Metal ion-catalyzed oxidative degradation of Orange II by H2O2. High catalytic activity of simple manganese salts
Erika
Ember
,
Sabine
Rothbart
,
Ralph
Puchta
and
Rudi
van Eldik
*
Inorganic Chemistry, Department of Chemistry and Pharmacy, University of Erlangen-Nürnberg, Egerlandstr. 1, 91058, Erlangen, Germany
First published on 17th November 2008
Abstract
In an effort to develop new routes for the clean oxidation of non-biodegradable organic dyes, a detailed study of some environmentally friendly Mn(II) salts that form very efficient in situcatalysts for the activation of H2O2 in the oxidation of substrates such as Orange II under mild reaction conditions, was performed. The studied systems have advantages from the viewpoint of green chemistry in that simple metal salts can be used as very efficient catalyst precursors and H2O2 is used as a green oxygen donor reagent. Oxidations were carried out in a glass reactor over a wide pH range in aqueous solution at room temperature. Under optimized conditions it was possible to degrade Orange II in a carbonate buffer solution in less then 100 s using 0.01 M H2O2 in the presence of only 2 × 10−5 M Mn(II) salt. To gain insight into the manganese catalyzed oxidation mechanism, the formation of the active catalyst was followed spectrophotometrically and appears to be the initiating step in the oxidative degradation of the dye. High valent manganese oxo species are instable in the absence of a stabilizing coordinating ligand and lead to a rapid formation of catalytically inactive MnO2. In this context, the role of the organic dye and HCO3− as potential stabilizing ligands was studied in detail. In situUV-Vis spectrophotometric measurements were performed to study the effect of pH and carbonate concentration of the buffer solution on the formation of the catalytically active species. Electrochemical measurements and DFT (B3LYP/LANL2DZp) calculations were used to study the in situ formation of the catalytic species. The catalytic cycle could be repeated several times and demonstrated an excellent stability of the catalytic species during the oxidation process. A mechanism that accounts for the experimental observations is proposed for the overall catalytic cycle.
Introduction
Nowadays, one of the major environmental problems concerns the strong increase in xenobiotic and organic substances that are persistent in the natural ecosystem. Most of these compounds have an aromatic structure, which makes them highly stable and thus difficult to degrade.1 A significant source of environmental pollution is industrial dye waste due to their visibility and recalcitrance, since dyes are highly coloured and designed to resist chemical, biochemical and photochemical degradation.2 About half of the global production of synthetic dyes (700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 t per year) are classified as aromatic azo compounds that have a –N
000 t per year) are classified as aromatic azo compounds that have a –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– unit as chromophore in their molecular structure. Over 15% of textile dyes are lost in waste water streams during the dyeing operation.3 Azo dyes are known to be largely non-biodegradable under aerobic conditions and to be reduced to more hazardous intermediates under anaerobic conditions.4 The decolorization of wastewater has acquired increasingly importance in recent years, however, there is no simple solution to this problem because the conventional physicochemical methods are costly and lead to the accumulation of sludges.5
N– unit as chromophore in their molecular structure. Over 15% of textile dyes are lost in waste water streams during the dyeing operation.3 Azo dyes are known to be largely non-biodegradable under aerobic conditions and to be reduced to more hazardous intermediates under anaerobic conditions.4 The decolorization of wastewater has acquired increasingly importance in recent years, however, there is no simple solution to this problem because the conventional physicochemical methods are costly and lead to the accumulation of sludges.5
One approach to solve these problems would be to develop low-cost, highly efficient, and environmental friendly oxidation catalysts on the basis of transition metal complexes.6,7 Recently, photodegradation methods based on TiO2 as a photocatalyst,8 beside Fenton systems,9 emerged as one of the most promising technologies and received increasing attention due to their practical and potential value in environmental protection. However, in some cases they are only successful under specific pH and temperature conditions.
Several studies were performed during the last few years in order to find good catalysts for the oxidative degradation of different organic dyes. From an environmental point of view, first row transition metals are the most challenging. Highly effective Fe,10,14Co,11Cr12 and Mn13 based oxidation catalyst were developed. In combination with different oxidizing agents, the decomposition of stable organic substances was possible. A novel highly active and environmental benign catalytic system based on Fe-TAML (TAML = tetraamido macrocyclic ligand) was recently reported by Chahbane et al.14 In many cases tremendous synthetic efforts are required to obtain an effective catalytic system and in addition the presence of high concentrations of oxidizing agents is needed. Among the possible oxidizing agents, H2O2 is one of the most commonly used owing to its eco-friendly nature. The use of H2O2 as a green oxidizing agent in these reactions is justified by a low organic content of the wastewater to be treated and a low reaction temperature, thus requiring the presence of an adequate catalyst due to the high kinetic activation barrier of such reactions. Commonly used methods for activation of H2O2 include the formation of reactive peroxyacids from carboxylic acids and peroxycarboximidic acid from acetonitrile (Payne oxidation),15 the generation of peroxyisourea from carbodiimide in the presence of either a weak acid or a mild base,16 or the use of percarbonate, persulfate or perborate in strongly basic solution.17 In order to achieve fast oxidative transformations, the use of large amounts of co-catalyst additives is often required.18
Among these, the use of percarbonate, a versatile oxidizing agent, is preferred for environmental reasons.19,20Oxidation using environmentally benign oxidants has aroused much interest,7,21 because chemical industry continues to require cleaner oxidation, which is an advance over environmentally unfavoured oxidations and a step up from more costly organic peroxides.22
In this report, we propose a fast and clean catalytic oxidative degradation of Orange II as model substrate by H2O2 in aqueous carbonate solution under mild reaction conditions, pH 8–10 and 25 °C, eqn (1).
 | (1) |
 | (2) |
In the present study, the formation of catalytically inactive Mn(OH)2 species was observed at higher pH, leading to deactivation of the produced Mn intermediates. The activation of H2O2 in the presence of manganese salts as a function of pH and carbonate concentration was therefore monitored using UV-Vis spectrophotometry. In situ formed, high valent manganese intermediates are known to be highly unstable in the absence of a spectator ligand. As the study progressed, it was of importance to investigate the role of the azo dye as a potential coordinating ligand to stabilize the produced intermediate under different reaction conditions. Electrochemical measurements and DFT calculations were used to develop a better understanding of the coordination chemistry of Orange II. The successful implementation of such catalytic systems becomes a worthwhile objective when issues such as environmental compatibility, high atom economy, availability, and expenses are considered.24
Experimental
Chemicals
Orange II, certified [Acid Orange 7, C.I. 15510, sodium 4-(2-hydroxy-1-naphthylazo)benzenesulfonate], 99% was supplied by Sigma–Aldrich and recrystallised from a Et2O/H2O mixture at 4 °C. 2,4,6-Tri-tert-butylphenol (TTBP) 96% was purchased from Sigma–Aldrich and recrystallised several times from EtOH/H2O (9 : 1) mixtures prior to use. Hydrogen peroxide 35 wt% as well as different manganese salt hydrates used in the experiments, were of analytical grade and provided by Acros Organics (Germany). Carbonate buffer solutions were prepared using Millipore Milli-Q purified water.General procedure
The manganese salts were freshly dissolved in water before use. To a freshly prepared sodium carbonate solution, an adequate amount of NaOH was added to adjust the pH of the solution. Under isothermal conditions, the desired amount of a concentrated manganese solution was added together with Orange II, previously dissolved in an aqueous carbonate solution, and H2O2. In typical measurements, 0.01 M H2O2 was prepared from a 35 wt% solution of H2O2. In addition, to gain more information on the activation mode of the catalyst, two further experimental procedures based on different activation and stabilization modes of the activated catalyst, were followed. In one, the catalytic active species was generated in situ in the carbonate buffer solution by addition of the desired amount of H2O2, followed by the addition of the corresponding quantity of Orange II to the reaction mixture. In the other, Orange II was added to the manganese solution and the formation of an Orange II⋯MnII complex was observed. The decomposition of the dye was initiated through the subsequent addition of H2O2. It is important to note that the catalytic oxidation of the dye by H2O2 could only be performed in an aqueous carbonate buffer solution. No other buffer at the same pH, viz. TRIS, TAPS, HEPES or phosphate, showed the observed catalytic reaction.Kinetic study of the manganese catalysed oxidative degradation of Orange II by H2O2
All kinetic data were obtained by recording time-resolved UV-Vis spectra using a Hellma 661.502-QX quartz Suprasil immersion probe attached via optical cables to a 150 W Xe lamp and a multi-wavelength J & M detector, which records complete absorption spectra at constant time intervals. In a thermostated open glass reactor vessel equipped with a magnetic stirrer, a 2 × 10−5 M freshly prepared catalyst solution and 0.01 M H2O2 were added to 40 ml of 5 × 10−5 M dye at a pH ranging from 8 to 10 at 25 °C. All kinetic measurements were carried out under pseudo-first order conditions (i.e. 50 ≤ [H2O2]/[Mn2+] ≤ 1000). The pH of the aqueous carbonate solution was carefully measured using a Mettler Delta 350 pH meter previously calibrated with standard buffer solutions at two different pH values (4 and 10). The kinetics of the oxidation reaction was monitored at 480 nm. First order rate constants, where possible, were calculated using Specfit/32 and Origin (version 7.5) software. To estimate the effect of the catalyst and H2O2 concentrations on the catalytic reaction at different carbonate concentrations, stopped-flow kinetic measurements were carried out using an SX.18MV stopped-flow instrument from Applied Photophysics.Spectrophotometric titration
UV-Vis spectra were recorded on a Shimadzu UV-2101 spectrophotometer at 25 °C. A 0.88 cm path length tandem cuvette with two separate compartments (0.44 cm path length each), was filled with 1 ml 5 × 10−5 M Orange II stock solution in one, and different concentrations of an aqueous Mn(NO3)2 solution in the other compartment. The cuvette was placed in the thermostated cell holder of the spectrophotometer for 10 min. UV-Vis spectra were recorded before and after mixing the solutions. The resulting spectrum presents the sum of the two individual spectra before, and that of the reaction mixture, after mixing. The observed spectral change is a result of complex-formation between Mn(II) and Orange II.Cyclovoltammetric measurements
Cyclovoltammetric (CV) measurements were performed in a one-compartment three-electrode cell using a gold working electrode (Metrohm) with a geometrical surface of 0.7 cm2 connected to a silver wire pseudo-reference electrode and a platinum wire serving as counter electrode (Metrohm). Measurements were recorded with an Autolab PGSTAT 30 unit at room temperature. The working electrode surface was cleaned using 0.05 μm alumina, sonicated and washed with water every time before use. The working volume of 10 ml was deaerated by passing a stream of high purity N2 through the solution for 15 min prior to the measurements and then maintaining an inert atmosphere of N2 over the solution during the measurements. All CVs were recorded for the reaction mixture with a sweep rate of 50 mV s−1 at 25 °C. Potentials were measured in a 0.5 M NaCl/NaOH electrolyte solution and are reported vs. an Ag/AgCl electrode.IR measurements
IR spectra were recorded as KBr pellets using a Mattson Infinity FTIR instrument (60 AR) at 4 cm−1 resolution in the 400–4000 cm−1 range.Elemental analysis
The measurements were carried out on an elemental analyzer Euro EA 3000 instrument from Hekaltech Gmbh. The analytical method is based on the complete instantaneous oxidation of the sample by “flash combustion” at 1000 °C, which converts all organic and inorganic substances into combustion products. The resulting combustion gases are swept into the chromatographic column by the carrier gas (He) where they are separated and detected by a thermal conductivity detector.DFT calculations
Unrestricted B3LYP/LANL2DZp hybrid density functional calculations,25a–ci.e., with pseudo-potentials on the heavy elements and the valence basis set25d–f augmented with polarization functions,25g were carried out using the Gaussian 0326 suite of programs. The relative energies were corrected for zero point vibrational energies (ZPE). The resulting structures were characterized as minima by computation of vibrational frequencies, and the wave functions were tested for stability.Synthesis of insoluble MnCO3
In a 150 ml round flask 3.36 g (0.4 M) NaHCO3 was dissolved in 100 ml doubly distilled water and the pH of the solution was set at 8.5 upon addition of small amounts of concentrated NaOH solution. To the freshly prepared carbonate solution 1 g (0.04 M) Mn(NO3)2 was added. The mixture was stirred at room temperature for 15 min during which MnCO3⋯H2O formed as a white precipitate. The product was filtered and washed several times with large amounts of water. Yield: 0.44 g MnCO3, 96.2%. IR (KBr pellets): ν (cm−1) 3421 (m), 1416 (vs), 862 (s), 725 (m). Elemental analysis (%) for MnCH2O4: calc.: C 9.03, H 1.52; found: C 9.38, H 1.52.Synthesis of Orange II⋯MnII complex
In a 50 ml Schlenk tube 0.014 g (2 × 10−3 M) Orange II was dissolved in 20 ml doubly distilled water and an aqueous solution of 0.01 g (2 × 10−3 M) Mn(NO3)2 was added dropwise under continuous stirring. The solution mixture was kept for several hours at room temperature. The formed precipitate was filtered and dried at room temperature. Yield: 0.018 g Orange II⋯MnII, 87.1%. IR (KBr pellets): ν (cm−1) 3527 (vs), 1619 (s), 1511 (s), 1383 (vs), 1262 (m), 1171 (s), 1120 (s), 1034 (s), 1007 (s), 829 (s), 759 (s), 696 (m), 644 (m), 595 (m). Elemental analysis (%) for MnC16H18O10N3SNa: calc.: C 36.79, H 3.47, N 8.04, S 6.14, O 30.63; found: C 29.44, H 3.39, N 8.05, S 4.77, O 30.31.Synthesis of Orange II⋯MnII⋯Orange II complex
An aqueous solution of 0.005 g (1 × 10−3 M) Mn(NO3)2 was added under continuous stirring to a 0.014 g (2 × 10−3 M) Orange II water solution at room temperature. The pale yellow precipitate was collected by filtration and dried in air. Yield: 0.017 g Orange II⋯MnII⋯Orange II, 93.7%. IR (KBr pellets): ν (cm−1) 3390 (s), 1619 (s), 1570 (m), 1554 (m), 1520 (vs), 1393 (m), 1260 (m), 1169 (vs), 1119 (vs), 1033 (vs), 1007 (s), 828 (s), 758 (s), 695 (m), 644 (m), 593 (m). Elemental analysis (%) for MnC32H32O15N5S2Na2: calc.: C 43.1, H 3.62, N 7.23, S 7.19, O 26.91; found: C 42.99, H 3.75, N 7.23, S 7.00, O 25.67.Results and discussion
General observations
A series of experiments were performed in order to investigate the in situ generation of the highly reactive manganese catalyst in the oxidative degradation of Orange II by H2O2 under mild reaction conditions starting with a simple Mn(II) salt. Oxidation reactions are in general affected by the protonation state of the substrate, catalyst and oxidant, and the solvent used. It is further important to note that the studied organic dye (Orange II) can exist in either one of two tautomeric forms, or in an equilibrium mixture, depending on the process parameters. This kind of rapid dynamic equilibrium is relevant as one dye species may be more reactive than the other. Azo dyes containing a hydroxyl group in the ortho position to the azo group within naphthyl or higher fused ring systems, can exist as azo and hydrazone tautomers,27 with the relative amounts varying with reaction parameters such as solvent and temperature.28 Furthermore, in aqueous solution these species are in a pH dependent equilibrium with a common anion, in which the negative charge is delocalised throughout the molecule (see Scheme 1).29 These are chemically distinct forms which have characteristically different visible spectra, the azo form absorbs typically at 400–440 nm and the hydrazone form at 475–510 nm (see Fig. 1).30 | ||
| Scheme 1 Orange II: R = SO3Na; pKA = 11.4, λmax = 480 nm. | ||
 | ||
| Fig. 1 UV-Vis spectrum of 10−4 M Orange II in carbonate buffer solution at pH 8.5. | ||
The absorption spectrum of Orange II in an aqueous carbonate solution shows under the selected reaction conditions (Fig. 1) one main band at 480 nm, which correspond to the n →π* transition of the azo form. The other two bands at 300 and 270 nm are attributed to the π→π* transition of the benzene and naphthalene rings, respectively.31 Orange II, due to the presence of aromatic groups, is very stable, and in the presence of a powerful bleaching agent such as H2O2, degradation of dye solutions occurs slowly under specific reaction conditions. Surprisingly, the oxidation rate was tremendously accelerated by addition of a simple manganese salt. The reactivity of the in situ formed intermediate was comparable with the catalytic activity of some earlier postulated, well known manganese bleach catalysts13,32 and manganese porphyrins.33 In our work, the formation and stabilization of the active catalyst was studied in a carbonate buffer solution.
Complex-formation between Orange II and Mn2+
ortho-Hydroxy aromatic azo dyes, which are bidentate complexing agents are of considerable practical and theoretical interest because of their ability to form stable chelate complexes with some metal ions.34 It is known that Orange II can act as a chelating agent since the hydroxy and sulfonate groups allow a stabilized complex to be formed.35 Addition of Orange II to a freshly prepared aqueous carbonate solution of a MnII salt results in significant changes in the UV-Vis spectrum of Orange II as shown in Fig. 2. | ||
| Fig. 2 (Blue curve) UV-Vis spectrum of a 5 × 10−5 M Orange II carbonate (0.1 M HCO3−) solution at pH 8.5 before mixing with a 5 × 10−5 M Mn(NO3)2 solution at pH 8.5. (red curve) UV-Vis spectrum recorded directly after mixing (ca. 5 s delay). | ||
UV-Vis spectra recorded before and after mixing (ca. 5 s delay) of 5 × 10−5 M Orange II with 5 × 10−5 M Mn(NO3)2 showed a significant increase in absorbance at 480, 310 and 228 nm, respectively. The differences before and after mixing are not profound at low Mn2+ concentrations. On increasing the Mn2+ concentration, a continuous increase in ΔAλ=480 nm = A(dye+Mn(II))−Adye was observed, indicating the formation of an Orange II⋯Mn2+ species according to eqn (3). It should be noted that at higher Mn2+ concentration, a precipitate started to form. The value of Keq was determined through a constant variation of the Mn2+ concentration. For a correct determination of the complex-formation constant, independent measurements were performed at constant manganese concentration where the Orange II concentration was continuously varied (see Fig. 3C). Independent measurements were repeated between five and eight times. Selected data are shown in Fig. 3A, where the solid line represents a fit of eqn (4) to the data.
 | (3) |
| Ax−A0 = (A∞−A0)Keq[Orange II]/(1 + Keq[Orange II]) |
| ΔA = ΔA∞Keq[Orange II]/(1 + Keq[Orange II]) | (4) |
The values of A0 and A∞ represent the absorbances of Orange II and of the complex Orange II⋯MnII, respectively, and Ax is the absorbance at any MnII concentration. The value of Keq was calculated from eqn (4) to be (2.9 ± 0.9) × 104 M−1, indicating a relatively weak coordination of the dye to the metal center. Experimentally, through addition of a 4 × 10−5 M Mn(NO3)2 solution to a 5 × 10−5 M Orange II aqueous carbonate solution (0.2 M HCO3−), a decrease in the pH of the solution from 8.5 to 8.3 was observed, which suggests phenolic proton release due to Mn(II) coordination to Orange II with the formation of a six-membered ring structure instead of coordination to the terminal sulfonato group. At higher concentrations (above ca. 10−3 M) Orange II forms dimers and higher aggregates in aqueous solutions,30,36 and has a marked effect on the observed spectra, particularly UV-Vis and NMR.37 A Benesi-Hildebrand treatment of the optical data to determine Keq could not be applied since the concentration of Orange II and MnII were close to each other.38 Using Job’s method,39 the stoichiometry of the formed complex could be determined. According to the data shown in Fig. 3B and D, at lower MnII concentration the formation of a complex with a stoichiometry of 1 : 1 can be assumed. On increasing the Orange II concentration further, complexes with a higher stoichiometry are possibly formed (see Fig. 4A and B).
 | ||
| Fig. 3 (A) Change in absorbance at 480 nm on addition of different concentrations of Mn2+ to 5 × 10−5 M Orange II in aqueous carbonate solution (0.2 M HCO3−) at pH 8.5 and 22 °C. (B) Job plot analysis for complex-formation between Orange II and Mn2+ in aqueous carbonate solution (0.2 M HCO3−) at pH 8.5. (C) Spectral changes at 480 nm on addition of different concentrations of Orange II to a freshly prepared 5 × 10−5 M Mn(NO3)2 carbonate solution (0.2 M HCO3−) at pH 8.5 and 22 °C. (D) Job plot analysis for the complex-formation in aqueous carbonate solution (0.2 M HCO3−). | ||
 | ||
| Fig. 4 (A) Proposed structure for a 1 : 1 Orange II⋯Mn2+ complex formed in a carbonate buffer solution at a low concentration of Mn2+. (B) Proposed structure for a 1 : 2 Orange II⋯Mn⋯Orange II complex formed in a carbonate buffer solution at a high concentration of Orange II. | ||
Similar structures have been reported earlier by Nadtochenko and Kiwi when a Fe3+ salt was added to an Orange II solution in acidic medium.40 Bauer also reported a TiIV complex, where TiIV is coordinated by two oxygen atoms from the sulfonato group and the oxygen of the carbonyl group of the hydrazone tautomer.41 In the enzyme manganese peroxidase, the double role of Orange II as a stabilizer, forming a complex with MnIII, and as a substrate that permits the regeneration of MnII, was recently postulated by López et al.35 Although, the coordination of organic dyes, viz.Alizarin, Alizarin S42 and Orange II,3 to several transition metal centres has been known for years, comparatively little has appeared on their use as potential stabilizing ligands in oxidative degradation of organic dyes.
The formed Orange II⋯MnII complex was isolated and the validity of its composition was confirmed by elemental analysis. In control experiments the reactivity of the isolated 1 : 1 Orange II⋯MnII and 2 : 1 complexes were studied. The isolated complexes exhibit the same catalytic activity and stability under the experimental conditions employed for the in situ generation of the complex. Due to the weak coordination mode of the ligand, no differences between the catalytic activity of the 1 : 1 and 2 : 1 complex were found.
Beside UV-Vis measurements and DFT calculations, electrochemical measurements were used to study the in situ formation of highly reactive MnII catalytic species in the presence of Orange II under the selected experimental conditions.
CV studies on the complex-formation between Orange II and Mn2+
CV measurements of a 4 × 10−5 M Mn2+ solution in the presence of different Orange II concentrations were performed in order to determine the interaction between the fully aquated Mn2+ ions and Orange II present in the reaction mixture. Fig. 5 shows the results of MnII⋯Orange II complex formation in NaCl electrolyte solution, performed using a standard three electrode electrochemical setup as described above. | ||
| Fig. 5 CVs of a 4 × 10−5 M Mn2+ electrolyte (0.1 M NaCl) solution in the presence of different Orange II concentrations at pH 8.5 (adjusted by addition of NaOH) and 22 °C. | ||
To avoid the oxidation of MnII to MnIV, which precipitates as MnO2, the potential scan was discontinued at +1.0 V, after which the reverse scan from +1.0 to −0.8 V was started. The CVs of Mnaq2+ in the absence of any coordinating substrate exhibit one quasi-reversible oxidation peak at E = +0.59 V vs.Ag/AgCl and one quasi-reversible reduction peak at E = +0.35 V, corresponding to the one electron Mn3+/Mn2+ redox couple. In addition, CV measurements on a freshly prepared 4 × 10−5 M Orange II electrolyte solution at pH 8.5 and 22 °C were performed. Orange II, as it can be seen in Fig. 5, undergoes two electrochemically quasi-reversible, one-electron reductions with CV half-wave potentials at Ered1 = −0.19 V and Ered2 = +0.11 V (vs.Ag/AgCl) with a difference between the cathodic and anodic wave of 0.02 and 0.204 V, respectively. Furthermore, the reduction potential of Mn3+ decreased from +0.35 V to +0.28 V when Orange II was added to the solution, indicating the stabilization of Mn3+ ions. In the presence of a chelating substrate, the generated Mn3+ complex becomes more stable and the redox potentials attain lower values.43 When the concentration of Orange II was increased up to 2 × 10−5 M, the presence of further reduction peaks along with changes in the oxidation peak intensity were observed, indicating the formation of other manganese–Orange II species as specified above.
DFT calculations
To assess the coordination mode of Orange II to the Mn(II) center, DFT (B3LYP/LANL2DZp) calculations were performed for a series of plausible complexes. Orange II in aqueous solution under the selected experimental conditions dissociates into an anionic sulfonate group and a cationic sodium ion. In the presence of an unsolvated SO3−group involving charge transfer from the electron-rich sulfonate group onto the rest of the molecule, may in general not give satisfactory DFT results.28Solvent Yellow 14, a model compound for Orange II containing no sulfonate group was selected for the DFT study of the interaction between the Mn(II) and the chosen azo dye. A picture of the calculated conformers of the model compound 1 is shown in Fig. 6.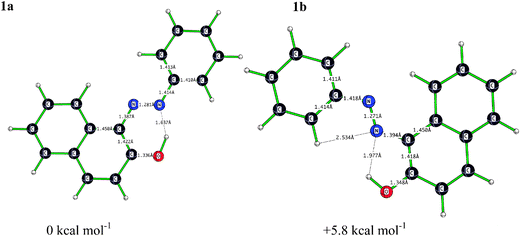 | ||
| Fig. 6 Optimized (B3LYP/LANL2DZp) structures of 1a and 1b with a planar geometry and dihedral angles of (a) 180.0° and (b) 178.7° about the azo group, C–N–N–C. | ||
The optimized geometry of 1a was calculated to be ca. 5.8 kcal mol−1 lower in energy than that of 1b. Furthermore, the calculated structure of 1a was compared with X-ray structural data of Solvent Yellow 14.44 A good agreement between calculated and crystallographically determined structure was found.
According to the UV-Vis and electrochemical data presented above, Orange II can coordinate to a fully aquated Mn2+ center. Different plausible interaction modes of Solvent Yellow 14⋯MnII (2) and Solvent Yellow 14⋯MnII⋯Solvent Yellow 14 (3) were studied in detail. Optimized structures of 2 adopting different coordination modes are presented in Fig. 7.
 | ||
| Fig. 7 Optimized structures of complex 2a, b and c (B3LYP/LANL2DZp). | ||
The studied organic dye can coordinate to aquated Mn2+ ion by forming two new bonds, one between Mn2+ and the deprotonated phenolic OH-group of 1a and the second between Mn2+ and one of the azo nitrogen atoms, leading to the formation of either a planar six-membered (2a) or five-membered (2b) chelate complex. Furthermore, a second interaction mode for 2 involving a hydrogen bond between one coordinated water molecule and the azo nitrogen atom (2c) was taken into consideration. The calculated energies indicate that 2a is energetically favoured over 2b by about 3 kcal mol−1. The N–N bond length of 1.30 Å for 2a is nearly identical to that found in the free model molecule 1a (1.28 Å), indicating a weak interaction between the nitrogen atoms and the positively charged manganese center.
In addition to these structures, DFT calculations were performed for a further possible interaction of a second dye molecule with the Mn(II) center leading to the formation of chelated Mn(II) inner-sphere complexes. Similar transition metal complexes of ortho-hydroxy azo dyes were prepared and characterised by Drew and Landquist.45 The introduction of a second dye molecule is expected to have certain advantages. In addition to the usual stabilization by the chelate-effect, the introduction of a second molecule of 1a could result in a protecting effect on the coordination framework. The optimized structure of 3 is presented in Fig. 8.
 | ||
| Fig. 8 Optimized structure of 3 (B3LYP/LANL2DZp). | ||
The calculated structure of 3 shows a C2-symmetry and the axial positions are nearly equivalent. The calculated Mn–N bond lengths in the equatorial plane for the energetically favoured 2a (2.15 Å) and 3 (2.30 Å) are comparable with the X-ray structural data for Mn(II) complexes with nitrogen containing ligands such as 1,2-bis(imidazol-1-yl)ethane (bim) (2.213–2.294 Å),46a2-[N,N-bis(2-pyridylmethyl)amoniumethyl]-6-[N-(3,5-di-tert-butyl-2-oxidobenzyl)-N-(2-pyridylamino)aminomethyl]-4-methylphenol (H2Ldtb) (2.118–2.237 Å)46b and 1,4,7-triazacyclononane (tacn) (2.118–2.146 Å).46c
As expected, upon coordination of two dye molecules in 3, the N–N bond distance becomes longer (1.29 Å) than observed in the crystal structure of 1a due to the partial neutralization of the delocalized negative charge of the nitrogen atom. The elongation of the Mn–O bond trans to the azo group (Mn–O = 2.38 Åvs. 2.06–2.27 Å for 2, and Mn–O = 2.27/2.26 Åvs. 1.81/2.11 Å for 3) exerts a significant trans influence opposite to the Mn–N bond. The increased lability of the axial ligand allows the subsequent interaction of the substituted transition metal atom with an oxidant, leading to the rapid formation of active oxidizing species. Moreover, DFT calculations performed by Blomberg et al.47 suggest that in the presence of weak-field ligands for Mn(II) and Mn(III), five-coordination is also accessible whereas Mn(IV) has a much stronger preference for six-coordination.47
Complex-formation between bicarbonate and Mn(II)
The reactions between bicarbonate ions (HCO3−) and different manganese species have been studied for several years, since aquated Mn2+ cations themselves are actually not able to catalyze H2O2 disproportionation. Depending on the HCO3− concentration in the reaction mixture, MnII⋯HCO3− complexes of different stoichiometry can be formed. Recently, it was suggested that only the neutral MnII(HCO3−)2 complex can facilitate H2O2 disproportionation.48 In this study the complex-formation reaction between Mn2+ and HCO3− was monitored using UV-Vis spectrophotometric beside CV measurements as a function of carbonate concentration at pH 8.5. UV-Vis spectra recorded before and after addition of HCO3− to an aqueous Mn2+ solution showed the formation of a new broad band at 300 nm as illustrated in Fig. 9A. The time course of the absorption band formation is shown in Fig. 9B. | ||
| Fig. 9 (A) UV-Vis spectra of an aqueous 4 × 10−4 M Mn2+ solution before (black curve) and after (red curve) addition of 0.4 M HCO3− at pH 8.5 and 25 °C. (B) Time course of the band formation at 300 nm of an aqueous 2 × 10−4 M Mn2+ solution containing different amounts of HCO3−. | ||
It can be seen from Fig. 9B that the rate of formation of the manganese carbonate intermediate is enhanced at higher carbonate concentration. The observed first order rate constants following the induction period in Fig. 9B, are directly proportional to the [HCO3−] in the range 0.01–0.1 M (see Fig. 10) with a second order rate constant of (3.6 ± 0.2) × 10−2 M−1 s−1 at 25 °C. Moreover, the observed induction period is probably related to the displacement of water from the first coordination sphere of the fully aquated Mn2+ ion by HCO3− and subsequent rearrangement of the coordinated ligand, viz. formation of bidentate carbonate complexes. It should be noted that under these experimental conditions (high carbonate concentration and pH 8.5) insoluble MnCO3 is formed as a very fine white precipitate at longer reaction times. Its composition was confirmed by elemental analysis and IR spectroscopy.
 | ||
| Fig. 10 Plot of observed first order rate constant (kobs) for the formation of Mn2+⋯HCO3−vs. the bicarbonate concentration in the presence of 4 × 10−4 M Mn2+ at pH 8.5 and 25 °C. | ||
The reactivity of the produced intermediate was tested in the oxidative degradation of Orange II by H2O2 at pH 8.5 and 25 °C. During the first 200 s, no change in the reactivity of the in situ formed manganese intermediate occurs. A significant time dependent loss in catalytic efficiency of the formed MnII⋯HCO3− intermediate was observed. An irreversible deactivation of the catalyst occurs within less than 20 min. On the other hand, no precipitate formation as well as no deactivation of the catalytically active manganese intermediate could be observed in the presence of a coordinating organic substrate, i.e. Orange II, over a long period of time (1–4 days) in a high carbonate (0.5 M) containing buffer solution under these conditions. Moreover, the stabilization of the in situ formed active catalyst in the presence of an organic substrate is of considerable practical interest, because its successful implementation could offer a more efficient alternative for clean oxidation reactions.
CV measurements of freshly prepared aqueous Mn(NO3)2 solutions were performed in the presence of different carbonate concentrations in a 0.1 M NaCl electrolyte solution at pH 8.5 (adjusted by careful addition of NaOH) and 22 °C. In the presence of a coordinating substrate, the displacement of a coordinated water molecule from the manganese coordination sphere takes place. By coordination of a negatively charged ligand such as HCO3− to a positively charged metal, the peak potentials are shifted to more negative potentials compared to the fully aquated Mn2+ (see Fig. 11A and 12).41 On increasing the carbonate concentration in solution a decrease in the peak current intensity occurs concomitantly with peak broadening because of complexation by carbonate. Typical multiple scan CVs of a 4 × 10−5 M Mn2+ solution in the presence of 0.2 M NaHCO3 and 0.1 M NaCl at pH 8.5 and 25 °C is presented in Fig. 11B. In the presence of a chelating substrate, the generated Mn3+ complex becomes more stable and the redox potentials attain lower values. Moreover, at higher carbonate concentrations in the reaction mixture the presence of a second oxidation peak at E = +0.41 V, attributed to the formation of further complexes such as proposed in eqn (5), was observed.
 | (5) |
By plotting the peak potential E as a function of the hydrogen carbonate concentration (see Fig. 12), the presence of different complex species at different carbonate concentrations is revealed.
 | ||
| Fig. 11 (A) Cyclovoltammograms for 4 × 10−5 M Mn2+ solution in an aqueous solution of 0.1 M NaCl and different concentrations of HCO3−. (B) Typical multiple scan CVs of a 4 × 10−5 M Mn2+ solution in the presence of 0.2 M NaHCO3 and 0.1 M NaCl at pH 8.5 and 22 °C. | ||
![Plot of peak potential E as function of [HCO3−]; E vs.Ag/AgCl electrode, [Mn2+] = 4 × 10−5 M, [HCO3−] = 0.1–05 M in 0.1 M NaCl electrolyte solution at pH 8.5 and 22 °C.](/image/article/2009/NJ/b813725k/b813725k-f12.gif) | ||
| Fig. 12 Plot of peak potential E as function of [HCO3−]; E vs.Ag/AgCl electrode, [Mn2+] = 4 × 10−5 M, [HCO3−] = 0.1–05 M in 0.1 M NaCl electrolyte solution at pH 8.5 and 22 °C. | ||
Carbonate concentration dependence of the catalytic reaction course
The effect of the carbonate concentration on the oxidative degradation of the dye was studied at a constant pH of 8.5. The total carbonate concentration was varied between 0.05 and 0.5 M.In the present case, the catalytic reaction leads to a square dependence of kobs on the HCO3− concentration (Fig. 13) with a third rate constant (8.3 ± 0.3) × 10−2 M−2 s−1, suggesting that 2 equivalents of HCO3− are involved in the oxidation mechanism. It is suggested, among other possibilities, that one equivalent of HCO3− is required for the formation of the more reactive [MnII(H2O)5(HCO3−)]+ intermediate, and the second equivalent of HCO3− is required for the formation of the more reactive peroxocarbonate species, known to be a versatile oxidizing agent. It should also be noticed that no oxidation of Orange II by H2O2 was observed in the absence of a carbonate buffer. In the view of these findings we decided to study the influence of carbonate on the manganese catalyzed oxidation of Orange II by H2O2 and HCO4−, respectively.
 | ||
| Fig. 13 Second-order carbonate concentration dependence of kobs. Experimental conditions: 2 × 10−5 M Mn(NO3)2, 5 × 10−5 M Orange II, 0.01 M H2O2, pH 8.5, 25 °C. | ||
The reaction of carbonate with H2O2 at pH 8.5 and 25 °C
Peroxycarbonate ions, known to be several orders of magnitude more reactive toward nucleophilic substrates than H2O2 itself,49 are formed in a relatively fast pre-equilibrium (K = 0.32 ± 0.02 M−1)40 between hydrogen carbonate ions and H2O2 shown in eqn (6).| HCO3− + H2O2⇌ HCO4− + H2O | (6) |
By performing the oxidation reactions in the presence of peroxycarbonate instead of H2O2 in a 0.5 M carbonate containing buffer solution at pH 8.5, no difference in the reactivity was observed (Fig. 14A).
 | ||
| Fig. 14 (A) Spectral changes observed at 480 nm for the 2 × 10−5 M Mn(NO3)2 catalyzed oxidative degradation of 2.5 × 10−5 M Orange II in the presence of (black curve) 0.01 M H2O2 and (red curve) 0.01 M HCO4−, respectively, at pH 8.5 and 0.5 M total carbonate concentration. (B). Comparison of the absorbance changes at 480 nm vs. time for the 2 × 10−5 M Mn2+ catalyzed oxidative degradation of 5 × 10−5 M Orange II by 0.01 M H2O2 at pH 8.5 and different carbonate concentrations. | ||
The Mn(II) catalyzed oxidative degradation of Orange II by using HCO4− as an oxidizing agent could be significantly enhanced through increasing the total carbonate concentration in the reaction mixture. This can be explained in terms of the equilibrium formulated in eqn (6). Based on our experimental observations and aspects reported in the literature41 for the Mn(II) catalyzed oxidation reaction by H2O2 in a carbonate containing solution, the reaction sequence presented in Scheme 2 can be suggested to occur.
 | ||
| Scheme 2 In situ formation of catalytically active Mn intermediates in the presence of hydrogen peroxide in a carbonate containing aqueous solution at pH 8.5 and 25 °C. | ||
Complex-formation between Mn2+ and H2O2 in an aqueous carbonate solution
Addition of H2O2 to hexaaqua Mn2+ in a carbonate solution leads to significant spectral changes in the UV-Vis spectra during the reaction (see Fig. 15A). The initial rapid increase of the intensity of the broad band at 300 nm, as it is illustrated in Fig. 15A, is attributed to the fast formation of [MnII(H2O)5(HCO3)]+. An isosbestic point at 330 nm suggests the formation of a new manganese intermediate by addition of an oxidizing agent, i.e. H2O2. According to our spectroscopic observations the formed complex with an absorption band at 400 nm could be attributed to a MnIV–η2-peroxycarbonate intermediate.53 Based on spectroscopic observations and data reported in the literature,54 the formed intermediate can be regarded as most likely to be a high valent manganese complex. Similar Rh,55Pt56 and Fe57 peroxycarbonate complexes have been isolated before and were characterized spectroscopically. The time course of the absorption band at 400 nm at different pH is illustrated in Fig. 15B. | ||
| Fig. 15 (A) UV-Vis spectra recorded for the reaction of 2 × 10−4 M Mn(NO3)2 with 0.01 M H2O2 in a 0.5 M HCO3− containing solution at pH 8.4 and 25 °C. (B) Comparison of typical absorbance at 400 nm vs. time plots at pH 8.5 (black curve) and 9.5 (red curve). | ||
In the absence of any stabilizing ligand, the formed complex rapidly decomposes with the formation of catalytically inactive MnIVO2 that precipitates from solution (see Fig. 15B). The decomposition of the active intermediate is accelerated at higher pH (see Fig. 15B). To ascertain that the formulated reaction steps in Scheme 2 are valid under our reaction conditions, a systematic spectroscopic investigation at different pH values was performed. Representative data for the reaction course at 400 nm at pH 8.5 and 9.5 are presented in Fig. 15B. Contrary to our expectations, an increase of one unit in pH resulted in an increase of the induction period and a decrease in the manganese peroxycarbonate complex formation rate under the mentioned reaction conditions. This could be partly due to subsequent formation of Mn(OH)2 precipitates at higher pH and to deprotonation of HCO3− that becomes significant at pH above 8 to 9.51 This results in a decrease in the HCO3− concentration in the equilibrium presented in eqn (6), reducing the concentration of peroxycarbonate present in solution.
Reactivity profile as function of pH
The reactivity of the catalytic system is generally influenced by the protonation state of the substrate, the catalyst and oxidizing agent. In our work the kinetics was studied in 0.4 M HCO3− containing buffer solution in the pH range between 8.0 and 9.5 at 25 °C. The pH of the carbonate buffer solution was adjusted carefully using small amounts of concentrated NaOH solution to avoid dilution. A typical manganese catalyzed oxidative degradation of Orange II by H2O2 in a carbonate buffer solution is presented in Fig. 16A. The catalytic degradation is usually complete within 1–10 min depending on the pH of the solution, the catalyst concentration, and the H2O2 and carbonate concentrations. The decomposition of the dye was followed by monitoring the spectral changes at 480 nm. The depletion of the band at 480 nm is in general correlated with cleavage (heterolytic or homolytic) of the azo group leading to colorless oxidation products due to the induced discontinuity in the conjugation of the π-system in the dye molecule. The inset in Fig. 16A shows the first spectrum of Orange II before the addition of the catalyst and H2O2, and the final spectrum recorded after 250 s. A decrease in the intensity of the two other bands at 270 and 300 nm was observed, showing that further bleaching also occurs under these reaction conditions. The isolation and characterization of reaction products is extremely difficult and requires large synthetic efforts, particularly as different reaction intermediates tend to react further under experimental conditions. A comparison of the reaction course at different pH values is shown in Fig. 16B.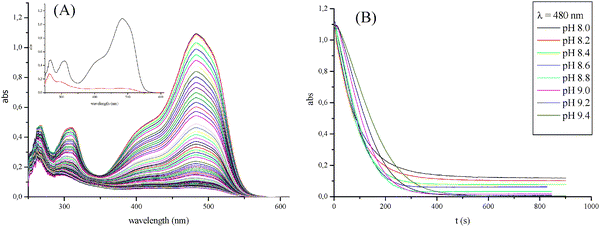 | ||
| Fig. 16 (A) UV-Vis spectra of a 2 × 10−5 M Mn(NO3)2 catalyzed oxidative degradation of 5 × 10−5 M Orange II by 0.01 M H2O2 in a 0.4 M total carbonate containing solution at pH 8.5 and 25 °C. The inset in Fig. 16A shows the first spectrum of Orange II before the addition of the catalyst and H2O2, and the final spectrum recorded after 250 s. (B) Comparison of absorbance at 480 nm vs. time plots for the 2 × 10−5 M Mn(NO3)2 catalyzed oxidative degradation of 5 × 10−5 M Orange II by 0.01 M H2O2 in a 0.4 M total carbonate containing solution at different pH values and 25 °C. | ||
The Mn(II) catalyzed decolorization and oxidative decomposition of Orange II was found to be sensitive to the pH of the solution. According to our experimental data, an increase in pH resulted in a slight decrease in the reaction rate under the above-mentioned reaction conditions and the highest reactivity is observed at a pH between 8.2 and 8.6 (see Fig. 17). Increasing the pH to >9 leads to a decrease in the oxidation rate for the bicarbonate-activated peroxide, which is presumably the result of the deprotonation of HOOCO2− to form CO42−, a less electrophilic oxidant.58 At even higher pH, the decomposition of the peroxide is accelerated and may reduce the oxidation reaction rate. Contrary to our expectations, the observed rate constants for the decolorization reaction of Orange II are similar to the destruction rate constants of naphthalene and benzene rings, long-lived intermediates, under the studied conditions (see Fig. 17). Thus, for a complete oxidation of these stable molecules higher concentrations of oxidant and catalyst are required.
 | ||
| Fig. 17 Plot of observed rate constant (kobs) calculated for the decoloring reaction followed at 480 and 300 nm, respectively. Experimental conditions: 2 × 10−5 M Mn(NO3)2, 5 × 10−5 M Orange II, 0.01 M H2O2, 0.4 M total carbonate and 25 °C. | ||
A similar screening using MnCl2, Mn(Ac)2 and Mn(SO4)2 showed identical catalytic activity in the oxidative degradation of Orange II by H2O2. In all cases, the manganese catalyzed oxidative degradation of Orange II is favored by moderate alkaline pH values and vanishes completely at very high or very low (strong acidic) values. According to the experimental observations mentioned above, the manganese catalyzed oxidative degradation of Orange II by H2O2 in a carbonate containing solution is considerably inhibited at higher pH values due to the lower formation of the high valent manganese η2-peroxycarbonate complex (see Fig. 15B).
Effect of the manganese concentration on the oxidative reaction course
To evaluate the effect of the catalyst concentration on the manganese catalyzed oxidative degradation of Orange II by H2O2 under catalytically relevant experimental conditions, kinetic studies were performed for solutions in which the carbonate containing water solution with various amounts of Mn(NO3)2 was added in the presence of 0.01 M H2O2 to a 5 × 10−5 M Orange II solution at 25 °C. The obvious accelerating ability of the HCO3− ions prompted us to study the catalytic reaction course in more detail at four different carbonate concentrations. The in situ produced catalyst concentration dependence was studied at 480 nm using in situUV-Vis spectroscopic measurements and the kinetic traces could be adequately fitted to a single exponential function. Plots of the observed rate constant as a function of [Mn2+] at different carbonate concentrations are presented in Fig. 18. | ||
| Fig. 18 Mn(NO3)2 concentration dependence of kobs. Reaction conditions: 5 × 10−5 M Orange II, 0.01 M H2O2, pH 8.5 and 25 °C. | ||
As it is evidenced in Fig. 18, the [Mn2+] dependences of the observed rate constants for the manganese catalyzed oxidative degradation of Orange II by H2O2 in a low carbonate concentration containing solution (0.1–0.3 M HCO3−) are strongly curved (higher K values, see Table 1) and reach a limiting value at higher catalyst concentration. In contrast, similar data at higher carbonate concentrations (0.4–0.5 M HCO3−) result in a less curved dependence of kobs on the catalyst concentration, i.e. lower K values (see Table 1). The observed rate profile can be explained by the general reaction mechanism proposed in Scheme 2 and simplified in Scheme 3. The observed rate law for the proposed reaction steps in Scheme 3 is given by eqn (7). The calculated k and K values from the non-linear concentration dependences in Fig. 18 are summarized in Table 1.
 | (7) |
 | ||
| Scheme 3 Proposed reactions steps for the formation of the catalytically active manganese intermediate in the presence of H2O2 in a carbonate containing solution. | ||
| [HCO3−]/M | k/s−1 | 10−3K/M−1 |
|---|---|---|
| 0.1 | 0.0033 | 34.6 |
| 0.3 | 0.032 | 17.6 |
| 0.4 | 0.051 | 17.8 |
| 0.5 | 0.138 | 15.2 |
Effect of the H2O2 concentration on the manganese-catalyzed oxidative degradation of Orange II
The effect of H2O2 on the oxidation reaction course was studied by varying its initial concentration over a wide range, between 5 and 30 mM (Fig. 19). At lower H2O2 concentrations (1 and 5 mM) a fast oxidation reaction occurs in the first few seconds followed by a rapid consumption of H2O2 resulting finally in a partial and inefficient decolorization of the dye. This prompted us to study the H2O2 concentration effect on the catalytic oxidation of the dye at higher concentrations of H2O2. The kobs values were calculated from a single exponential fit to the absorbance at 480 nm vs. time plots and showed a linear dependence on the initial H2O2 concentration over the studied concentration range. | ||
| Fig. 19 H2O2 concentration dependence of kobs. Reaction conditions: 5 × 10−5 M Orange II, 2 × 10−5 M Mn(NO3)2, pH 8.5 and 25 °C. | ||
Stability of the in situ formed catalyst
In control experiments the stability of the in situ generated catalyst was studied by repeated addition of dye and H2O2 to a solution of 2 × 10−5 M Mn(NO3)2 at pH 8.5 (0.4 M HCO3−) and 25 °C (see Fig. 20A and B). | ||
| Fig. 20 (A) Spectral changes observed at 480 nm for the repeated addition of 5 × 10−5 M Orange II to a 2 × 10−5 M Mn(NO3)2 solution in the presence of 0.01 M H2O2 at pH 8.5 and 0.4 M total carbonate concentration. (B) Spectral changes observed at 480 nm for a new addition of 5 × 10−5 M Orange II and 0.01 M H2O2 to a 48 h old reaction mixture containing the catalyst solution under the same experimental conditions as mentioned in A. | ||
As it can be seen in Fig. 20A, the catalytic cycle could be repeated several times without any significant loss of activity during the oxidation reaction, indicating an excellent stability of the in situ formed catalyst. After the fifth cycle the reaction solution containing the active catalyst was allowed to stay at ambient temperature for 48 h. Subsequently, the catalytic activity of the in situ formed manganese complex was evaluated again by performing the oxidation reaction in the presence of freshly added Orange II and H2O2. The experimental results illustrated in Fig. 20B provide clear evidence for the high efficiency of the in situ formed catalyst under the above mentioned experimental reaction conditions.
Mechanistic aspects of the manganese-catalyzed oxidative degradation of Orange II by H2O2 in carbonate solution
Throughout this study, the oxidation reactions were carried out in a thermostated open glass reactor vessel at ambient temperature in aqueous hydrogen carbonate containing solutions. The readily available manganese salts, the mild reaction conditions and the operation simplicity and practicability allow for an easy and green oxidative degradation of the studied organic dye. In control experiments the catalytic activity of the in situ generated manganese complex was investigated under an inert atmosphere. By performing the catalytic reaction in a closed glass reactor under inert reaction conditions no change in the decomposition reaction rate was noticed. A comparison of the reaction course carried out under different experimental conditions is illustrated in Fig. 21. | ||
| Fig. 21 Comparison of typical absorbance at 480 nm vs. time plots of a 2 × 10−5 M Mn(NO3)2 catalyzed oxidative degradation of 5 × 10−5 M Orange II by 0.01 M H2O2 in a 0.4 M HCO3− containing solution at pH 8.5 and ambient temperature performed in the presence of atmospheric oxygen (black curve), inert atmosphere (red curve) and TTBP (blue curve), respectively. | ||
By performing the reaction under inert reaction conditions no significant differences in the decomposition reaction rate was observed, indicating that HO˙ or HOO˙ radical formation is not prevalent for this oxidation reaction. This is further supported by the observation that addition of radical traps such as TTBP had no effect on the reaction course (see Fig. 21).
Taking into account all obtained spectroscopic and kinetic data, the following reaction schemes can be proposed for the Mn2+ catalyzed oxidative degradation of Orange II by H2O2 in carbonate solution under catalytically relevant experimental conditions.
A key feature of the proposed reaction mechanism outlined in Scheme 4 is that the overall oxidation of Orange II occurs in a two electron oxidation step leading to the formation of a relatively stable high-valent MnO intermediate and transfer of the oxo group to the substrate. Most of the earlier reported papers22,59 on the oxidation reaction catalyzed by several isolated and structurally well defined manganese complexes have emphasized the formation of a high-valent MnO intermediate by the reaction of manganese with the appropriate oxidant. According to our observations, HCO3− ions are involved in two catalytically relevant reactions. HCO3− ions react with aquated MnII present in solution to form a catalytically active Mn–HCO3− complex. HCO3− is also involved in a fast equilibrium with H2O2 to form HOOCO2−, a versatile heterolytic oxidant. In the following step, through nucleophilic attack of the oxidizing agent on the MnII center, a MnII–η2-peroxycarbonate complex is formed. The remaining coordination sites in the first shell will be occupied by water and hydroxyl at a pH between 8 and 10. The principal mode of the formation of relatively stable high-valent MnO intermediates is believed to involve the heterolytic cleavage of the peroxide bond, as shown in Scheme 4. An important role in the stabilization of the formed MnO species is played by the electron donating bicarbonate ions. This may also account for the unique requirement of HCO3− in the oxidative decomposition of Orange II catalyzed by simple manganese salts. The further coordination of the substrate followed by an oxygen transfer step along with the second electron, leads to the formation of several oxidation products and finally to the regeneration of the catalyst. It must be noted that in the absence of a catalyst, the oxidative degradation of Orange II by addition of an electrophilic bleaching agent, HOOCO2−, occurs very slowly under certain reaction conditions. The oxidation mechanism involves nucleophilic attack of the dye at the electrophilic oxygen of HOOCO2−. In aqueous solution, proton transfer can lead to the displacement of HCO3− and the slow formation of oxidized substrate.
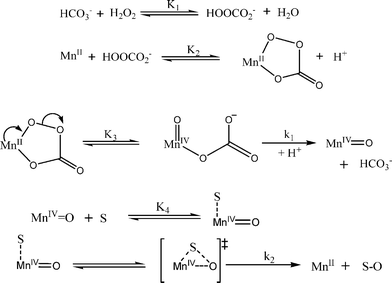 | ||
| Scheme 4 Proposed reaction mechanism for the Mn(II) catalyzed oxidative degradation of Orange II by H2O2 in a carbonate containing aqueous solution at pH between 8–9 and 25 °C. | ||
If substrate binding to MnII occurs before the addition of HOOCO2− to the catalyst solution, following reactions can be assumed to take place during the reaction cycle under the chosen experimental conditions.
In line with the concerns mentioned above, the first step in Scheme 5 involves the prior coordination of Orange II to MnII and formation of MnII–Orange II complexes of different stoichiometry, followed by nucleophilic attack of the oxidant on the MnII center leading to the formation of Orange II–MnII–peroxycarbonate species. The subsequent scission of the peroxo bond leads to the formation of high-valent oxo intermediates, as formulated in Scheme 4. In this case, the formed MnIVO intermediate is stabilized by Orange II, an electron rich organic molecule with chelating capacity. The importance of Orange II as an equatorial ligand is also to favor the heterolytic scission of the peroxo bond leading to the MnIVO intermediate and bicarbonate.
 | ||
| Scheme 5 Proposed reaction mechanism involving first substrate coordination to MnII in a pre-equilibrium step during the catalyzed oxidative degradation of Orange II by H2O2 in a carbonate containing aqueous solution at pH between 8–9 and 25 °C. | ||
Conclusions
A fast and environmentally benign method for the oxidative degradation of Orange II could be achieved using H2O2 in conjunction with catalytic amounts of relatively non-toxic manganese salts as catalyst precursors in a carbonate containing aqueous solution under mild reaction conditions. Screening and spectroscopic methods allowed us to study the catalytic reaction course and to identify some key features of the reaction that reflect upon its mechanism. Our study revealed that the oxidative degradation of the model substrate Orange II is catalytic only in carbonate containing aqueous solution. No other buffer containing aqueous solution could induce the oxidative degradation of Orange II by H2O2 and this led to the implication of peroxycarbonate as a key molecular entity. The reported experimental data suggests that the in situ formed high-valent manganese intermediate possessing one hydrogen carbonate ligand is able to activate H2O2, but decomposes rapidly with the formation of neutral MnCO3, which precipitates from solution as an insoluble white solid. One of the main factors affecting the process efficiency was the stabilization of the catalytically active Mn complex. Furthermore, by addition of Orange II, the formation of MnII⋯Orange II complexes with different stoichiometry was observed. The simultaneous σ,π-coordination of the organic dye is well-precedented, and recent DFT studies support this type of complex formation.28 The catalytic activity of the formed intermediates was tested under catalytic reaction conditions.The kinetic investigations performed at different pH could provide relevant information about the nature of the oxidizing agent involved in the reaction. It was found that the pH is a critical issue for the rate of the oxidation process due to its influence on the deprotonation of the bicarbonate ions, the formation of peroxycarbonate in solution, and the deprotonation of aquated Mn2+. The ongoing studies are presently complemented by investigations on different organic substrates with various functional groups in order to determine the influence of substrate modification on the catalytic reaction cycle. DFT studies beside further kinetic and spectroscopic investigations should contribute to a better understanding of the catalytic system.
Acknowledgements
The authors kindly acknowledge fruitful discussions with Dr Anette Nordskog and Dr Wolfgang von Rybinski, Henkel KGaA, Düsseldorf, Germany.References
- I. Mielgo, C. López, M. T. Mareira, G. Feijoo and J. M. Lema, Biotechnol. Prog., 2003, 19, 325 CrossRef CAS.
- J. Hastie, D. Bejan, M. Teutli–León and N. J. Bunce, Ind. Eng. Chem. Res., 2006, 45, 4898 CrossRef CAS.
- H. Park and W. Choi, J. Photochem. Photobiol., A, 2003, 159, 241 CrossRef CAS.
- G. L. Baughman and E. J. Weber, Environ. Sci. Technol., 1994, 28, 267 CrossRef CAS.
- T. Robinson, G. McMullan, R. Marchant and P. Nigam, Bioresource Technol., 2001, 77, 247 CrossRef CAS.
- T. J. Collins, Acc. Chem. Res., 1994, 27, 279 CrossRef CAS.
- R. Hage, J. E. Iburg, J. Kerschner, J. H. Koek, E. L. M. Lempers, R. J. Martens, U. S. Racherla, S. W. Russel, T. Swarthoff, M. R. P. van Vliet, J. B. Warnaar, L. van der Wolf and B. Krijnen, Nature, 1994, 369, 637 CrossRef CAS.
- A. Battacharyya, S. Kawi and M. B. Ray, Catal. Today, 2004, 98, 431 CrossRef CAS.
- J. H. Ramirez, F. J. Maldonado-Hódar, A. F. Pèrez-Cadenas, C. Moreno-Castilla, C. A. Costa and L. M. Madeira, Appl. Catal., B, 2007, 75, 312 CrossRef CAS.
- J. H. Ramirez, C. A. Costa, L. M. Madeira, G. Mata, M. A. Vicente, M. L. Rojas-Cervantes, A. J. López-Peinado and R. M. Martin-Aranda, Appl. Catal., B, 2007, 71, 44 CrossRef CAS.
- (a) Y. Zhiyong, M. Bensimon, D. Laub, L. Kiwi–Minsker, W. Jardim, E. Mielczarski, J. Mielczarski and J. Kiwi, J. Mol. Catal. A: Chem., 2007, 272, 11 CrossRef CAS; (b) X. Chen, X. Qiao, D. Wang, J. Lin and J. Chen, Chemosphere, 2007, 67, 802 CrossRef CAS.
- T. W. Kim, S. G. Hur, S.-J. Hwang, H. Park, W. Choi and J.-H. Choy, Adv. Funct. Mater., 2007, 17, 307 CrossRef CAS.
- T. Wieprecht, J. Xia, U. Heinz, J. Dannacher and G. Schlingloff, J. Mol. Catal. A: Chem., 2003, 203, 113 CrossRef CAS.
- N. Chahbane, D.-L. Popescu, D. A. Mitchell, A. Chanda, D. Lenoir, A. D. Ryabov, K.-W. Schramm and T. J. Collins, Green Chem., 2007, 9, 49 RSC.
- G. B. Payne, P. H. Deming and P. H. Williams, J. Org. Chem., 1961, 26, 659 CrossRef CAS.
- G. Majetich and R. Hicks, Synlett, 1996, 649 CrossRef CAS.
- A. McKillop and W. R. Sanderson, Tetrahedron, 1995, 51, 6145 CrossRef CAS.
- J. E. Barker and T. Ren, Tetrahedron Lett., 2004, 45, 4681 CrossRef CAS.
- J Oakes, Eur. Pat. Appl., 145091, 1985.
- X.-M. Feng, Z. Wang, N.-S. Bian and Z.-L. Wang, Inorg. Chim. Acta, 2007, 360, 4103 CrossRef CAS.
- (a) C. Kim, K. Chen, J. Kim and L. Que Jr, J. Am. Chem. Soc., 1997, 119, 5964 CrossRef CAS; (b) D. D. Vos and T. Bein, Chem. Commun., 1996, 917 RSC; (c) P. Battioni, J. P. Renaud, J. F. Bartoli, M. Rein-Artiles, M. Fort and D. Mansuy, J. Am. Chem. Soc., 1988, 110, 8462 CrossRef CAS.
- N. Nakayama, S. Tsuchiya and S. Ogawa, J. Mol. Catal. A: Chem., 2007, 277, 61 CrossRef CAS.
- C. F. Baes, J. R. Mesmer and R. E. Mesmer, Am. J. Sci., 1981, 281, 935 Search PubMed.
- S.-Y. Liu and D. G. Nocera, Tetrahedron Lett., 2006, 47, 1923 CrossRef CAS.
- (a) A. D. Becke, J. Phys. Chem., 1993, 97, 5648; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS; (c) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS; (d) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS; (e) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 284 CrossRef CAS; (f) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS; (g) Gaussian Basis Sets for Molecular Calculations, ed. S. Huzinaga, Elsevier, Amsterdam, 1984 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Wallingford, CT, 2004.
- H. Zollinger, Color Chemistry, Wiley-VCH, Weinheim Germany, 3rd edn, 2003, ch. 2, pp. 18–50 Search PubMed.
- L. C. Abbott, S. N. Batchelor, J. Oakes, B. C. Gilbert, A. C. Whitwood, J. R. Lindsay Smith and J. N. Moor, J. Phys. Chem., 2005, 109, 2894 Search PubMed.
- G. R. Hodges, J. R. Lindsay Smith and J. Oakes, J. Chem. Soc., Perkin Trans. 2, 1998, 617 RSC.
- J. Oakes and P. Gratton, J. Chem. Soc., Perkin Trans. 2, 1998, 1857 RSC.
- W. Feng, D. Nansheng and H. Helin, Chemosphere, 2000, 41, 1233 CrossRef CAS.
- J. R. Lindsay Smith, B. C. Gilbert, A. M. Payeras, J. Murray, T. R. Lowdon, J. Oakes, R. P. Prats and P. H. Walton, J. Mol. Catal. A: Chem., 2006, 251, 114 CrossRef.
- J. Tokuda, R. Oura, T. Iwasaki, Y. Takeuchi, A. Kashiwada and M. Nango, Coloration Technol., 2000, 116, 42 Search PubMed.
- T. Yoshida and S. Sawada, Bull. Chem. Soc. Jpn., 1975, 48, 345 CrossRef CAS.
- C. López, J. C. García-Monteagudo, M. T. Madeira, G. Feijoo and J. M. Lema, Enzyme Microb. Technol., 2007, 42, 70 CrossRef CAS.
- (a) R. L. Reeves, M. S. Maggio and S. A. Harkaway, J. Phys. Chem., 1979, 83, 2359 CrossRef CAS; (b) T. Asakura and M. Ishida, J. Colloid Interface Sci., 1989, 130, 184 CrossRef CAS.
- K. Murakami, Dyes Pigm., 2002, 53, 31 CrossRef CAS.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703 CrossRef CAS.
- P. Job, Compt. Rend., 1925, 180, 928 Search PubMed.
- V. Nadtochenko and J. Kiwi, J. Chem. Soc., Faraday Trans., 1997, 93, 2373 RSC.
- C. Bauer, P. Jacques and A. Kalt, Chem. Phys. Lett., 1999, 307, 397 CrossRef CAS.
- A. Ya. Sychev, U. Pfannmueller and V. G. Isak, Zh. Fiz. Khim., 1983, 8, 1974.
- (a) D. Lesht and J. E. Bauman, Inorg. Chem., 1978, 17, 3332 CrossRef CAS; (b) J. Dasgupta, A. M. Tyryshkin, Y. N. Kozlov, V. V. Klimov and G. C. Dismukes, J. Phys. Chem. B, 2006, 110, 5099 CrossRef CAS.
- A. C. Olivieri, R. B. Wilson, I. C. Paul and D. Y. Curtin, J. Am. Chem. Soc., 1989, 111, 5525 CrossRef CAS.
- H. D. K. Drew and J. K. Landquist, J. Chem. Soc., 1939, 292 RSC.
- (a) X. Zhu, B. Li, X. He and Y. Zhang, J. Chem. Crystallogr., 2005, 35, 443 CrossRef CAS; (b) A. J. Bortoluzzi, A. Neves, R. A. A. Couto and R. A. Perelta, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, C62, m27 CrossRef CAS; (c) T. H. Bennur, D. Srinivas, S. Sivasanker and V. G. Puranik, J. Mol. Catal. A: Chem., 2004, 219, 209 CrossRef CAS.
- M. R. Blomberg, P. E. M. Siegbahn, S. Styring, G. T. Babeock, B. Akermark and P. Korall, J. Am. Chem. Soc., 1997, 119, 8285 CrossRef CAS.
- K. G. Tikhonov, O. M. Zastrizhanaya, Yu N. Kozlov and V. V. Klimov, Biochemistry, 2006, 71, 1270 CAS.
- D. Swern, Organic Peroxides, Wiley, New York, 1970, pp. 313 Search PubMed.
- D. E. Richardson, H. Yao, K. M. Frank and D. A. Bennett, J. Am. Chem. Soc., 2000, 122, 1729 CrossRef CAS.
- B. S. Lane, M. Vogt, V. J. DeRose and K. Burgess, J. Am. Chem. Soc., 2002, 124, 11946 CrossRef CAS.
- D. E. Richardson, C. A. S. Regino, H. Yao and J. V. Johnson, Free Radical Biol. Med., 2003, 35, 1538 CrossRef CAS.
- MnIV in the presence of further MnII ions leads to the formation of MnIII , a strong but highly unstable oxidant, according to the reaction: MnIV + MnII⇌ 2MnIII.
- K. Hashimoto, S. Nagatomo, S. Fujinami, H. Furutachi, S. Ogo, M. Suzuki, A. Uehara, Y. Maeda, Y. Watanabe and T. Kitagawa, Angew. Chem., 2002, 114, 1251.
- M. Aresta, I. Tommasi, E. Quaranta, C. Fragale, J. Mascetti, M. Tranquille, F. Galan and M. Fouassier, Inorg. Chem., 1996, 35, 4254 CrossRef CAS.
- P. J. Hayward, D. M. Blake, G. Wilkinson and C. J. Nyman, J. Am. Chem. Soc., 1970, 92, 5873 CrossRef.
- K. Hashimoto, S. Nagatomo, S. Fujinami, H. Furutachi, S. Ogo, M. Suzuki, A. Uerhara, Y. Maeda, Y. Watanabe and T. Kitagawa, Angew. Chem., Int. Ed., 2002, 41, 1202 CrossRef CAS.
- B. Balagam and D. E. Richardson, Inorg. Chem., 2008, 47, 1173 CrossRef CAS.
- (a) A. M. Khenkin, D. Kumar, S. Shaik and R. Neumann, J. Am. Chem. Soc., 2006, 128, 15451 CrossRef CAS; (b) P. E. M. Siegbahn and R. H. Crabtree, J. Am. Chem. Soc., 1999, 121, 117 CrossRef CAS; (c) M. Kondo, T. Mitsui, S. Ito, Y. Kondo, S. Ishigure, T. Dewa, K. Yamashita, J. Nakamura, R. Oura and M. Nango, J. Colloid Interface Sci., 2007, 310, 686 CrossRef CAS; (d) G. B. Shuĺpin, M. G. Matthes, V. B. Romakh, M. I. F. Barbosa, J. L. T. Aoyagi and D. Mandelli, Tetrahedron, 2008, 64, 2143 CrossRef CAS; (e) A. E. Anastasi, P. H. Walton, J. R. Lindsay Smith, W. M. C. Sameera and J. E. McGrady, Inorg. Chim. Acta, 2008, 361, 1079 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2009 |