Rickards, P. C. Sill and S. J. Geib, Org. Lett., 2002, 4, 1931 CrossRef CAS; (b) For a computational discussion of the divergent pathways, see: A. S. Bayden, K. M. Brummond and K. D. Jordan, Organometallics, 2006, 25, 5204 Search PubMed.
- K. M. Brummond, H. Chen, P. Sill and L. You, J. Am. Chem. Soc., 2002, 124, 15186 CrossRef CAS.
- K. M. Brummond, H. Chen, B. Mitasev and A. D. Casarez, Org. Lett., 2004, 6, 2161 CrossRef CAS.
- (a) P. Dent and S. Grant, Cancer Biology & Therapy, 2004, 3, 1143 Search PubMed; (b) T. C. McMorris, Bioorg. Med. Chem., 1999, 7, 881 CrossRef CAS.
- (a) J. Gong, V. G. Vaidyanathan, X. Yu, T. W. Kensler, L. A. Peterson and S. J. Sturla, J. Am. Chem. Soc., 2007, 129, 2101 CrossRef CAS; (b) M. Movassaghi, G. Piizzi, D. S. Siegel and G. Piersanti, Angew. Chem., Int. Ed., 2006, 45, 5859 CrossRef CAS; (c) T. C. McMorris, M. D. Staake and M. J. Kelner, J. Org. Chem., 2004, 69, 619 CrossRef CAS.
- K. M. Brummond and J. Lu, J. Am. Chem. Soc., 1999, 121, 5087 CrossRef CAS.
- K. M. Brummond, J. Lu and J. Petersen, J. Am. Chem. Soc., 2000, 122, 4915 CrossRef CAS.
- K. M. Brummond, P. C. Sill and H. Chen, Org. Lett., 2004, 6, 149 CrossRef CAS.
- (a) S. F. Brady, M. P. Singh, J. E. Janso and J. Clardy, J. Am. Chem. Soc., 2000, 122, 2116 CrossRef CAS; (b) S. F. Brady, S. M. Bondi and J. Clardy, J. Am. Chem. Soc., 2001, 123, 9900 CrossRef CAS.
- (a) A. K. Miller, C. C. Hughes, J. J. Kennedy-Smith, S. N. Gradl and D. Trauner, J. Am. Chem. Soc., 2006, 128, 17057 CrossRef CAS; (b) S. Iimura, L. E. Overman, R. Paulini and A. Zakarian, J. Am. Chem. Soc., 2006, 128, 13095 CrossRef CAS; (c) S. V. Maifeld and D. Lee, Synlett, 2006, 1623 CAS; (d) W. D. Shipe and E. J. Sorensen, J. Am. Chem. Soc., 2006, 128, 7025 CrossRef CAS; (e) S. Lin, G. B. Dudley, D. S. Tan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2002, 41, 2188 CrossRef CAS; (f) For a review of much of the synthetic work through 2005, see: M. Mischne, Current Organic Synthesis, 2005, 2, 261 Search PubMed.
- K. M. Brummond and J. L. Kent, Tetrahedron, 2000, 56, 3263 CrossRef CAS.
- K. M. Brummond, D. Chen and M. M. Davis, J. Org. Chem., 2008, 73, 5064 CrossRef CAS.
- N. Kumagai, G. Muncipinto and S. L. Schreiber, Angew. Chem., Int. Ed., 2006, 45, 3635 CrossRef CAS.
- J. S. Lazo, J. J. Skoko, S. Werner, B. Mitasev, A. Bakan, F. Koizumi, A. Yellow-Duke, I. Bahar and K. M. Brummond, J. Pharmacol. Exp. Ther., 2007, 322, 940 CrossRef CAS.
- K. M. Brummond, T. O. Painter, D. A. Probst and B. Mitasev, Org. Lett., 2007, 9, 347 CrossRef CAS.
- O. Kwon, S. B. Park and S. L. Schreiber, J. Am. Chem. Soc., 2002, 124, 13402 CrossRef CAS.
- (a) Y. Zhang and J. S. Panek, Org. Lett., 2007, 9, 3141 CrossRef CAS; (b) J. T. Lowe, I. E. Wrona and J. S. Panek, Org. Lett., 2007, 9, 327 CrossRef CAS.
- (a) M. D'Ambrosio, M. Tato, G. Pocsfalvi, C. Debitus and F. Pietra, Helv. Chim. Acta, 1999, 82, 347 CrossRef CAS; (b) M. D'Ambrosio, A. Guerriero, C. Debitus and F. Pietra, Helv. Chim. Acta, 1996, 79, 51 CrossRef CAS.
- (a) O. A. Ulanovskaya, J. Janjic, M. Suzuki, S. S. Sabharwal, P. T. Schumacker, S. J. Kron and S. A. Kozmin, Nat. Chem. Biol., 2008, 4, 418 CrossRef CAS; (b) P. A. Evans and W. J. Andrews, Angew. Chem., Int. Ed., 2008, 47, 5426 CrossRef CAS; (c) L. J. Van Orden, B. D. Patterson and S. D. Rychnovsky, J. Org. Chem., 2007, 72, 5784 CrossRef CAS; (d) L. Ferrie, S. Reymond, P. Capdevielle and J. Cossy, Org. Lett., 2007, 9, 2461 CrossRef CAS; (e) D. R. Williams, S. V. Plummer and S. Patnaik, Angew. Chem., Int. Ed., 2003, 42, 3934 CrossRef CAS; (f) I. Paterson and M. Tudge, Angew. Chem., Int. Ed., 2003, 42, 343 CrossRef CAS; (g) M. T. Crimmins and P. Siliphaivanh, Org. Lett., 2003, 5, 4641 CrossRef CAS; (h) P. Wipf and J. T. Reeves, Chem. Commun., 2002, 2066 RSC; (i) Y. Wang, J. Janjic and S. A. Kozmin, J. Am. Chem. Soc., 2002, 124, 13670 CrossRef CAS; (j) A. Fettes and E. M. Carreira, Angew. Chem., Int. Ed., 2002, 41, 4098 CrossRef CAS; (k) K. R. Hornberger, C. L. Hamblett and J. L. Leighton, J. Am. Chem. Soc., 2000, 122, 12894 CrossRef CAS; (l) Much of the preceding work is summarized in a review of recent approaches to the synthesis of macrolides: K.-S. Yeung and I. Paterson, Chem. Rev., 2005, 105, 4237 Search PubMed.
- A. E. Wright, J. C. Botelho, E. Guzman, D. Harmody, P. Linley, P. J. McCarthy, T. P. Pitts, S. A. Pomponi and J. K. Reed, J. Nat. Prod., 2007, 70, 412 CrossRef CAS.
- (a) D. W. Custar, T. P. Zabawa and K. A. Scheidt, J. Am. Chem. Soc., 2008, 130, 804 CrossRef CAS; (b) For subsequent formal and total syntheses, see: H. Fuwa, S. Naito, T. Goto and M. Sasaki, Angew. Chem., Int. Ed., 2008, 47, 4737 Search PubMed; (c) S. K. Woo, M. S. Kwon and E. Lee, Angew. Chem., Int. Ed., 2008, 47, 3242 CrossRef CAS; (d) V. V. Vintonyak and M. E. Maier, Org. Lett., 2008, 10, 1239 CrossRef CAS.
- G. Chiosis, E. C. Lopes and D. Solit, Curr. Op. Invest. Drugs, 2006, 7, 534 Search PubMed.
- (a) J. S. Panek, F. Xu and A. C. Rondon, J. Am. Chem. Soc., 1998, 120, 4113 CrossRef; (b) J. S. Panek and F. Xu, J. Am. Chem. Soc., 1995, 117, 10587 CrossRef CAS.
- I. E. Wrona, A. E. Garbada, G. Evano and J. S. Panek, J. Am. Chem. Soc., 2005, 127, 15026 CrossRef CAS.
- (a) A. B. Beeler, D. E. Acquilano, Q. Su, F. Yan, B. L. Roth, J. S. Panek and J. A. Porco, Jr., J. Comb. Chem., 2005, 7, 673 CrossRef CAS; (b) Q. Su, A. B. Beeler, E. Lobkovsky, J. A. Porco and J. S. Panek, Org. Lett., 2003, 5, 2149 CrossRef CAS.
- S. Dandapani, P. Lan, A. B. Beeler, S. Beischel, A. Abbas, B. L. Roth, J. A. Porco, Jr. and J. S. Panek, J. Org. Chem., 2006, 71, 8934 CrossRef CAS.
- Y. Chen, J. A. Porco, Jr. and J. S. Panek, Org. Lett., 2007, 9, 1529 CrossRef CAS.
- C. Li, R. P. Johnson and J. A. Porco, Jr., J. Am. Chem. Soc., 2003, 125, 5095 CrossRef CAS.
- M.-C. Liang, S. Bardhan, J. A. Porco, Jr. and T. D. Gilmore, Cancer Letters (Amsterdam, Netherlands), 2006, 241, 69 Search PubMed; M.-C. Liang, S. Bardhan, C. Li, E. A. Pace, J. A. Porco, Jr. and T. D. Gilmore, Mol. Pharmacol., 2003, 64, 123 CrossRef CAS.
- (a) C. Li, S. Bardhan, E. A. Pace, M.-C. Liang, T. D. Gilmore and J. A. Porco, Jr., Org. Lett., 2002, 4, 3267 CrossRef CAS; (b) C. Li and J. A. Porco, Jr., J. Org. Chem., 2005, 70, 6053 CrossRef CAS.
- M. Shoji, H. Imai, M. Mukaida, K. Sakai, H. Kakeya, H. Osada and Y. Hayashi, J. Org. Chem., 2005, 70, 79 CrossRef CAS.
- C. Li and J. A. Porco, Jr., J. Am. Chem. Soc., 2004, 126, 1310 CrossRef CAS.
- X. Lei, R. P. Johnson and J. A. Porco, Jr., Angew. Chem., Int. Ed., 2003, 42, 3913 CrossRef CAS.
- E. Marris, Nature, 2006, 442, 492 CrossRef CAS.
- B. Schlegel, A. Hartl, H.-M. Dahse, F. A. Gollmick, U. Grafe, H. Dorfelt and B. Kappes, J. Antibiot., 2002, 55, 814 CAS.
- J. J. La Clair, Angew. Chem., Int. Ed., 2006, 45, 2769 CrossRef CAS.
- See, for example: http://pipeline.corante.com/archives/2006/07/23/hexacyclinol_rides_again.php (accessed on August 15, 2008).
- (a) S. D. Rychnovsky, Org. Lett., 2006, 8, 2895 CrossRef CAS; (b) For a subsequent analysis, see: A. J. Williams, M. E. Elyashberg, K. A. Blinov, D. C. Lankin, G. E. Martin, W. F. Reynolds, J. A. Porco, C. A. Singleton and S. Su, J. Nat. Prod., 2008, 71, 581 Search PubMed.
- J. A. Porco, Jr., S. Su, X. Lei, S. Bardhan and S. D. Rychnovsky, Angew. Chem., Int. Ed., 2006, 45, 5790 CrossRef CAS.
- G. Mehta and S. Roy, Tetrahedron Lett., 2008, 49, 1458 CrossRef CAS.
- X. Lei, N. Zaarur, M. Y. Sherman and J. A. Porco, Jr., J. Org. Chem., 2005, 70, 6474 CrossRef CAS.
- L. F. Peng, S. S. Kim, S. Matchacheep, X. Lei, S. Su, W. Lin, W. Runguphan, W.-H. Choe, N. Sakamoto, M. Ikeda, N. Kato, A. B. Beeler, J. A. Porco, Jr., S. L. Schreiber and R. T. Chung, Antimicrob. Agents Chemother., 2007, 51, 3756 CrossRef CAS.
- (a) N. Castagnoli, Jr., J. Org. Chem., 1969, 34, 3187 CrossRef CAS; (b) N. Castagnoli, Jr. and M. Cushman, J. Org. Chem., 1971, 36, 3404 CrossRef.
- (a) M. Cushman, J. Gentry and F. W. Dekow, J. Org. Chem., 1977, 42, 1111 CrossRef CAS; (b) M. Cushman and E. J. Madaj, J. Org. Chem., 1987, 52, 907 CrossRef CAS.
- For the only comprehensive review of this reaction and related chemistry, see: (a) M. González-López and J. T. Shaw, Chem. Rev., 2008, 108 DOI:10.1021/cr8002714. For selected examples, see: (b) M. A. Haimova, N. M. Mollov, S. C. Ivanova, A. I. Dimitrova and V. I. Ognyanov, Tetrahedron, 1977, 33, 331 CrossRef CAS; (c) Y. Ni, V. Krchnak and M. Lebl, Molecular diversity, 2002, 5, 153 Search PubMed; (d) M. Lebl, V. Krchnak, G. Ibrahim, J. Pires, C. Burger, Y. Ni, Y. Chen, D. Podue, P. Mudra, V. Pokorny, P. Poncar and K. Zenisek, Synthesis, 1999, 1971 CrossRef CAS; (e) R. D. Clark and M. Souchet, Tetrahedron Lett., 1990, 31, 193 CrossRef CAS.
- P. Y. Ng, C. E. Masse and J. T. Shaw, Org. Lett., 2006, 8, 3999 CrossRef.
- J. Wei and J. T. Shaw, Org. Lett., 2007, 9, 4077 CrossRef CAS.
- C. E. Masse, P. Y. Ng, Y. Fukase, M. Sanchez-Rosello and J. T. Shaw, J. Comb. Chem., 2006, 8, 293 CrossRef CAS.
- K. Yamada, T. Kubo, H. Tokuyama and T. Fukuyama, Synlett, 2002, 231 CrossRef CAS.
- A. Klapars, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421 CrossRef CAS.
- (a) W. H. B. Sauer and M. K. Schwarz, J. Chem. Inf. Comp. Sci., 2003, 43, 987 CrossRef CAS; (b) For a related DOS strategy, see: J. M. Mitchell and J. T. Shaw, Angew. Chem., Int. Ed., 2006, 45, 1722 Search PubMed.
- P. Y. Ng, Y. Tang, W. M. Knosp, H. S. Stadler and J. T. Shaw, Angew. Chem., Int. Ed., 2007, 46, 5352 CrossRef CAS.
- K. M. Brummond and B. Mitasev, Org. Lett., 2004, 6, 2245 CrossRef CAS.
- S. Werner, D. Kasi and K. M. Brummond, J. Comb. Chem., 2007, 9, 677 CrossRef CAS.
- S. Mao, D. Probst, S. Werner, J. Chen, X. Xie and K. M. Brummond, J. Comb. Chem., 2008, 10, 235 CrossRef CAS.
- S. Werner, D. M. Turner, P. G. Chambers and K. M. Brummond, Tetrahedron, 2008, 64, 6997 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |