Aplyronine A, a potent antitumour macrolide of marine origin, and the congeners aplyronines B–H: chemistry and biology
Kiyoyuki
Yamada
*a,
Makoto
Ojika
b,
Hideo
Kigoshi
c and
Kiyotake
Suenaga
d
aGraduate School of Science, Nagoya University, Chikusa, Nagoya 464-8602, Japan
bGraduate School of Bioagricultural Sciences, Nagoya University, Chikusa, Nagoya 464-8601, Japan
cGraduate School of Pure and Applied Sciences, University of Tsukuba, Tsukuba, 305-8571, Japan
dDepartment of Chemistry, Faculty of Science and Technology, Keio University, Kohoku, Yokohama 223-8522, Japan
First published on 9th September 2008
Abstract
Covering: up to 2007
Aplyronines A–H are cytotoxic macrolides isolated from the sea hare Aplysia kurodai. Aplyronine A is the major constituent among the aplyronines, and this review concentrates on the results of chemical and biological research into this natural product. The isolation, determination of stereostructure and enantioselective total synthesis of the aplyronines are covered, together with discussion of their antitumour activity and structure–activity relationships, and the three-dimensional X-ray structure of the actin–aplyronine A complex.
 Kiyoyuki Yamada | Kiyoyuki Yamada completed his Ph.D. degree at Nagoya University, Japan, under the direction of Professor Yoshimasa Hirata in 1962. He joined the faculty at Nagoya University in 1961, and did postdoctoral work at Stanford University with Professor Eugene E. van Tamelen from 1962 to 1964. He was promoted to Associate Professor at Nagoya University in 1966 and then to Professor in 1979. His research interests are in the fields of natural products chemistry and bio-organic chemistry. He was awarded the Chemical Society of Japan Award in 1996. Since 1997 he has been Professor Emeritus at Nagoya University. |
 Makoto Ojika | Makoto Ojika completed his Ph.D. degree in 1987 under the direction of Professor Kiyoyuki Yamada at Nagoya University. He became Assistant Professor at Nagoya University in 1982, and was a postdoctoral fellow at Columbia University with Professor Koji Nakanishi in 1988–1989. He was promoted to Associate Professor (Graduate School of Bioagricultural Sciences) at Nagoya University in 1995 and then to Professor in 2001. His research interests are in the fields of organic and bio-organic chemistry on natural products. He was awarded the Chemical Society of Japan Award for Young Chemists in 1990. |
 Hideo Kigoshi | Hideo Kigoshi completed his doctoral work in 1989 under the direction of Professor Kiyoyuki Yamada at Nagoya University. He was a postdoctoral fellow with Professor Elias J. Corey (Harvard University) in 1990–1991. He became Assistant Professor at Nagoya University in 1984 and was promoted to Associate Professor in 1994. He moved to University of Tsukuba in 2000 as Professor of Chemistry. His research interests lie in the field of chemistry and chemical biology of the bioactive natural products. He received the Chemical Society of Japan Award for Young Chemists in 1993. |
 Kiyotake Suenaga | Kiyotake Suenaga received his Ph.D. degree in 1997 from Nagoya University under the direction of Professor Kiyoyuki Yamada, and became Assistant Professor at Nagoya University in 1995. He moved to the University of Shizuoka in 2001 and then to the University of Tsukuba in 2003. He was appointed Associate Professor (Faculty of Science and Technology) at Keio University in 2006. His research interests span the fields of natural products chemistry and bio-organic chemistry. He was awarded the Chemical Society of Japan Award for Young Chemists in 2003. |
1 Introduction
A large number of bioactive substances have been isolated from marine organisms such as cyanobacteria, algae, sponges, corals, tunicates, coelenterates, bryozoans and molluscs. Sea hares, belonging to the opisthobranch group of molluscs, are shell-less and slow-moving marine animals that feed on a variety of algae and sponges. These molluscs are thought to have chemical defense substances that appear to deter predators; the poisonous properties of sea hare secretions were recorded as long ago as Roman times. Sea hares have been known to be a rich source of bioactive metabolites; these are generally present in minute amounts and are considered to be of dietary origin and/or to be produced by symbiotic microbes.Since the epoch-making report on the isolation of aplysin, a bromine-containing sesquiterpene, by Hirata and Yamamura in 1963,1 investigations into the secondary metabolites of sea hares of the genus Aplysia were carried out to isolate various compounds, most of which were halogenated terpenes and halogenated cyclic ethers. Bioactive compounds obtained from sea hares, including those of the genus Aplysia, have been reviewed.2–4
In 1993, Yamada and co-workers discovered, isolated and characterized cytotoxic compounds named aplyronines from Japanese specimens of the sea hare Aplysia kurodai.5 This review concentrates on the results of chemical and biological research into aplyronine A, the major aplyronine present in Aplysia kurodai, together with some of the chemical and biological studies into the congeners aplyronines B–H.
2 Isolation of aplyronines A–H
Specimens of the sea hare Aplysia kurodai were collected along the Pacific coast of the Shima Peninsula, Mie Prefecture, Japan, and after homogenization they were extracted with methanol. The methanol extract was partitioned between ethyl acetate and water. The ethyl acetate extract showed strong cytotoxicity against the tumour cell line HeLa-S3. Originally, aplyronine A (1)5,6 was isolated by an eight-step chromatographic separation of the ethyl acetate extract guided by cytotoxic assay using HeLa-S3 cells. Subsequently, a more efficient method for the isolation of 1 was developed (Scheme 1): the ethyl acetate extract was separated by solvent partition (twice) and by five-step chromatography (column chromatography, three times; preparative layer chromatography, once; reversed-phase HPLC, once). This efficient method enabled us to isolate not only a substantial quantity of 1 but also seven minor congeners, aplyronines B (2) and C (3)5,6 and D–H (4–8).3 Aplyronines A–H (1–8) are colorless and amorphous.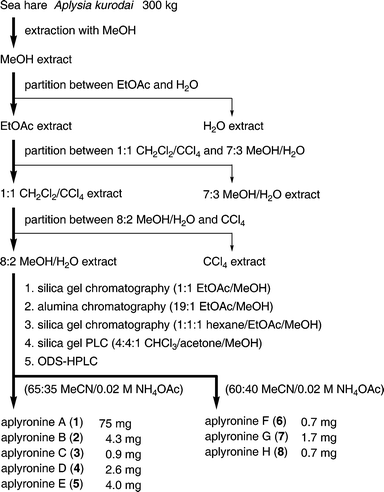 | ||
| Scheme 1 Isolation procedure for aplyronines A–H (1–8). | ||
3 Structure of aplyronine A
Aplyronine A (1) possesses the following physical and spectral properties: molecular formula C59H101N3O14; [α]28D +32 (c 0.26, MeOH); IR (CHCl3) 3690, 3500, 1730, 1690, 1655 cm−1; HR-FAB-MS m/z 1076.7360 (M + H); for 1H and 13C NMR spectra, see ref. 5 and 6.3.1 Gross structure
The IR spectrum of 1 indicated the presence of ester, amide, and hydroxyl groups: furthermore, the presence of hydroxyl groups was confirmed by acetylation of 1 to afford the diacetate 9. The analysis of 1D NMR data of 1 showed the presence of an acetate ester, three additional esters, five olefins, three methoxy groups, an N-methyl group, a formyl group and two dimethylamino groups. Owing to the rotamers at the N-methyl-N-vinylformamide terminus (2 : 1 ratio) and the epimers at the two amino acid portions (1.1 : 1 and 3 : 1 ratios for the N,N,O-trimethylserine and N,N-dimethylalanine moieties, respectively), the NMR spectra seemed to be those of a complex mixture of isomers. However, the detailed analysis of DQF-COSY, 13C--1H COSY and HMQC spectra of 1 made it possible to assign most signals to each component in this isomeric mixture, to construct eight partial structures: C2–C9, C10–C11, C12–C13, C14–C17, C18–C26, C28–C34, C2′–C3′ and C2″–C3″ (Fig. 1). The low-field shifts observed for H9 and H25 of the diacetate 9 indicated the locations of two hydroxyl groups in 1. Long-range H–C correlations were then obtained by HMBC experiments of 1 to connect not only the above partial structures but also the four ester carbonyl (C1, C1′, C1″ and an acetate), the formyl and the heteroatom-bearing methyl groups. The E geometry of all the double bonds was shown by the large trans coupling constants (ca. 15 Hz) or NOEs (H13/H15 and 14-Me/H16). On the basis of these findings the gross structure of aplyronine A was established as 1, a 24-membered macrolide possessing two amino acid esters and an N-methyl-N-vinylformamide terminus.5,6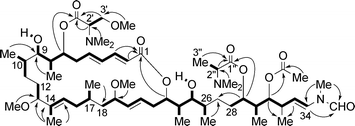 | ||
| Fig. 1 Gross structure of 1 elucidated by DQF-COSY (bold bonds) and HMBC (arrows from H to C) correlations. | ||
3.2 Stereostructure
The absolute stereostructure of aplyronine A (1), possessing 17 stereocentres, was determined by NMR spectral analysis and enantioselective synthesis of the fragments obtained from chemical degradation of aplyronine A (1). The fragments 10–14 derived from 1 are shown in Scheme 2. Note that a sequence of degradation reactions afforded the fragment 12 and its diastereomer 14-epi-12, 12 being the major product.6,7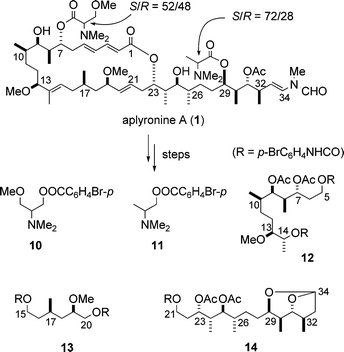 | ||
| Scheme 2 Fragments obtained from chemical degradation of aplyronine A (1). | ||
3.2.1 Absolute stereochemistry of amino acid units. The enantiomerically pure authentic p-bromobenzoates (R)-10 and (S)-11 were synthesized from L-serine and L-alaninol, respectively. Chiral HPLC analyses revealed that both natural 10 and 11 were enantiomeric mixtures with S/R ratios of 52 : 48 and 72 : 28, respectively.6,7
3.2.2 Relative stereochemistry. The stereochemistry of the dioxabicyclo[3.2.1]octane moiety in fragment 14 was determined by NOE spectroscopy and the coupling constant depicted in Fig. 2. The relative stereochemistry of the four contiguous stereocentres (C29–C32) was thus established to be syn–anti–anti.
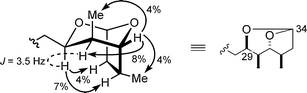 | ||
| Fig. 2 NOEs in the bicyclic portion of 14. | ||
For the determination of the relative stereochemistry of the other sets of the contiguous stereocentres, C7–C10 and C23–C26, the fragments 12 and 14 were converted to the acetonides 15 and 16, respectively (Fig. 3). Subsequently, the 1H coupling constants and chemical shifts of their contiguous stereocentres were compared with those of all the eight possible diastereomers of the model compounds 17a–h6,8 (Fig. 4), which were prepared by applying Kishi's methods for the asymmetric synthesis of polypropionates.9
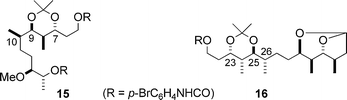 | ||
| Fig. 3 Acetonide derivatives of the fragments 12 and 14. | ||
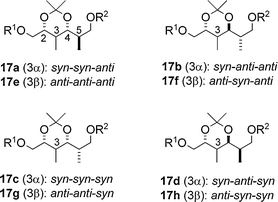 | ||
| Fig. 4 Eight possible diastereomers 17a–h possessing four contiguous stereocentres: R1 = Me3CCO, R2 = Si(t-Bu)Ph2. | ||
The eight diastereomers were classified into four groups based on the differences of the coupling constants (ΔJ) of the protons (H2–H4) of 17a–h as indicated in the difference graphs (Fig. 5), that is, 2,3-syn-3,4-syn type (17a and 17c), 2,3-syn-3,4-anti type (17b and 17d), 2,3-anti-3,4-anti type (17e and 17g) and 2,3-anti-3,4-syn type (17f and 17h). Thus, the relative stereochemistry on the acetonide portions of the natural fragments 15 and 16 are estimated to be 7,8-anti-8,9-syn and 23,24-syn-24,25-anti, respectively. However, the relative stereochemistry of the C9–C10 positions of 15 and the C25–C26 positions of 16 was still unclear, because the differences of the stereochemistry at these positions are not reflected in the coupling constant J4,5 in the eight isomers of 17 at all. Fortunately, the difference graphs (Fig. 5) obtained from the chemical shifts of the two secondary methyl groups made it possible to distinguish the stereochemistry of the C4–C5 positions in the eight isomers of 17. Thus, the chemical shift differences (Δδ) of the secondary methyl groups between 17f (4,5-anti) and 15 are less than those between 17h (4,5-syn) and 15, indicating the 9,10-anti stereochemistry of 15.
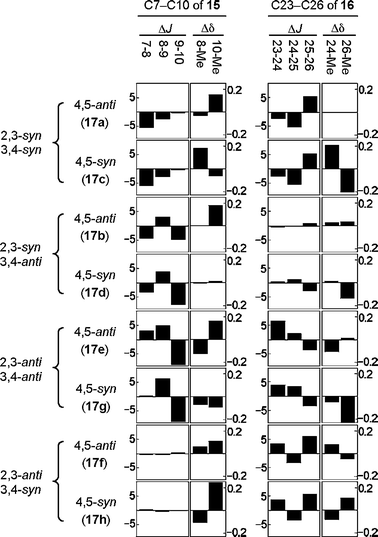 | ||
| Fig. 5 Difference graphs of the 1H NMR data between the model compounds 17a–h and the natural fragments 15 and 16. The ΔJ and Δδ values are ‘J(17) − J(fragment)’ and ‘δ(17) − δ(fragment)’, respectively, at the indicated positions of the fragments 15 and 16 and the corresponding positions C2–C5 of 17. | ||
In the same manner, the 25,26-anti stereochemistry of 16 was deduced. On the basis of these findings the relative stereochemistry of C7–C10 of 12 and C23–C26 of 14 was estimated to be anti–syn–anti and syn–anti–anti, respectively.6–8
3.2.3 Absolute stereochemistry. Subsequently, the absolute stereochemistry of the three fragments 12–14 were determined by the enantioselective synthesis of possible stereoisomers for each of the fragments based on the aforementioned relative stereochemistry.
First, we examined the absolute stereochemistry of the C5–C14 fragment 12, which contained five unassigned stereocentres (C7, C8, C9, C10 and C13). Since the relative stereochemistry of the four contiguous stereocentres C7–C10 was established to be anti–syn–anti, the C5–C14 fragment must be one of four possible stereoisomers, i.e.12, 13-epi-12 and their enantiomers. We chose ent-12 and ent-13-epi-12 as the synthetic targets and carried out their synthesis, which is illustrated in Scheme 3.6,10 By comparison of the spectroscopic data and optical rotations, the synthetic urethane ent-12 was found to be identical with the natural C5–C14 fragment 12 except for the sign of the optical rotation, elucidating the absolute stereochemistry at C7, C8, C9, C10 and C13.6,10
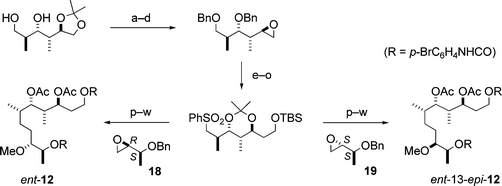 | ||
| Scheme 3 Enantioselective synthesis of ent-12 and ent-13-epi-12. Reagents and conditions: (a) BnBr, NaH; (b) 2 M HCl; (c) TsCl, pyridine; (d) K2CO3, MeOH; (e) 1,3-dithiane, BuLi; (f) BnBr, NaH; (g) CuO, CuCl2, acetone–H2O; (h) NaBH4, EtOH; (i) t-BuMe2SiCl (TBSCl), imidazole; (j) Li, liq. NH3, i-PrOH; (k) pivaloyl chloride (PivCl), pyridine; (l) Me2C(OMe)2, CSA; (m) LiAlH4; (n) (PhS)2, Bu3P; (o) m-CPBA; (p) BuLi, then 18 (or 19); (q) 6% Na(Hg), Na2HPO4; (r) MeI, NaH; (s) Bu4NF; (t) Na, liq. NH3; (u) p-BrC6H4NCO, pyridine; (v) AcOH, H2O; (w) Ac2O, DMAP, pyridine. | ||
Second, we examined the absolute stereochemistry of the C15–C20 fragment 13, containing two unassigned stereocentres at C17 and C19. To determine the absolute stereochemistry of these stereocentres, the synthesis of two possible diastereomeric urethanes, 13a and 13b, was carried out (Scheme 4). The stereochemistry of two diastereomeric lactones with the siloxymethyl group (Scheme 4) was determined by NMR spectral analysis. The 1H NMR spectra of the synthetic urethane 13a and the natural C15–C20 fragment 13 were identical, and the CD spectra of both 13a and 13 showed a negative maximum at 251 nm. These results disclosed the absolute stereochemistry at C17 and C19 in fragment 13.6,10
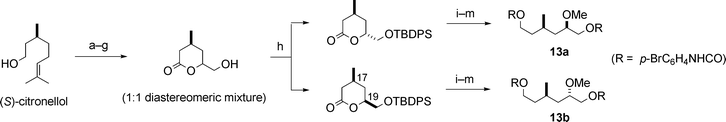 | ||
| Scheme 4 Synthesis of 13a and 13b. Reagents and conditions: (a) PDC, DMF; (b) CH2N2; (c) O3, MeOH, then NaBH4; (d) o-NO2C6H4SeCN, Bu3P; (e) 30% H2O2; (f) OsO4, pyridine, then NaHSO3; (g) TsOH; (h) t-BuPh2SiCl (TBDPSCl), imidazole, then silica gel chromatographic separation; (i) LiAlH4; (j) TBDPSCl, imidazole; (k) CH2N2, silica gel; (l) Bu4NF; (m) p-BrC6H4NCO, pyridine. | ||
Third, the absolute stereochemistry of the C21–C34 fragment 14 was determined as follows. Since the relative stereochemistry for each of the two parts, C23–C26 and C29–C32, in the fragment 14 was elucidated to be syn–anti–anti as described above, the fragment 14 must be one of four possible stereoisomers, i.e.14, 14a and their enantiomers. Thus, the enantioselective synthesis of the two urethanes 14a and ent-14, was performed, illustrated in Schemes 5 and 6, respectively. By comparison of the spectroscopic data for 14a and ent-14 with those of the natural fragment 14, the synthetic urethane ent-14 proved to be identical with the natural one 14 except for the sign of the optical rotation, thus establishing the absolute stereochemistry of eight stereocentres (C23, C24, C25, C26, C29, C30, C31 and C32) in the natural fragment 14.6,8
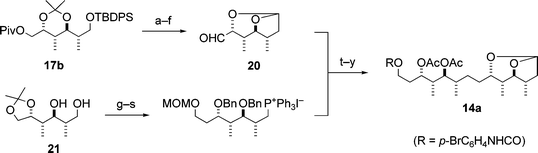 | ||
| Scheme 5 Synthesis of 14a. Reagents and conditions: (a) Bu4NF; (b) TsCl, pyridine; (c) NaCN; (d) DIBAL; (e) AcOH–H2O; (f) Swern oxidation; (g) t-BuPh2SiCl (TBDPSCl), imidazole; (h) BnBr, NaH; (i) AcOH–EtOH–H2O; (j) TsCl, pyridine; (k) K2CO3, MeOH; (l) 1,3-dithiane, BuLi; (m) BnBr, NaH; (n) CuCl2, CuO, acetone–H2O; (o) NaBH4, EtOH; (p) MeOCH2Cl (MOMCl), i-Pr2NEt; (q) Bu4NF; (r) I2, PPh3, imidazole; (s) PPh3; (t) NaN(TMS)2; (u) H2, Pd/C, EtOH; (v) HCl, MeOH–H2O; (w) TrCl, pyridine, then Ac2O; (x) AcOH–H2O; (y) p-BrC6H4NCO, pyridine. | ||
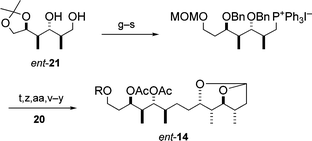 | ||
| Scheme 6 Synthesis of ent-14. Reagents and conditions: As for Scheme 5, with (z) Ca, liq. NH3; (aa) H2, Rh/Al2O3, EtOAc. | ||
With the absolute stereochemistry of the three fragments 12, 13 and 14 in hand, we were able to define the complete absolute stereostructure of aplyronine A, as shown by structure 1. Aplyronine A (1) was shown to be an inseparable mixture of four diastereomers of two amino acid esters, which were epimerized partially or almost completely. Later, this was confirmed by the enantioselective total synthesis of 1 as a diastereomeric mixture of the amino acid esters with the same ratios as in the case of natural 1.
4 Structures of aplyronines B–H
4.1 Aplyronines B and C
Aplyronine B (2)5,6 was shown to be an isomer of aplyronine A (1) on the basis of the same molecular formula C59H101N3O14 as that of 1 and the 1H NMR data similar to those for 1 (Fig. 6). The NMR signals were assigned by DQF-COSY and HMQC experiments. Marked differences between 2 and 1 were observed in the chemical shifts due to H7 and H9, namely a high-field shift (Δδ −1.08) of the signal at H7 and a low-field shift (Δδ +1.64) of the signal at H9 in 2 in comparison with 1, indicating that the N,N,O-trimethylserine ester part is at C9 in 2, whereas the same group is at C7 in 1. The gross structure of 2 was confirmed by the long-range H–C correlation in the HMBC spectrum. The split NMR signals arising from the amino acid parts, N,N,O-trimethylserine and N,N-dimethylalanine, showed that these parts epimerized in the ratios of ca. 1 : 1 and ca. 4 : 1, respectively.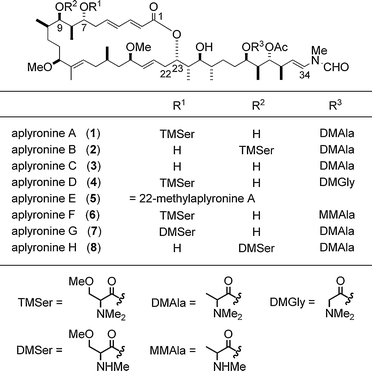 | ||
| Fig. 6 Structures of aplyronines A–H (1–8). | ||
The structure of aplyronine C (3) was shown to be the de-O-trimethylseryl analogue of aplyronine A (1): in the 1H NMR data the differences between 3 and 1 were the lack of signals due to the N,N,O-trimethylserine ester part and the high-field shift of the signal due to H7 in 3 (Fig. 6). Analysis of the DQF-COSY spectrum along with the HMQC and HMBC experiments on 3 supported the aforementioned structure for 3. The NMR spectra of 3 revealed that this compound was also a diastereomeric mixture at the N,N-dimethylalanine ester in a ratio of ca. 2 : 1.
The structures including the absolute stereochemistry for aplyronines B (2) and C (3) were confirmed by the enantioselective synthesis of 2 and 3.11,12 The synthesis of 2 and 3 proved the predominance of the S configuration (L-form) of the amino acid moieties, suggesting that the original L-amino acids epimerized during the aplyronines' biosynthesis or in the isolation process.
4.2 Aplyronines D–H
The structural elucidation of aplyronines D–H (4–8) was performed on the basis of the FABMS data and detailed analysis of the NMR (1D and 2D) spectral data, which enabled us to deduce their structures (Fig. 6). While aplyronine E (5) was inferred to be 22-methylaplyronine A, aplyronine D (4), F (6), G (7) and H (8) were deduced to contain two amino acid residues, one of which was different from both of those in aplyronine A (1). In addition, the location of one of the two amino acid residues in aplyronine H (8) was shown to be different from that of one of the two amino acid residues in aplyronine A (1). In short, the structural diversity of the aplyronines results from variation in the amino acid residues, as well as difference in their locations.5 Enantioselective total synthesis of aplyronines A–C
The enantioselective total synthesis of aplyronine A (1) achieved by the Yamada group12–14 resulted in the following benefits. First, the enantioselective synthesis of 1 unambiguously confirmed its absolute stereostructure. Second, since the supply of the sample of 1 from the natural source was quite limited, the synthesis provided a sufficient amount of 1 to enable further biological evaluation. Thirdly, the structure–activity relationships of 1 could be examined by using the analogues and the intermediates prepared in the course of the synthesis of 1. In addition, the enantioselective synthesis of aplyronines B (2) and C (3) was performed, which firmly established their absolute stereostructures deduced by spectroscopic means.5.1 Aplyronine A
The strategic disconnections employed in the enantioselective synthesis of aplyronine A (1) are shown in Scheme 7. The strategy included the following key operations: (1) construction of three of the four contiguous stereocentres, C7–C10 in the C5–C11 segment 24, C23–C26 in the C21–C27 segment 27 and C29–C32 in the C28–C34 segment 28, by the Evans aldol reaction and the Sharpless asymmetric epoxidation; (2) synthesis of the C5–C20 segment 22 by connecting the three segments 24, 25 and 26 in sequential order; (3) preparation of the C21–C34 segment 23 by connecting the two segments 27 and 28; (4) Julia olefination between the C5–C20 segment 22 and the C21–C34 segment 23 followed by Horner–Wadsworth–Emmons (HWE) reaction and Yamaguchi macrolactonization.15,16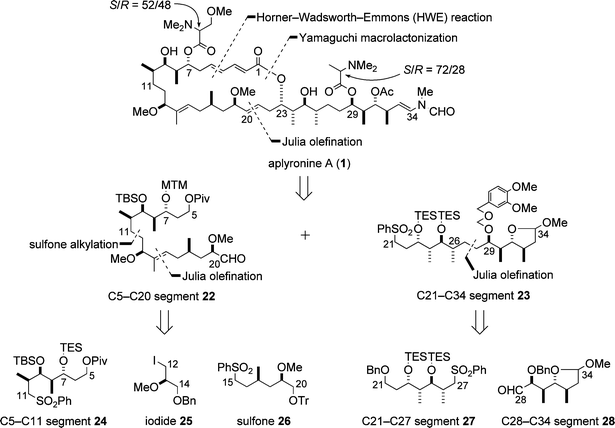 | ||
| Scheme 7 Strategy for the enantioselective synthesis of aplyronine A (1). | ||
5.1.1 C5–C20 segment. Scheme 8 illustrates the synthesis of the C5–C20 segment 22, which began with the Evans aldol reaction between the aldehyde 29 and the imide 30. In the synthetic sequence, the oxidation of hydroxyl groups by the Dess–Martin procedure was frequently employed, which was mild enough to avoid undesired reactions such as epimerization of the methoxy group in the synthetic intermediates.
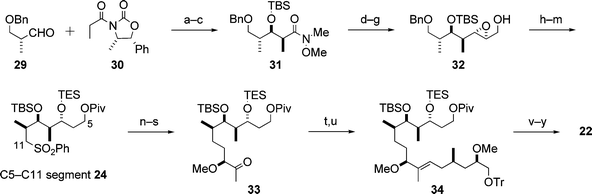 | ||
| Scheme 8 Enantioselective synthesis of the C5–C20 segment 22. Reagents and conditions: (a) 30, Bu2BOTf, Et3N, then 29; (b) MeONHMe·HCl, Me3Al; (c) t-BuMe2SiOTf (TBSOTf), 2,6-lutidine, 84% (3 steps); (d) DIBAL; (e) (i-PrO)2P(O)CH2COOEt, t-BuOK; (f) DIBAL; (g) Sharpless AE, 96% (4 steps); (h) Red-Al; (i) pivaloyl chloride (PivCl), pyridine; (j) H2, Pd/C, EtOH; (k) (PhS)2, Bu3P; (l) Et3SiCl (TESCl), imidazole; (m) m-CPBA, NaHCO3, 66% (6 steps); (n) 24, LDA, HMPA, then 25; (o) 5% Na(Hg), Na2HPO4; (p) H2, Pd/C, NaHCO3, EtOH; (q) Dess–Martin periodinane, pyridine; (r) Me2CuLi; (s) Dess–Martin periodinane, pyridine, 63% (6 steps); (t) 26, BuLi, then 33; (u) 6% Na(Hg), Na2HPO4, 44% (2 steps); (v) AcOH–H2O; (w) DMSO, Ac2O, AcOH; (x) HCO2H; (y) Dess–Martin periodinane, pyridine, 60% (4 steps). | ||
5.1.2 C21–C34 segment. The synthesis of the C21–C34 segment 23 is shown in Scheme 9. The synthesis was carried out by connection of the C21–C27 and C28–C34 segments 27 and 28, which have the syn–anti–anti sequences with regard to the four contiguous stereocentres and are enantiomeric. Thus, as starting compounds, antipodal Evans chiral auxiliaries, 30 and ent-30, were employed for the synthesis of two segments 28 and 27, respectively (Scheme 9). Both 27 and 28 were prepared by a synthetic strategy similar to that for the C5–C11 segment 24 (cf.Scheme 8). Selection of a protecting group for the C29 hydroxyl group in the C21–C34 segment 23 was crucial to the synthesis of aplyronine A (1): the [(3,4-dimethoxybenzyl)oxy]methyl ether group at C29 could be removed under mild conditions at a later stage of the synthesis without decomposition of the α,β,γ,δ-unsaturated lactone moiety in the synthetic intermediates, whereas the [(4-methoxybenzyl)oxy]methyl ether group that is often employed was removed with the concomitant decomposition of the α,β,γ,δ-unsaturated lactone moiety in the intermediates.
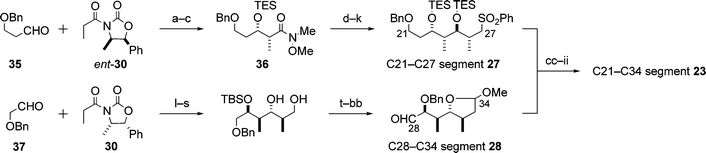 | ||
| Scheme 9 Enantioselective synthesis of the C21–C34 segment 23. Reagents and conditions: (a) ent-30, Bu2BOTf, Et3N, then 35; (b) MeONHMe·HCl, Me3Al; (c) Et3SiCl (TESCl), imidazole, 84% (3 steps); (d) DIBAL; (e) (i-PrO)2P(O)CH2COOEt, t-BuOK; (f) DIBAL; (g) Sharpless AE; (h) Me2CuLi; (i) (PhS)2, Bu3P; (j) TESCl, imidazole; (k) m-CPBA, NaHCO3, 69% (8 steps); (l) 30, Bu2BOTf, Et3N, then 37; (m) MeONHMe·HCl, Me3Al; (n) t-BuMe2SiCl (TBSCl), imidazole; (o) DIBAL; (p) (i-PrO)2P(O)CH2COOEt, t-BuOK; (q) DIBAL; (r) Sharpless AE; (s) Me2CuLi, 60% (8 steps); (t) TsCl, pyridine; (u) NaCN; (v) DIBAL, then silica gel; (w) CSA, MeOH; (x) Na, liq. NH3; (y) t-BuPh2SiCl (TBDPSCl), imidazole; (z) BnBr, NaH; (aa) Bu4NF; (bb) Swern oxidation, 50% (9 steps); (cc) 27, BuLi, then 28; (dd) 5% Na(Hg), Na2HPO4; (ee) Ca, liq. NH3, i-PrOH; (ff) H2, Rh/Al2O3, EtOH; (gg) (PhS)2, Bu3P; (hh) m-CPBA, NaHCO3; (ii) 3,4-(MeO)2C6H3CH2OCH2Cl, i-Pr2NEt, 71% (7 steps). | ||
5.1.3 Macrolactonization. With the C5–C20 and C21–C34 segments 22 and 23 in hand, the stage was set for the task of connecting these two segments, which is illustrated in Scheme 10. One of the key reactions in the synthesis of aplyronine A (1) is the macrolactonization of the seco-acid 39, which was carried out by the modified Yamaguchi method15,16 to afford the 24-membered lactone 40 and the 26-membered lactone 41 in a ratio of ca. 4 : 3. The latter lactone 41 was isomerized to 40 under equilibrium conditions (40/41 = ca. 2.5 : 1) in the presence of Ti(O-i-Pr)4.
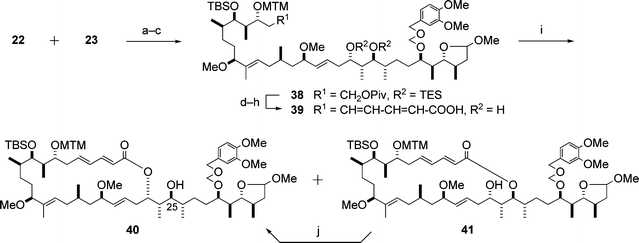 | ||
Scheme 10 Enantioselective synthesis of the macrolactone 40. Reagents and conditions: (a) 23, BuLi, then 22; (b) Ac2O, DMAP, pyridine; (c) 5% Na(Hg), Na2HPO4, 88% (3 steps); (d) DIBAL; (e) Dess–Martin periodinane, pyridine; (f) LDA, (EtO)2P(O)CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCOOEt; (g) HF·pyridine, pyridine; (h) LiOH, MeOH–H2O, 76% (5 steps); (i) 2,4,6-trichlorobenzoyl chloride, DMAP, Et3N, CHCl3, 40 (40%) and 41 (28%); (j) Ti(O-i-Pr)4, CH2Cl2, 65%. CHCOOEt; (g) HF·pyridine, pyridine; (h) LiOH, MeOH–H2O, 76% (5 steps); (i) 2,4,6-trichlorobenzoyl chloride, DMAP, Et3N, CHCl3, 40 (40%) and 41 (28%); (j) Ti(O-i-Pr)4, CH2Cl2, 65%. | ||
5.1.4 Synthesis of aplyronine A. The synthesis of aplyronine A (1) is illustrated in Scheme 11. The terminal N-methyl-N-vinylformamide structure was constructed by application of the Pattenden procedure17 to the aldehyde 42. As for the introduction of N,N-dimethylalanine into the alcohol 43, the following phenomena were observed: (i) (S)-N,N-dimethylalanine differs from (R)-N,N-dimethylalanine with regard to the rate of the esterification reaction with 43, and (ii) the activated forms of (S)- and (R)-N,N-dimethylalanines are interconvertible with each other. The same phenomena as described above were observed for the introduction of the N,N,O-trimethylserine moiety into the hydroxyl group at C7 (Scheme 11, step l). In consideration of these phenomena, the hydroxyl group of 43 was esterified with N,N-dimethylalanine (S/R = 3 : 2) under Keck conditions18 to give a diastereomeric mixture of dimethylalanine esters (S/R = 4 : 1), which, via a three-step sequence, led to aplyronine A (1). Thus, the enantioselective synthesis of aplyronine A (1) was achieved by a convergent route in 0.39% overall yield based on the longest linear sequence (47 steps); the average yield for each step was 89%.
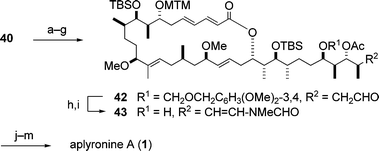 | ||
| Scheme 11 Completion of the enantioselective synthesis of aplyronine A (1). Reagents and conditions: (a) t-BuMe2SiCl (TBSCl), imidazole; (b) HCl, H2O–DME; (c) NaBH(OMe)3, MeOH; (d) TrCl, pyridine; (e) Ac2O, DMAP, pyridine; (f) HCOOH; (g) Dess–Martin periodinane, pyridine, 51% (7 steps); (h) MeNHCHO, PPTS, hydroquinone, 3 Å molecular sieves, benzene; (i) DDQ, buffer (pH 6), CH2Cl2, t-BuOH, 42% (2 steps); (j) N,N-dimethylalanine (S/R = 3 : 2), DCC, CSA, DMAP; (k) AgNO3, 2,6-lutidine, H2O–THF; (l) N,N,O-trimethylserine (S/R = 5 : 2), DCC, CSA, DMAP; (m) HF·pyridine, pyridine, 23 °C, 65% (4 steps). | ||
5.2 Aplyronines B and C
The enantioselective synthesis of aplyronine B (2) was performed by the Yamada group,11,12 and is outlined in Scheme 12. As the starting compound, the synthetic intermediate 40 of aplyronine A (1) (Scheme 10) was employed. Selection of the protecting group for the C25 hydroxyl group in 40 was important for the manipulation of the hydroxyl groups at C7, C9 and C29 in the later stages of the synthesis. A sequence of seven steps involving the protection of the C25 hydroxyl group as the MTM ether was required to transform the lactone 40 into the aldehyde 44, which was further converted to the alcohol 45 by two steps. Introduction of two kinds of amino acids into 45via a four-step sequence led to aplyronine B (2).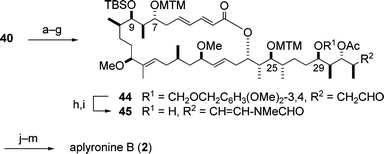 | ||
| Scheme 12 Enantioselective synthesis of aplyronine B (2). Reagents and conditions: (a) DMSO, Ac2O, AcOH; (b) HCl, H2O–DME; (c) NaBH(OMe)3, MeOH; (d) TrCl, pyridine; (e) Ac2O, DMAP, pyridine; (f) HCOOH; (g) Dess–Martin periodinane, pyridine, 18% (7 steps); (h) MeNHCHO, PPTS, hydroquinone, 3 Å molecular sieves, benzene; (i) DDQ, buffer (pH 6), CH2Cl2, t-BuOH, 29% (2 steps); (j) N,N-dimethylalanine (S/R = 8 : 5), DCC, CSA, DMAP; (k) HF·pyridine, pyridine; (l) N,N,O-trimethylserine (S/R = 1 : 0), DCC, CSA, DMAP; (m) AgNO3, 2,6-lutidine, H2O–THF, 50% (4 steps). | ||
The enantioselective synthesis of aplyronine C (3) achieved by Yamada and co-workers11,12 is illustrated in Scheme 13. The synthetic intermediate 43 of aplyronine A (1) (Scheme 11) was transformed into aplyronine C (3) via a three-step sequence.
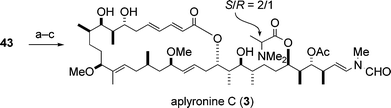 | ||
| Scheme 13 Enantioselective synthesis of aplyronine C (3). Reagents and conditions: (a) N,N-dimethylalanine (S/R = 1 : 1), DCC, CSA, DMAP; (b) AgNO3, 2,6-lutidine, H2O–THF; (c) HF·pyridine, pyridine, 27% (3 steps). | ||
6 Synthetic studies on the aplyronines
Studies directed towards the synthesis of aplyronines have also been carried out: in the following sections, the results of the synthetic studies are summarized with the emphasis on strategies, key reactions and novel methods.6.1 Paterson’s approach
Paterson proposed a synthetic strategy for aplyronine A (1), which is outlined in Scheme 14.19,20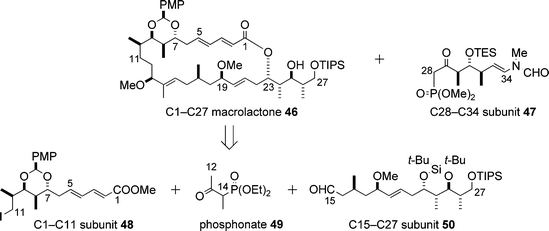 | ||
| Scheme 14 Strategy proposed by Paterson for the synthesis of aplyronine A (1). | ||
A key part of the synthesis of the C1–C11 subunit 48 was the temporary masking of the C1–C5 (E,E)-diene ester as an alkyne ester, making it easy to execute a boron-mediated anti-aldol reaction21 (Paterson aldol reaction) for the installation of the C7 and C8 stereocentres (Scheme 15: 51 + 52 → 53).20 After stereoselective reduction of the keto group in 53 under Evans–Tishchenko conditions, the alkyne ester part of 54 was isomerized to the (E,E)-diene ester moiety in the product 55, which was converted to the C1–C11 subunit 48via a four-step sequence.
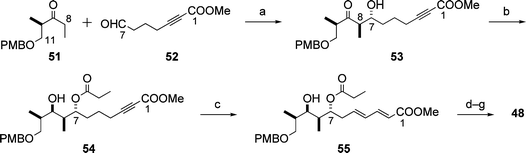 | ||
| Scheme 15 Synthesis of the C1–C11 subunit 48. Reagents and conditions: (a) 51, (c-Hex)2BCl, Et3N, then 52, 96%; (b) SmI2, EtCHO, then 53, 77%; (c) PPh3, PhOH, 95%; (d) K2CO3, MeOH; (e) p-MeOC6H4CH(OMe)2, PPTS; (f) DDQ, buffer (pH 7); (g) I2, PPh3, imidazole, 55% (4 steps). | ||
For the synthesis of the C15–C27 subunit 50 a tin(II)-mediated syn-aldol reaction22 (Paterson aldol reaction) between the aldehyde 56 and the ketone 57 was executed to afford the adduct 58, which was transformed into the aldehyde 59 in four steps involving the anti-reduction of the keto group in 58 (Scheme 16). The Horner–Wadsworth–Emmons (HWE) reaction between 59 and 60 gave the (E)-enone 61. 1,2-Reduction of 61 was effected by the use of Corey's oxazaborolidine (S)-63 to afford the alcohol 62, which led to the C15–C27 subunit 50via a three-step sequence.
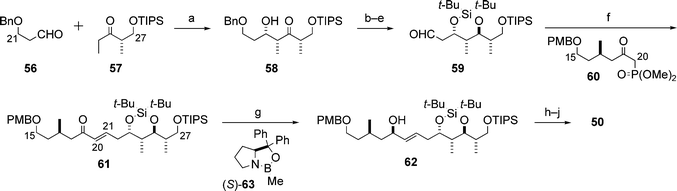 | ||
| Scheme 16 Synthesis of the C15–C27 subunit 50. Reagents and conditions: (a) 57, Sn(OTf)2, Et3N, then 56, 88%; (b) Me4NBH(OAc)3, AcOH; (c) (t-Bu)2Si(OTf)2, 2,6-lutidine; (d) H2, Pd/C, EtOAc; (e) Dess–Martin periodinane, 78% (4 steps); (f) 60, Ba(OH)2, then 59, 94%; (g) (S)-63, BH3·SMe2, 98%; (h) MeI, NaH; (i) DDQ, buffer (pH 7); (j) Dess–Martin periodinane, 85% (3 steps). | ||
With the two subunits 48 and 50 in hand, Paterson and co-workers carried out the synthesis of the C1–C27 macrolactone 46 on the basis of the efficient three-component coupling of the subunits 48, 49 and 50, and subsequent macrolactonization and isomerization (Scheme 17).23 It is notable that, in the HWE olefination reaction between 50 and the phosphonate 65, complete stereoselectivity for the formation of the (E)-trisubstituted double bond in the product 66 was realised. The Paterson group have developed a novel method for the selective oxidation/deprotection of allylic silyl ethers by DDQ under neutral conditions in the presence of other silyl protecting groups:24 this method was applied for the conversion of the intermediate 67 into the aldehyde 68, which led to the 26-membered macrolactone 69 in three steps. Isomerization of 69 afforded the desired C1–C27 macrolactone 46 (Scheme 17).
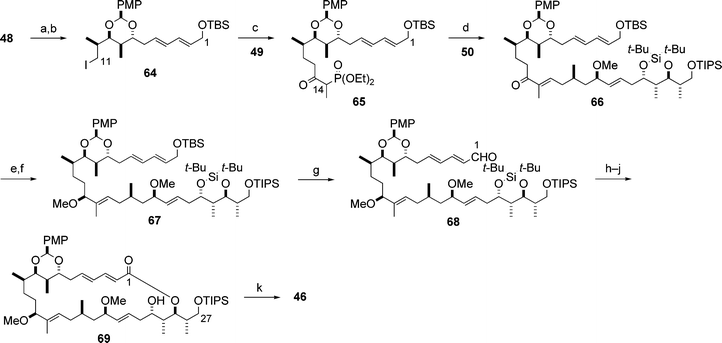 | ||
| Scheme 17 Synthesis of the C1–C27 subunit 46. Reagents and conditions: (a) DIBAL; (b) t-BuMe2SiOTf (TBSOTf), 2,6-lutidine, 94% (2 steps); (c) 49, NaH/BuLi, then 64, 95%; (d) 65, Ba(OH)2, then 50, 80%; (e) (R)-63, BH3·SMe2, 83%; (f) MeI, NaH, 90%; (g) DDQ, buffer (pH 7), 92%; (h) NaClO2, NaH2PO4, 2-methylbut-2-ene; (i) HF·pyridine, pyridine; (j) 2,4,6-trichlorobenzoyl chloride, DMAP, Et3N, 65% (3 steps); (k) Ti(O-i-Pr)4, CH2Cl2, 80%. | ||
Furthermore, the Paterson group reported a synthesis of the C28–C34 subunit 76 that contained the terminal N-methyl-N-vinylformamide group (Scheme 18).25 The synthesis of 76 began with the Paterson anti-aldol reaction between the aldehyde 70 and the ketone 71. The N-methyl-N-vinylformamide part was introduced by a Wittig olefination26 of the aldehyde 73 with the ylide generated from the phosphonium salt 74.
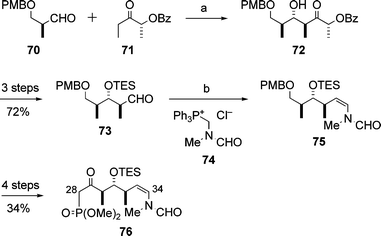 | ||
| Scheme 18 Synthesis of the C28–C34 subunit 76. Reagents and conditions: (a) 71, (c-Hex)2BCl, Me2NEt, then 70, 94%; (b) 74, LiHMDS, then 73, 72%. | ||
6.2 Marshall’s approach
The Marshall group disclosed the synthesis of the C5–C20 and C21–C34 subunits, 85 and 87, of aplyronine A (1) (Schemes 19 and 20).27 These subunits contained all the 15 stereocentres of the core structure of 1. The key features of the synthesis were the construction of three contiguous stereocentres (stereotriad units) by means of enantioselective and diastereoselective additions of in situ-generated chiral allenylindium reagents to α-methyl-β-oxygenated propionaldehydes28 (e.g., 77 + 78 → 79 in Scheme 19). The products of these addition reactions were transformed into the desired C5–C20 and C21–C34 subunits, 85 and 87, respectively, by a sequence of reactions involving the Sharpless asymmetric epoxidation.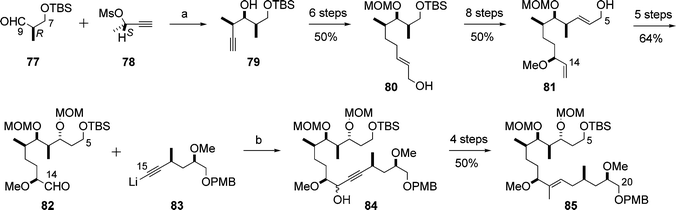 | ||
| Scheme 19 Synthesis of the C5–C20 subunit 85. Reagents and conditions: (a) Pd(dppf)Cl2, InI, 63–75%; (b) THF, 70%. | ||
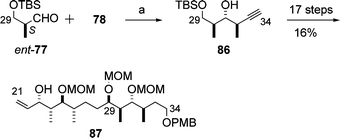 | ||
| Scheme 20 Synthesis of the C21–C34 subunit 87. Reagents and conditions: (a) Pd(dppf)Cl2, InI, 79%. | ||
6.3 Calter’s approach
In 2002, the Calter group reported the synthesis of the C21–C34 subunit 94 of aplyronine A (1) using the methylketene dimer 88, available in both enantiomeric forms by asymmetric catalysis (Scheme 21).29 A key feature of Calter's synthesis of the C21–C34 subunit 94 was the opening of the methylketene dimer 88, followed by the aldol reaction of the resulting lithium enolate,30 which provided the syn-dipropionate aldol adduct 89. Similarly, the enantiomer ent-88 afforded the adduct 91. Diastereoselective reduction and functional group manipulation of 89 and 91 yielded the phosphonium salt 90 and the aldehyde 93, respectively. The Wittig coupling reaction between 90 and 93, and hydrogenation of the resulting olefin, afforded the C21–C34 subunit 94.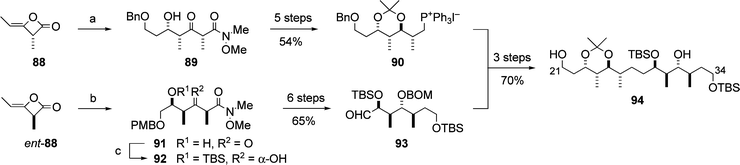 | ||
| Scheme 21 Synthesis of the C21–C34 subunit 94. Reagents and conditions: (a) LiN(OMe)Me, then BnOCH2CH2CHO, 57%; (b) LiN(OMe)Me, then PMBOCH2CHO; (c) t-BuMe2SiOTf (TBSOTf), 2,6-lutidine, then KBEt3H, 29% (2 steps). | ||
Subsequently, the Calter group reported the synthesis of the C1–C27 subunit 102 (Scheme 22):31 the synthesis began with reduction of the methylketene dimer ent-88, followed by acylation. The resulting β-acyloxyl ketone 95 underwent boron-mediated aldol reaction32 to give anti-aldol adduct 96, which was converted to the phosphonate 97. The assembly of 97 and the aldehyde 98 prepared from D-glucose (9 steps) was effected by the Paterson procedure23 to yield the silyl ether 99, which was transformed into the aldehyde 100. The coupling reaction between the aldehyde 100 and the sulfone 101 derived from the aldol 89 (Scheme 21) afforded the C1–C27 subunit 102.
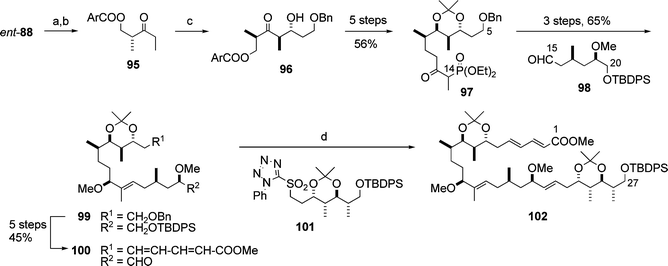 | ||
| Scheme 22 Synthesis of the C1–C27 subunit 102. Reagents and conditions: (a) LiAlH4; (b) (ArCO)2O (Ar = 3,4-dimethoxyphenyl), DMAP, pyridine, 62% (2 steps); (c) (c-Hex)2BCl, Et3N, then BnOCH2CH2CHO, 81%; (d) 101, KHMDS, then 100, 59%. | ||
6.4 Fuchs’ approach
The Fuchs group developed a novel method for the stereoselective construction of the four contiguous stereocentres (stereotetrads), which are contained in the structure of aplyronine A (1).33 The synthesis of one stereotetrad 106 corresponding to the C7–C10 part of aplyronine A (1) is illustrated in Scheme 23 as an example. The key features of the Fuchs synthesis are two sequential chemo- and stereoselective Lawton SN2′ reactions to establish the desired product stereochemistry. Oxidative cleavage of the stereotetrad 106 affords the termini-differentiated acyclic heptanyl stereotetrad.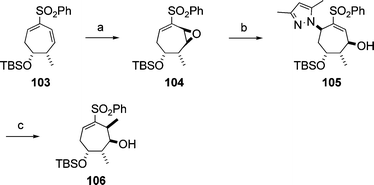 | ||
| Scheme 23 Synthesis of the stereotetrad 106. Reagents and conditions: (a) H2O2, Jacobsen salen–Mn catalyst, 79%; (b) 3,5-dimethylpyrazole, 90%; (c) MeMgBr, 95%. | ||
In 2007, Fuchs and co-workers achieved asymmetric synthesis of all eight diastereomeric seven-carbon dipropionate stereotetrads including the above-mentioned compound 106, and made possible access to all 16 enantiomers of these eight diastereomeric tetrads.34 The Fuchs group reported a novel method for the conversion of cyclic vinyl sulfones to transposed vinyl phosphonates in 2007, and this method was applied for the preparation of a complementary set of termini-differentiated ester-aldehydes, 107 and 110, corresponding to the C15–C20 part of aplyronine A (1) (Scheme 24).35
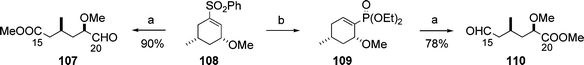 | ||
| Scheme 24 Synthesis of termini-differentiated ester-aldehydes 107 and 110. Reagents and conditions: (a) O3, NaHCO3, CH2Cl2–MeOH, then PPh3; (b) HP(O)(OEt)2, NaHMDS, 93%. | ||
7 Biology of aplyronines
The results of biological studies on aplyronines are described in the following sections.7.1 Cytotoxicity
Aplyronines A–H (1–8) exhibit strong cytotoxicities against HeLa-S3 cells in vitro as shown in Table 1.3,12 Five of the aplyronines, 1, 4, 5, 6 and 7, show particularly strong cytotoxicities: the common structural feature for these five aplyronines is the presence of the methylated serine ester group at C7 of the macrolide unit. The cytotoxicities of the aplyronines depend primarily on this methylated serine ester group: (i) aplyronine A (1) is much more cytotoxic (nearly 50 times) than aplyronine C (3), which lacks the methylated serine ester group; (ii) the location of the methylated serine ester group on the macrolide part also affects the cytotoxicity (the cytotoxicity of aplyronine A (1) is considerably stronger than that of aplyronine B (2) or aplyronine H (8)).| Compound | IC50/ng mL−1 |
|---|---|
| Aplyronine A (1) | 0.48 |
| Aplyronine B (2) | 3.11 |
| Aplyronine C (3) | 21.2 |
| Aplyronine D (4) | 0.075 |
| Aplyronine E (5) | 0.201 |
| Aplyronine F (6) | 0.201 |
| Aplyronine G (7) | 0.127 |
| Aplyronine H (8) | 10.4 |
7.2 Antitumour activity
Aplyronine A (1) exhibited exceedingly potent antitumour activities in vivo against five tumour cell lines, which are summarized in Table 2.5,6 The antitumour activities of 1 were shown to be dose-dependent. In particular, the antitumour activities against P388 leukemia, Lewis lung carcinoma and Ehrlich carcinoma are remarkable. Notably, the number of survivors at the end of the experimental term (after 60 days) was large in the case of Lewis lung carcinoma and P388 leukemia. Aplyronine A (1) is thus regarded as a candidate antitumour agent.36| Tumour | Routea | Dose/mg kg−1 day−1 | Survival time | No. of survivors after 60 days | |
|---|---|---|---|---|---|
| Median time/days | Test/control (%) | ||||
| a Schedule: i.p. days 1–5. Aplyronine A (1) was dissolved in DMSO (0.08 mg mL−1) and then diluted with a physiological solution of NaCl. | |||||
| P388 leukemia | i.p. | 0.08 | 59.9 | 545 | 4/6 |
| 0.04 | 46.0 | 418 | 2/6 | ||
| 0.02 | 17.3 | 157 | 0/6 | ||
| Control | — | 11.0 | 100 | 0/7 | |
| Colon C26 carcinoma | i.p. | 0.08 | 40.0 | 255 | 0/6 |
| 0.04 | 39.0 | 248 | 1/6 | ||
| 0.02 | 25.0 | 159 | 1/6 | ||
| Control | — | 15.7 | 100 | 0/10 | |
| Lewis lung carcinoma | i.p. | 0.08 | 9.3 | 86 | 0/6 |
| 0.04 | 60.1 | 556 | 6/6 | ||
| 0.02 | 59.9 | 555 | 4/6 | ||
| Control | — | 10.8 | 100 | 0/8 | |
| B16 melanoma | i.p. | 0.08 | 10.1 | 43 | 0/6 |
| 0.04 | 46.8 | 201 | 0/6 | ||
| 0.02 | 43.0 | 185 | 0/6 | ||
| Control | — | 23.3 | 100 | 1/9 | |
| Ehrlich carcinoma | i.p. | 0.08 | 12.0 | 80 | 0/6 |
| 0.04 | 59.7 | 398 | 2/6 | ||
| 0.02 | 33.0 | 220 | 1/6 | ||
| Control | — | 15.0 | 100 | 0/8 | |
7.3 Interaction with actin
The target biomolecule of aplyronine A (1) was revealed to be actin by Karaki, Yamada and co-workers in 1996.37 Actin is one of the most abundant and common proteins in the cytoskeleton and regulates various cell functions such as muscle contraction, cell motility and cell division. Aplyronine A (1) was found to form a 1 : 1 complex with globular actin (G-actin, monomer), and it was shown that aplyronine A (1) inhibited polymerization of G-actin to fibrous actin (F-actin, polymer) and depolymerized F-actin to G-actin by severing.37 Prior to the studies on the actin–aplyronine A (1) interaction, Karaki, Fusetani and co-workers reported in 1994 that mycalolide B (111),38,39 a cytotoxic macrolide isolated from a marine sponge, depolymerized F-actin by severing and formed a 1 : 1 complex with G-actin.40,41 Considering the fact that the side chain structure of aplyronine A (1) is remarkably similar to that of mycalolide B (111), it was suggested that the side chain part of aplyronine A (1) might play an important role in its binding activity towards actin.37,41In the 1980s, the Kashman group identified latrunculins (e.g., latrunculin A (112)) of marine origin and investigated their biological properties:42,43 latrunculins were the first actin-binding substances to be isolated from a marine source. Since then, a considerable number of cytotoxic marine natural products that interact with actin have been identified. Three marine macrolides structurally related to aplyronine A (1) are shown in Fig. 7; kabiramide C (113),44–46 scytophycin C (114)47 and reidispongiolide A (115);48,49 all three have been shown to exhibit actin-binding activity. For comprehensive details about actin-targeting natural products, three excellent reviews are available.19,50,51
 | ||
| Fig. 7 Actin-binding marine macrolides structurally related to aplyronine A (1). | ||
7.4 Structure–activity relationships
Based on the synthesis of aplyronine A (1), we prepared a variety of synthetic analogues, which enabled us to examine the structure–activity relationships of 1. The bioactivities of eighteen synthetic analogues and natural aplyronines were evaluated.12,52,53 Selected data on the cytotoxicity and actin depolymerizing activity of the natural and synthetic compounds are shown in Fig. 8.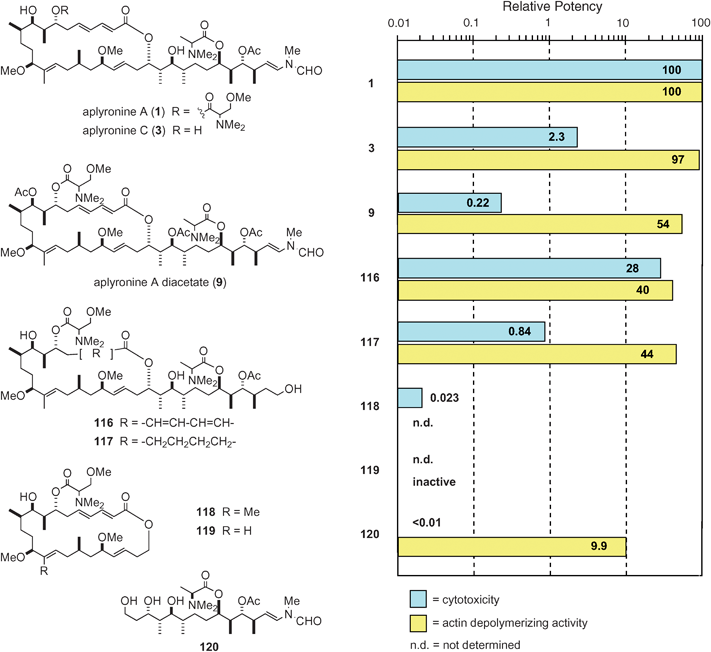 | ||
| Fig. 8 Structure–activity relationships of aplyronine A (1) and its analogues. | ||
7.4.1 Structure–cytotoxicity relationships. Some of the functional groups of the macrolide part of aplyronine A (1) were found to have a strong influence on the cytotoxicity (Fig. 8): (i) Comparison of the cytotoxicity of aplyronines A (1) and C (3) indicated that the N,N,O-trimethylserine ester moiety plays an important role in the strong cytotoxicity of 1: aplyronine A (1) is about 50-fold more cytotoxic than 3 (see Table 1); (ii) The two hydroxyl groups in 1 were shown to be responsible for the strong cytotoxicity through comparison of the cytotoxicities of 1 and its diacetate 9: the former is nearly 500-fold more cytotoxic than the latter; (iii) Comparison of the cytotoxicities of the two synthetic analogues 116 and 117 revealed that the conjugated diene moiety is responsible for the strong cytotoxicity of 1.
In 1996, the side chain part of aplyronine A (1) was found to be essential for its strong cytotoxicity.12 In further studies53 the effect of the side chain part of 1 on cytotoxicity was evaluated in more detail: not only the presence of the side chain part but also its length is crucial. For example, the cytotoxicity of the macrolactone 118 without the side chain was extremely weak. It should be noted that the synthetic analogue 120 only consisting of the side chain was almost non-cytotoxic.
To sum up, in order for aplyronine A (1) to exhibit strong cytotoxicity, the combination of the macrolide part and the side chain part is essential, and furthermore, the presence of the N,N,O-trimethylserine ester, the conjugated diene and two hydroxyl groups is necessary, while the C14 methyl, the N-methyl-N-vinylformamide and the N,N-dimethylalanine ester groups are not important.
7.4.2 Structure–actin depolymerizing activity relationships. Some derivatives of aplyronine A (1) (e.g., 9 in Fig. 8) exhibited strong actin depolymerizing activity comparable to that of 1, whereas those with a shorter side chain (e.g., 121) are approximately 100 times less active than 1, and the synthetic analogue 119 without a side chain is totally inactive (Fig. 8).
It is notable that the synthetic analogue 120 only comprising the side chain portion of 1 exhibits relatively strong activity despite the small size of the molecule (Fig. 8). These results indicate that (i) the side chain part in 1 plays a key role in its activity, (ii) the macrolide part enhances the actin depolymerizing activity of the side chain portion to some extent. In addition, the C14 methyl group in aplyronine A (1) was not important for the strong activity of 1, as is the case of the cytotoxicity of 1. In connection with the synthetic analogue 120, the analogue 122 only consisting of the side chain part of mycalolide B (111) was prepared and the actin depolymerizing activity of 122 was evaluated to be stronger than that of 120.54,55
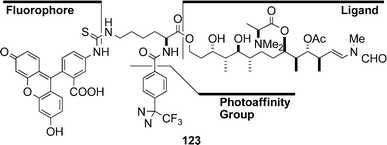
Chemical evidence of the interaction between actin and aplyronine A (1) was obtained by photoaffinity labelling experiments:56 actin was photolabelled with a probe 123 containing the side chain portion of 1, prepared from the synthetic analogue 120. Since aplyronine A (1) competitively inhibited the binding of the probe 123 to actin, the probe 123 was revealed to bind to actin specifically at the binding site of aplyronine A (1). The results indicate that the side chain part of 1 possesses actin-binding capability. Mycalolide B (111) also inhibited the binding of the probe 123 to actin, indicating that mycalolide B (111) binds to actin at the same site as the probe 123: thus it became clear that aplyronine A (1) and mycalolide B (111) bind to actin at the same site.
7.4.3 Relation between cytotoxicity and actin depolymerizing activity. As described in Sections 7.4.1 and 7.4.2, the side chain portion of aplyronine A (1) proved to be crucial for both cytotoxicity and actin depolymerizing activity. On the other hand, cytotoxicity of 1 turned out to be influenced to a large extent by the N,N,O-trimethylserine ester, the conjugated diene and two hydroxyl groups (three of these groups being situated on the macrolide part, except for one hydroxyl group), whereas these functional groups were shown to affect actin depolymerizing activity to a quite small extent, if any.
7.5 Structure of the actin–aplyronine A complex
While the studies on the structure–activity relationships of aplyronine A (1) were performed by using natural and synthetic analogues as described above, the three-dimensional structure of the actin–aplyronine A (1) complex (referred to as the AA complex) was determined by X-ray crystallographic analysis at atomic resolution, providing important details about the molecular interactions between actin and aplyronine A (1).57 The structure of the AA complex is shown in Fig. 9. Aplyronine A (1) binds to the hydrophobic cleft composed of subdomains 1 and 3 of actin (the barbed end) by intercalating the side chain of 1 into actin. This hydrophobic interaction in the AA complex is the same as in a complex between actin and another actin depolymerizing agent, viz. the actin–kabiramide C (113) complex reported by the Rayment group.45 Consequently, the hydrophobic interactions between the cleft of actin and aplyronine A (1) play a key role in the actin depolymerizing activity. Furthermore, the N-methyl-N-vinylformamide group, at the end of the side chain of aplyronine A (1), interacts with actin through the hydrogen bond, and this interaction seems to be important for the actin depolymerizing activity.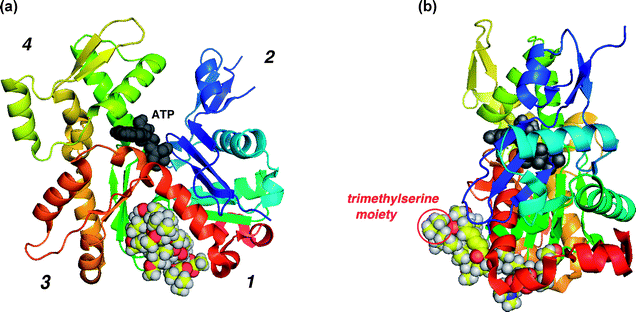 | ||
| Fig. 9 (a) Structure of the actin–aplyronine A (1) complex; (b) the same complex rotated by 90°. | ||
Both the structures of the AA complex and that of the actin–kabiramide C (113) complex45 are similar, as far as the side chain portions of two compounds are concerned. On the other hand, the macrolide part of aplyronine A (1) binds to subdomain 3 of actin in the AA complex, whereas the macrolide portion of kabiramide C (113) in the actin–kabiramide C (113) complex interacts with subdomain 1. This difference is caused by the opposite absolute stereochemistry of the carbon bearing the ethereal oxygen of the lactone moiety of each compound: the α-configuration of the oxygen at C23 in 1 and the β-configuration of the oxygen at C24 in 113. In addition, there are characteristic interactions involving a hydrogen bond between the hydroxyl group at C9 of 1 and Glu334 of actin: these interactions stabilize the conformation of the macrolide ring of aplyronine A (1) within subdomain 3.
One significant feature of the structure of the AA complex is that the N,N,O-trimethylserine moiety (S/R = 1 : 1) protrudes toward the bulk solvent region from the molecular surface of actin and does not interact with actin (Fig. 9(b)). Additionally, the aforementioned interactions between actin and aplyronine A (1) seem to function as an anchor to retain the protruding N,N,O-trimethylserine moiety in the AA complex.
Among the characteristics of the structure of the AA complex the following two points are worthy of note: (i) the macrolide part of 1 is situated in subdomain 3 of actin; and (ii) the N,N,O-trimethylserine moiety protrudes toward the bulk solvent region. These structural features are considered to be associated with the cytotoxicity of aplyronine A (1). It should be noted that the cytotoxicities of aplyronine C (3) (which lacks the N,N,O-trimethylserine moiety) and aplyronine A diacetate (9) (which has no hydroxyl group at C9) are weak (Fig. 8).
To sum up, the cytotoxic analogues of aplyronine A (1) necessarily have actin depolymerizing activity, whereas the analogues possessing actin depolymerizing activity are not necessarily cytotoxic. Thus, the actin depolymerizing activity is a necessary condition for the cytotoxicity of aplyronine A (1) but is not a sufficient condition.
From the above findings it is probable that aplyronine A (1) first binds to actin to afford the actin–aplyronine A complex, which subsequently binds to another biomolecule in order to exhibit cytotoxicity: the protruding N,N,O-trimethylserine moiety of aplyronine A (1) might play an important role in this second stage.
A comprehensive review of actin-targeting natural products by Rayment is available, which includes the precise structures of natural product–actin complexes.51
8 Concluding remarks
Following the discovery of the strongly cytotoxic aplyronines A–H (1–8) from the sea hare Aplysia kurodai, the absolute stereostructure of aplyronine A (1) was determined by spectroscopic means in conjunction with the enantioselective synthesis of the fragments obtained from chemical degradation, and enantioselective total synthesis. The structures of aplyronines B–H (2–8) were also elucidated, and the absolute stereostructures of aplyronines B (2) and C (3) determined by enantioselective total synthesis. Biological studies of the aplyronines have shown aplyronine A (1) to exhibit exceedingly potent antitumour activity in vivo. The target biomolecule of aplyronine A (1) has been shown to be actin, with aplyronine A (1) inhibiting polymerization of G-actin and depolymerizing F-actin. X-Ray crystallographic analysis has enabled detailed elucidation of the structure of the actin–aplyronine A complex, with the results showing in detail the nature of the interactions between actin and aplyronine A (1).9 Acknowledgements
The authors are grateful to Dr Tomohisa Takita, Dr Hisao Ekimoto, Dr Masayuki Arakawa, Ms Rie Tanaka and Ms Mutsuko Kimura (Nippon Kayaku Co. Ltd) for biological tests (cytotoxicity and antitumour activity), to Professor Hirokazu Hotani, Dr Tomohiko Itoh and Dr Kingo Takiguchi (Nagoya University), Professor Hiroshi Ozaki and Professor Masatoshi Hori (University of Tokyo) for donation of actin and their helpful advice, and to Dr Masaki Takata and Dr Kunio Hirata (JASRI/SPring-8) for collaboration for the X-ray crystallographic studies.10 References
- S. Yamamura and Y. Hirata, Tetrahedron, 1963, 19, 1485–1496 CrossRef CAS.
- K. Yamada and H. Kigoshi, Bull. Chem. Soc. Jpn., 1997, 70, 1479–1489 CAS.
- K. Yamada, M. Ojika, H. Kigoshi and K. Suenaga, in Drugs from the sea, ed. N. Fusetani, Karger, Basel, 2000, pp. 59–73 Search PubMed.
- H. Kamiya, R. Sakai and M. Jimbo, in Molluscs: from chemo-ecological study to biotechnological application, ed. G. Cimino and M. Gavagnin, Springer-Verlag, Berlin/Heidelberg, 2006, pp. 215–239 Search PubMed.
- K. Yamada, M. Ojika, T. Ishigaki, Y. Yoshida, H. Ekimoto and M. Arakawa, J. Am. Chem. Soc., 1993, 115, 11020–11021 CrossRef CAS.
- M. Ojika, H. Kigoshi, Y. Yoshida, T. Ishigaki, M. Nisiwaki, I. Tsukada, M. Arakawa, H. Ekimoto and K. Yamada, Tetrahedron, 2007, 63, 3138–3167 CrossRef CAS.
- M. Ojika, H. Kigoshi, T. Ishigaki and K. Yamada, Tetrahedron Lett., 1993, 34, 8501–8504 CrossRef CAS.
- M. Ojika, H. Kigoshi, T. Ishigaki, M. Nisiwaki, I. Tsukada, K. Mizuta and K. Yamada, Tetrahedron Lett., 1993, 34, 8505–8508 CrossRef CAS.
- H. Nagaoka and Y. Kishi, Tetrahedron, 1981, 37, 3873–3888 CrossRef , and references cited therein.
- M. Ojika, H. Kigoshi, T. Ishigaki, I. Tsukada, T. Tsuboi, T. Ogawa and K. Yamada, J. Am. Chem. Soc., 1994, 116, 7441–7442 CrossRef CAS.
- K. Suenaga, T. Ishigaki, A. Sakakura, H. Kigoshi and K. Yamada, Tetrahedron Lett., 1995, 36, 5053–5056 CAS.
- H. Kigoshi, K. Suenaga, T. Mutou, T. Ishigaki, T. Atsumi, H. Ishiwata, A. Sakakura, T. Ogawa, M. Ojika and K. Yamada, J. Org. Chem., 1996, 61, 5326–5351 CrossRef CAS.
- H. Kigoshi, M. Ojika, K. Suenaga, T. Mutou, J. Hirano, A. Sakakura, T. Ogawa, M. Nisiwaki and K. Yamada, Tetrahedron Lett., 1994, 35, 1247–1250 CrossRef CAS.
- H. Kigoshi, M. Ojika, T. Ishigaki, K. Suenaga, T. Mutou, A. Sakakura, T. Ogawa and K. Yamada, J. Am. Chem. Soc., 1994, 116, 7443–7444 CrossRef CAS.
- J. Inanaga, K. Hirata, H. Saeki, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989–1993 CAS.
- M. Hikota, Y. Sakurai, K. Horita and O. Yonemitsu, Tetrahedron Lett., 1990, 31, 6367–6370 CrossRef CAS.
- M. J. Kiefel, J. Maddock and G. Pattenden, Tetrahedron Lett., 1992, 33, 3227–3230 CrossRef CAS.
- E. P. Boden and G. E. Keck, J. Org. Chem., 1985, 50, 2394–2395 CrossRef CAS.
- K.-S. Yeung and I. Paterson, Angew. Chem., Int. Ed., 2002, 41, 4632–4653 CrossRef.
- I. Paterson, C. J. Cowden and M. D. Woodrow, Tetrahedron Lett., 1998, 39, 6037–6040 CrossRef CAS.
- I. Paterson, J. M. Goodman and M. Isaka, Tetrahedron Lett., 1989, 30, 7121–7124 CrossRef CAS.
- I. Paterson and R. D. Tillyer, Tetrahedron Lett., 1992, 33, 4233–4236 CrossRef CAS.
- I. Paterson, M. D. Woodrow and C. J. Cowden, Tetrahedron Lett., 1998, 39, 6041–6044 CrossRef CAS.
- I. Paterson, C. J. Cowden, V. S. Rahn and M. D. Woodrow, Synlett, 1998, 915–917 CrossRef CAS.
- I. Paterson, S. B. Blakey and C. J. Cowden, Tetrahedron Lett., 2002, 43, 6005–6008 CrossRef CAS.
- I. Paterson, C. J. Cowden and C. Watson, Synlett, 1996, 209–211 CrossRef CAS.
- J. A. Marshall and B. A. Johns, J. Org. Chem., 2000, 65, 1501–1510 CrossRef CAS.
- J. A. Marshall, J. Org. Chem., 2007, 72, 8153–8166 CrossRef CAS.
- M. A. Calter and X. Guo, Tetrahedron, 2002, 58, 7093–7100 CrossRef CAS.
- M. A. Calter, X. Guo and W. Liao, Org. Lett., 2001, 3, 1499–1501 CrossRef CAS.
- M. A. Calter and J. Zhou, Tetrahedron Lett., 2004, 45, 4847–4850 CrossRef CAS.
- M. A. Calter, W. Song and J. Zhou, J. Org. Chem., 2004, 69, 1270–1275 CrossRef CAS.
- A. El-Awa and P. L. Fuchs, Org. Lett., 2006, 8, 2905–2908 CrossRef CAS.
- A. El-Awa, X. M. du Jourdin and P. L. Fuchs, J. Am. Chem. Soc., 2007, 129, 9086–9093 CrossRef CAS.
- M. N. Noshi, A. El-Awa, E. Torres and P. L. Fuchs, J. Am. Chem. Soc., 2007, 129, 11242–11247 CrossRef CAS.
- P. Crews, W. H. Gerwick, F. J. Schmitz, D. France, K. W. Bair, A. E. Wright and Y. Hallock, Pharm. Biol., 2003, 41 Search PubMed Supplement, 39–52.
- S. Saito, S. Watabe, H. Ozaki, H. Kigoshi, K. Yamada, N. Fusetani and H. Karaki, J. Biochem., 1996, 120, 552–555 CAS.
- N. Fusetani, K. Yasumuro, S. Matsunaga and K. Hashimoto, Tetrahedron Lett., 1989, 30, 2809–2812 CrossRef CAS.
- S. Matsunaga, P. Liu, C. A. Celatka, J. S. Panek and N. Fusetani, J. Am. Chem. Soc., 1999, 121, 5605–5606 CrossRef CAS.
- S. Saito, S. Watabe, H. Ozaki, N. Fusetani and H. Karaki, J. Biol. Chem., 1994, 269, 29710–29714 CAS.
- S. Saito and H. Karaki, Clin. Exp. Pharmacol. Physiol., 1996, 23, 743–746 CrossRef CAS.
- Y. Kashman, A. Groweiss and U. Shmueli, Tetrahedron Lett., 1980, 21, 3629–3632 CrossRef CAS.
- I. Spector, N. R. Shochet, Y. Kashman and A. Groweiss, Science, 1983, 219, 493–495 CrossRef CAS.
- S. Matsunaga, N. Fusetani, K. Hashimoto, K. Koseki and M. Noma, J. Am. Chem. Soc., 1986, 108, 847–849 CrossRef CAS.
- V. A. Klenchin, J. S. Allingham, R. King, J. Tanaka, G. Marriott and I. Rayment, Nat. Struct. Biol., 2003, 10, 1058–1063 CrossRef CAS.
- J. Tanaka, Y. Yan, J. Choi, J. Bai, V. A. Klenchin, I. Rayment and G. Marriott, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13851–13856 CrossRef CAS.
- M. Ishibashi, R. E. Moore, G. M. L. Patterson, C. Xu and J. Clardy, J. Org. Chem., 1986, 51, 5300–5306 CrossRef CAS.
- M. V. D'Auria, L. G. Paloma, L. Minale, A. Zampella, J.-F. Verbist, C. Roussakis, C. Debitus and J. Patissou, Tetrahedron, 1994, 50, 4829–4834 CrossRef CAS.
- J. S. Allingham, A. Zampella, M. V. D'Auria and I. Rayment, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 14527–14532 CrossRef CAS.
- G. Fenteany and S. Zhu, Curr. Top. Med. Chem., 2003, 3, 593–616 CrossRef CAS.
- J. S. Allingham, V. A. Klenchin and I. Rayment, Cell. Mol. Life Sci., 2006, 63, 2119–2134 CrossRef CAS.
- K. Suenaga, N. Kamei, Y. Okugawa, M. Takagi, A. Akao, H. Kigoshi and K. Yamada, Bioorg. Med. Chem. Lett., 1997, 7, 269–274 CrossRef CAS.
- H. Kigoshi, K. Suenaga, M. Takagi, A. Akao, K. Kanematsu, N. Kamei, Y. Okugawa and K. Yamada, Tetrahedron, 2002, 58, 1075–1102 CrossRef CAS.
- K. Suenaga, S. Miya, T. Kuroda, T. Handa, K. Kanematsu, A. Sakakura and H. Kigoshi, Tetrahedron Lett., 2004, 45, 5383–5386 CrossRef CAS.
- K. Suenaga, T. Kimura, T. Kuroda, K. Matsui, S. Miya, S. Kuribayashi, A. Sakakura and H. Kigoshi, Tetrahedron, 2006, 62, 8278–8290 CrossRef CAS.
- T. Kuroda, K. Suenaga, A. Sakakura, T. Handa, K. Okamoto and H. Kigoshi, Bioconjugate Chem., 2006, 17, 524–529 CrossRef CAS.
- K. Hirata, S. Muraoka, K. Suenaga, T. Kuroda, K. Kato, H. Tanaka, M. Yamamoto, M. Takata, K. Yamada and H. Kigoshi, J. Mol. Biol., 2006, 356, 945–954 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |