Beryllium uptake and related biological effects studied in THP-1 differentiated macrophages
Jian
Ding
,
Lin
Lin
,
Wei
Hang
and
Xiaomei
Yan
*
Department of Chemical Biology, College of Chemistry and Chemical Engineering, The Key Laboratory for Chemical Biology of Fujian Province, Key Laboratory of Analytical Science, Xiamen University, Xiamen 361005, China. E-mail: xmyan@xmu.edu.cn; Fax: +86-592-2189959; Tel: +86-592-2184519
First published on 14th September 2009
Abstract
Investigation of cellular uptake of metal compounds is important in understanding metal-related toxicity and diseases. Inhalation of beryllium aerosols can cause chronic beryllium disease, a progressive, granulomatous fibrosis of the lung. Studies in laboratory animals and cultured animal cells indicate that alveolar macrophages take up beryllium compounds and participate in a hypersensitivityimmune response to a beryllium-containing antigen. In the present work, human monocytecell line THP-1 was induced with phorbol myristate acetate to differentiate into a macrophage. This cell with characteristics of human alveolar macrophages was employed to study cellular beryllium uptake and related biological effects. Morphological changes, phagocytosis of fluorescent latex beads, and cell surface CD14 expression were used to verify the successful differentiation of THP-1 monocytes into macrophages. An improved mass spectrometry method for quantitative analysis of intracellular beryllium as opposed to the traditional radioisotopic approach was developed using ICP-MS. The influence of the solubility of beryllium compounds, exposure duration, and beryllium concentration on the incorporation of beryllium was studied. Our data indicated that the uptake of particulate BeO was much more significant than that of soluble BeSO4, suggesting the major cellular uptake pathway is phagocytosis. Nevertheless, subsequent DAPI nuclear staining and PARP cleavage study indicated that beryllium uptake had a negligible effect on the apoptosis of THP-1 macrophages compared to the unstimulated macrophage control. Meanwhile, no substantial variation of tumour necrosis factor-alpha production was observed for THP-1 macrophages upon beryllium exposure. These data imply alveolar macrophages could have some level of tolerance to beryllium and this may explain why most Be-exposed individuals remain healthy throughout life.
Introduction
Beryllium, the lightest stable metal, possesses many unique physical and chemical properties that make it an ideal choice as a material for high-technology industries, such as aerospace, nuclear defense, and electronics. However, environmental exposure to beryllium, even at very low concentrations, can trigger beryllium sensitization (BeS) and chronic beryllium disease (CBD), a granulomatous lung disease resulting from a delayed-hypersensitivity, cell-mediated immune response to beryllium.1 BeS represents the proliferative responses of beryllium-specific T cells in the blood and/or bronchoalveolar lavage (BAL) of beryllium-exposed workers, and the rate of progression from BeS to CBD has been estimated to be about 6% to 8% per year.2 Depending on the nature of beryllium exposure and the individual’s genetic susceptibility, CBD will eventually develop in 2-16% of these subjects.3–5 The current understanding of how CBD develops is that inhaled beryllium is taken up by alveolar macrophages and processed by the MHC class II pathway, possibly via binding to an as-yet-unidentified peptide. This Be-peptide complex is subsequently presented by the HLA-DP heterodimers formed on the surface of the antigen-presenting cells and recognized by some Be-specific CD4+ T cell clones, which initiate a host immune response.6–10 However, the true identity of the beryllium antigen remains only speculative and it is not completely clear how the beryllium initially enters the cell.Information about the interaction of beryllium with lung macrophages is important because these cells play central roles in handling environmental burdens, generating chronic inflammatory responses, and participating in the sensitization of T lymphocytes in cell-mediated immune responses.11 In the respiratory tract, alveolar macrophage cells are among the first lung cells to be exposed to inhaled foreign substances, such as particulate beryllium. However, due to the inconvenience and difficulty of obtaining an adequate amount of BAL cells, very limited investigation on cellular beryllium uptake using alveolar macrophages has been reported.11,12 These studies, which used laboratory animals, along with reports on intravenous administration to rat liver cells13,14 and studies of cultured animal cells,15,16 indicate that particulate forms (e.g., beryllium phosphate) are taken up to a markedly greater extent than soluble salts (e.g., beryllium sulfate). Yet, the results obtained from animal cells cannot be directly extrapolated to humans because of species differences.
The human monocytic leukemia cell line THP-1 develops macrophage functions following inducement by phorbol myristate acetate (PMA), and has been shown to be a good model for in vitro-differentiated, monocyte-derived macrophages.17,18 The THP-1 differentiated macrophage has been extensively characterized,19,20 and its many cytochemical and immunological characteristics, such as lysozyme production, phagocytic activity, and capacity of response to T-lymphocyte, resemble alveolar macrophages of human origin. Due to metabolic and morphological similarities, THP-1 differentiated macrophages have been widely used as an in vitro model in the study of alveolar macrophage involvement in inflammatory diseases.21–24 Considering cell culture offers the advantages of no donor variability in macrophage function and that large numbers of cells can be grown reproducibly, in this study, we seek to use THP-1 differentiated macrophages to mimic human alveolar macrophages for a cellular beryllium uptake and related biological effects study.
In all the aforementioned cellular beryllium uptake studies using laboratory animals or cultured animal cells, radioactive isotope labeling (7Be, half-life of 53 d) has been used exclusively to ensure detection sensitivity. However, radioisotopes have inherent problems associated with safety hazards, legal requirements, and regulations for storage and disposal. Technology development in atomic spectroscopy has enabled accurate and sensitive measurement of metal concentrations by more convenient and eco-friendly means. Inductively coupled plasma mass spectrometry (ICP-MS) is a powerful tool for the elemental analysis of biological samples.25–27 In the present work, a new analytical approach for measuring cellular beryllium content was developed using ICP-MS in place of conventional radioactive isotope labeling. The influence of the solubility of beryllium compounds, exposure duration, and beryllium concentration on the incorporation of beryllium was studied. Apoptosis and tumor necrosis factor-alpha (TNF-α) production were investigated to evaluate the biological effects of beryllium on THP-1 differentiated macrophages.
Materials and methods
Chemicals and reagents
PMA (Sigma, St. Louis, MO, USA) was prepared as a solution of 2 mg/mL in dimethyl sulfoxide, aliquoted and frozen. A PMA aliquot was then diluted with cell culture medium to obtain a stock solution of 1 μg/mL, to be used at a final concentration of 100 ng/mL for THP-1 cell inducement. A stock solution of 100 mM BeSO4 (Alfa Aesar China, Tianjing) and a stock suspension of 25 mM BeO (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) were prepared in deionized water. Before being added into the cell culture medium, the BeO stock suspension was sonicated for 5–10 min. Due to the propensity of beryllium salts to form beryllium phosphate in the presence of inorganic phosphate, a 0.9% NaCl solution was used instead of PBS as the balanced salt solution. A 1-M sulfosalicylic acid (SSA, Sinopharm Chemical Reagent Co., Ltd.) solution was prepared in 0.9% NaCl solution, and when used, diluted to a concentration of 100 μM. A stock solution of 1 mg/mL 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI, Sigma) was prepared in deionized H2O and diluted to 1 μg/mL in PBS when used to stain the cells. Doxycycline was purchased from Sigma. All the other reagents were of analytical grade. The fluorescein isothiocyanate (FITC) labeled mouse anti-human CD14 monoclonal antibody (mAb) and the mouse IgG2b FITC/Isotype Control were purchased from Beijing CellChip Biotechnology Co., Ltd., Beijing, China. Poly-ADP-ribose polymerase (PARP) mAb, β-actin mAb, and goat anti-mouse IgG were purchased from BD Biosciences (NJ, USA), Santa Cruz Biotechnology (Santa Cruz, CA, USA), and Thermo Scientific (Rockford, IL, USA), respectively.Cellular culture and differentiation
The THP-1 cell line was purchased from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. THP-1 cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated, fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, 2.05 mM L-glutamine, 1 mM sodium pyruvate (all from Hyclone Co., Logan, UT, USA), 10 mM HEPES, 0.05 mM 2-mercaptoethanol, and 2.5 g/L D-glucose (all from Amresco Co., San Diego, CA, USA). Cells were maintained in continuous culture at 37 °C in 5% CO2 and 95% air. Before PMA inducement, cell viability as determined by trypan blue exclusion was >95% in all experiments. The cells were counted manually using a hemacytometer under the light microscope. Differentiation was achieved by plating 10 mL of the THP-1 monocytes suspended in growth medium containing 100 ng/mL PMA (5.0 × 105 cells/mL) into 100-mm Falcon culture dishes, and then culturing for 48 h or other indicated periods of time. After aspirating the medium, the differentiated THP-1 macrophage cells were washed twice with 0.9% NaCl solution. Optimal differentiation conditions were identified with CD14 expression measurement and a phagocytic activity test.Beryllium uptake protocol and ICP-MS measurement
Ten micromolar and one hundred micromolar BeSO4 or BeO supplemented in culture medium were used for macrophage treatments, as these concentrations have been reported to be within the optimal stimulatory concentration range.7,28,29 A schematic of the experimental design for the cellular beryllium uptake study is shown in Fig. 1. Briefly, THP-1 differentiated macrophages were stimulated with beryllium for 48 h or other indicated periods of time at 37 °C in a humidified, 5% CO2 atmosphere. The cells were washed five times with 0.9% NaCl solution. After the second wash, the cells were incubated in 0.9% NaCl containing 100 μM of SSA for 10 minutes. The final (fifth) washing solutions were collected. After the final plate washing, the macrophages were harvested (0.25% trypsin treated for 90–150 s, counted, and centrifuged) and resuspended in 150 μL of deionized water and 250 μL of concentrated, ultrapure-grade HNO3 (Alfa Aesar China) was added. After 5 min of sonication, the mixture was heated at 80 °C on a metallic heating plate for 2 h. The beryllium content of the digested cell samples and the collected washing solutions were analyzed on an Agilent 4500 ICP-MS. An equal concentration of Rh (10 ng/mL) was added to all blanks, standards, and samples to serve as an internal standard. The detection limit of Be was 0.3 pg/mL and the recoveries for Be-spiked samples were between 92% and 108%. The detected beryllium concentrations of the cell digests upon beryllium exposure were all greater than several hundred pg/mL, well above the detection limit. The average cellular beryllium content was calculated based on the ICP-MS-detected beryllium concentration of the cell digest, the digest sample volume, and the cell number. All experiments were conducted in triplicate from the point of PMA inducement onward. Experiments were repeated a minimum of three times, and representative data are presented.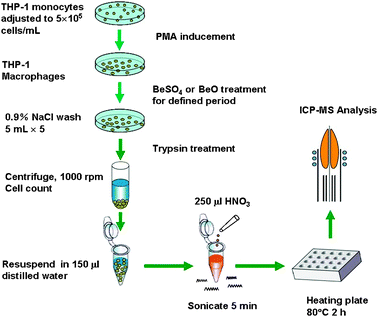 | ||
| Fig. 1 Experiment flowchart for the cellular beryllium uptake study. | ||
Phagocytic activity tests
To determine the phagocytic capability of THP-1 macrophages, carboxyl fluorescent yellow beads (Spherotech Inc., Lake Forest, IL, USA) were added to the medium of the negative control THP-1 cells and the differentiated macrophage cells at a bead-to-cell molar ratio of 45 : 1. After 2 h of incubation, the differentiated cells were washed twice with PBS to remove the free beads. The THP-1 control cells were centrifuged at 1000 rpm (Beckman Coulter Allegra X-15R Centrifuge) to remove the free beads. Then, the macrophages and THP-1 control cells were observed under a Ti-U Nikon fluorescent microscope.Flow cytometric assays
Flow cytometric measurements were made using a FACSAria flow cytometer (BD Biosciences, San Jose, CA, USA). Expression of CD14, a marker known to increase in differentiated macrophages, was analyzed by incubating a FITC-conjugated CD14-specific mAb with the differentiated macrophage cells and the THP-1 control cells. Mouse IgG2b-FITC was used as a negative isotype control. The cells were harvested, washed three times with ice-cold PBS, and resuspended in PBS at 1 × 106 cells/mL. Then the mAb or the control isotype was added and incubated at room temperature for 30 min. After three washes with PBS, the cells were resuspended in 200 μL of PBS. Flow cytometry analysis was performed by analyzing 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cell events. THP-1 cells were gated for analysis by a combination of forward scatter (FSC) and 90° side scatter (SSC). The percentage of cells positive for the CD14 marker was determined by setting a cursor on the negative isotope control to include 98–99% of the population. Data analysis was performed using BD FACSDiva software (BD Biosciences).
000 cell events. THP-1 cells were gated for analysis by a combination of forward scatter (FSC) and 90° side scatter (SSC). The percentage of cells positive for the CD14 marker was determined by setting a cursor on the negative isotope control to include 98–99% of the population. Data analysis was performed using BD FACSDiva software (BD Biosciences).
Nuclear staining with DAPI
For the apoptosis study, THP-1 macrophages were cultured at 1 × 106 cells/mL with 2 mL per well in NUNC six-well plates (#150628, Roskilde, Denmark). THP-1 macrophages were unstimulated, BeSO4-stimulated, BeO-stimulated, and doxycycline-stimulated for 48 h. After treating the cells with beryllium compounds, the THP-1 macrophages were washed with cold PBS and stained with DAPI solution for 15 min at room temperature. The nuclear morphology of the cells was examined with a fluorescent microscope (ZEISS AXIO Imager.A1, Germany). The unstimulated macrophages were the negative control, and the THP-1 macrophages induced to apoptosis by 80 μg/mL doxycycline were the positive control.30Western blot analysis
The unstimulated, Be-stimulated, and doxycycline-induced cells (cultured as described in “Nuclear staining with DAPI”) were harvested and lysed with RIPA buffer (150 mM sodium chloride, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris, pH 8.0). Protein concentrations were quantified using a Bio-Rad proteinassay (Bio-Rad Laboratories, Hercules, CA, USA), according to the manufacturer’s instructions. Equal amount of proteins were subjected to electrophoresis on 10% SDS-polyacrylamide gel and transferred to a poly(vinylidene difluoride) membrane (PVDF, GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). The blots were probed with anti-PARP and anti-β-actin mAbs for 12 h at 4 °C, incubated with the diluted enzyme-linked goat anti-mouse secondary antibody and visualized according to the recommended procedure.Quantification of TNF-α production
The supernatants from BeSO4-stimulated, BeO-stimulated, and unstimulated THP-1 macrophages and THP-1 monocytes (cultured as described in “Nuclear staining with DAPI”) were collected by centrifugation at 12000 rpm for 10 min. A standard sandwich ELISA assay was performed with a human TNF-α kit (eBioscience, Camarillo, CA, USA) following the manufacturer’s instructions. The measured detection limit of the kit was 4 pg/mL. Dilution was made to the supernatants to ensure that the detected value falls into the linear dynamic range of the standard calibration curve. For each sample, three replicates were carried out in parallel.
Results
Induction of THP-1 cell differentiation into macrophages
CD14 upregulation upon PMA treatment has been reported to be a surface marker for macrophages.17,31 In order to confirm the successful differentiation of THP-1 cells into macrophages, we analyzed CD14 expression on the cell surface after PMA inducement. After 24 h of PMA treatment, the confluent THP-1 cells become adherent and pseudopod spreading of the macrophages can be observed. However, as shown in Fig. 2, very few cells (∼17%) exhibited an upregulated CD14 expression after 24 h of inducement. With the treatment time increased to 48 h or longer, a marked increase in CD14 cell surface expression was identified. Further increase of PMA inducement duration to 72 h did not yield distinct enhancement of CD14 expression (65% at 48 h versus 70% at 72 h). Therefore, 48 h was chosen as the optimal PMA inducement time. The phagocytic capability of the THP-1 macrophages was further tested using fluorescent beads. After incubation with fluorescent beads for 2 h, bright fluorescence from beads engulfed within the macrophage was clearly observed under the microscope, and the bead number varied from several to 20 per macrophage cell. On the other hand, no bead uptake could be identified in the THP-1 monocytes (data not shown).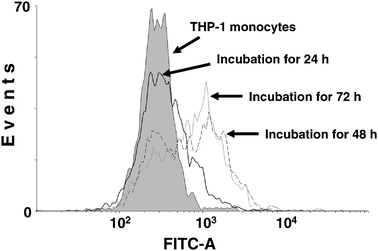 | ||
| Fig. 2 PMA-stimulated CD14 cell surface expression in THP-1 macrophages. THP-1 monocytes were induced with PMA for 24 h, 48 h, 72 h, or left untreated. Flow cytometric measurements were made using a FACSAria flow cytometer. | ||
Examination of cell surface adsorption of beryllium
One obstacle to measuring the uptake of metal compounds is extracellular binding.32 In order to examine whether cell surface adsorption of beryllium would occur and how this would affect the accurate measurement of the intracellular beryllium content, the extent of cell-surface-adsorbed beryllium was carefully quantified. Sulfosalicylic acid (SSA) is a well-known beryllium ligand and was used here to rinse off any beryllium that may have been nonspecifically adsorbed onto the cell surface.16 Upon beryllium treatment for 48 h, the macrophages were washed twice with 0.9% NaCl solution and then incubated with 100 μM SSA for 10 minutes, followed by two additional washes (five washes in total, 2 NaCl + 1 SSA + 2 NaCl). Cells not washed with SSA were processed in parallel as a control. The 5th washing solution and the harvested cells were analyzed on the ICP-MS. For beryllium sulfate treatment, the beryllium concentration of the 5th washing solution was marginal regardless of SSA washing. For beryllium oxide stimulation, the beryllium concentration of the 5th washing solution was below the detection limit of ICP-MS. These results indicated that five washes were adequate to remove the beryllium in the medium. Meanwhile, for cells treated with 10 μM BeSO4, the beryllium concentrations measured were 2.51 ± 0.05 ng/mL and 2.63 ± 0.01 ng/mL for the harvested macrophages with and without SSA washing, respectively. For cells treated with 100 μM BeSO4, the beryllium concentrations measured were 6.13 ± 0.09 ng/mL and 6.43 ± 0.02 ng/mL for the harvested macrophages with and without SSA washing, respectively. These data indicated that for cells treated with soluble beryllium sulfate, the cell-associated beryllium content was about 5% less with SSA washing than without SSA washing. This reduced value was defined as “nonspecific adsorption”, which is similar to the ∼5% cell surface adsorption rate reported in the literature.11 But for particulate forms, nonspecifically adsorbed beryllium was negligible in comparison to the significant cellular uptake of beryllium viaphagocytosis.Beryllium compound solubility, incubation duration, and concentration effects on cellular uptake
The THP-1 differentiated macrophages were cultured in medium supplemented with BeSO4 or BeO (both were tested at 10 μM and 100 μM) for 2 h, 4 h, 12 h, 24 h, or 48 h. The cellular beryllium content was measured by ICP-MS, with the results shown in Fig. 3. Clearly, cellular beryllium uptake was much more significant for particulate BeO than for soluble BeSO4. Within the initial 2 h, the measured cellular beryllium content levels were 0.21 ± 0.06, 0.23 ± 0.04, 4.61 ± 0.04, and 59.32 ± 1.01 nmol/106 cells for 10 μM BeSO4, 100 μM BeSO4, 10 μM BeO, and 100 μM BeO, respectively. After 48 h of treatment, cellular beryllium content reached 3.84 ± 0.26, 7.24 ± 0.50, 9.12 ± 0.16, and 78.17 ± 9.04 nmol/106 cells for 10 μM BeSO4, 100 μM BeSO4, 10 μM BeO, and 100 μM BeO, respectively. For THP-1 macrophages treated with 10 μM BeO, cellular beryllium uptake increased linearly with time; for treatment with 100 μM BeO, ∼75% of the total uptake was completed within the initial 2 h, indicating that BeO was readily taken up as a particle. For soluble BeSO4, the cellular beryllium uptake increased linearly with time, but the total amount taken up was much lower than for BeO. Moreover, the amount taken up was not proportional to the BeSO4 concentration administrated. Meanwhile, in the control experiment with THP-1 monocytes exposed to 100 μM BeSO4 for 24 h, the measured cellular beryllium content was 0.65 ± 0.03 nmol/106 cells, which was much lower than the 3.14 ± 0.41 nmol/106 cells for THP-1 macrophages. These results indicate that phagocytosis of particulate beryllium is the major pathway for cellular beryllium uptake.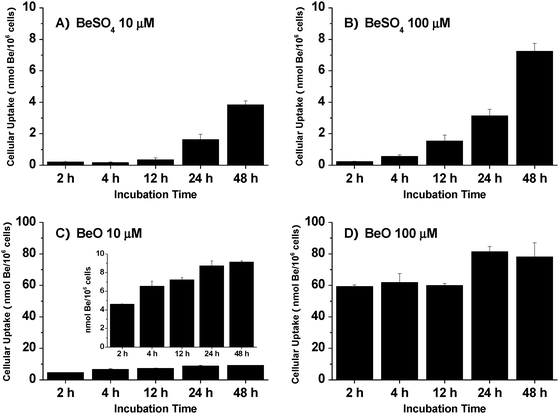 | ||
| Fig. 3 Uptake of beryllium by THP-1 macrophages as a function of beryllium solubility, incubation duration and beryllium concentration. (A), (B), (C), (D) Cellular beryllium content for incubation with 10 μM BeSO4, 100 μM BeSO4,10 μM BeO, and 100 μM BeO, respectively. The data represent the mean ± SD of three replicated experiments. | ||
Cytotoxic effects of cellular beryllium uptake
From the above cellular uptake experiment, it is clear that a considerable amount of beryllium was taken up by the THP-1 differentiated macrophage. The cytotoxicity of internalized beryllium on the THP-1 macrophages was examined using fluorescence microscopy of DAPI staining and Western blot analysis of the degradation of PARP. Results were compared for THP-1 macrophages that were untreated (negative control), BeSO4-stimulated (10 μM or 100 μM), BeO-stimulated (10 μM or 100 μM), and doxycycline-stimulated (positive control) for 48 h. Normally, live cells show uniformly DAPI-stained, intact nuclei. Late apoptotic cells can be detected by the fragmentation of DAPI-stained nuclei. As shown in Fig. 4A, very few apoptotic cells were detected in unstimulated THP-1 macrophages. Compared to untreated cells, administration of 10 μM or 100 μM of BeSO4 or BeO did not result in marked enhancements of the percentage of cells undergoing late apoptosis. On the other hand, obvious nuclei fragmentation can be identified for THP-1 macrophages treated with 80 μg/mL doxycycline.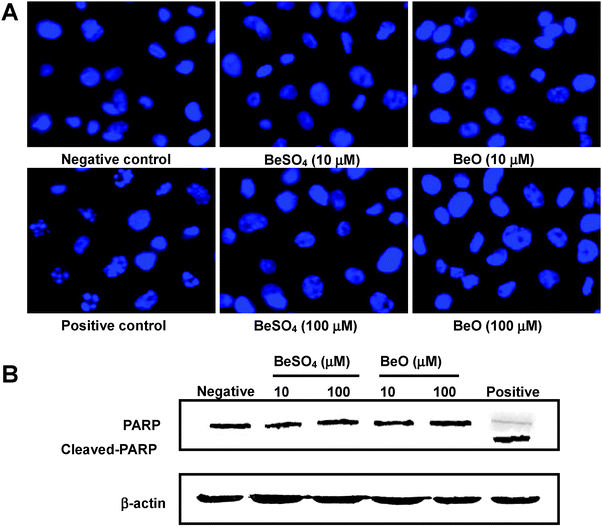 | ||
| Fig. 4 Cytotoxicity of cellular beryllium uptake on THP-1 macrophages. (A) The macrophages were treated with 10 μM or 100 μM of BeO or BeSO4, respectively, for 48 h, fixed and stained with DAPI. The nuclei were then photographed under fluorescence using a blue filter (400× magnification). (B) Western blot analysis of PARP from the THP-1 differentiated macrophages treated with 10 μM or 100 μM of BeO or BeSO4, respectively, for 48 h. THP-1 macrophages that were untreated were the negative control. Doxycycline treated THP-1 macrophages were the positive control. All DAPI-stained images and Western blots are representative of three independent experiments. | ||
One of the key initiation elements of the apoptotic pathway is the activation of caspases, followed by cleavage of the caspase substrates. The 113-kDa PARP, which is normally involved in DNA repair, DNA stability, and other cellular events, is cleaved by members of the caspase family during early apoptosis. Detection of an 89-kDa or 24-kDa caspase cleavage fragment of PARP has been used as a hallmark of apoptosis.33 To further examine the apoptotic effect of beryllium compounds on THP-1 macrophages, PARP cleavage was analyzed by immunoblotting. In Fig. 4B, all the samples loaded showed a similar amount of a housekeeping protein (β-actin). Compared to the untreated control, no distinct PARP cleavage could be observed for any of the Be-treated THP-1 macrophages while a cleaved-PARP band was observed for the positive control. Clearly, both DAPI staining and PARP cleavage studies indicated that cellular beryllium uptake had negligible cytotoxic effects on the THP-1 macrophages.
Effect of Be-stimulation on TNF-α production
TNF-α has been reported as a pro-inflammatory cytokine in specific response to Be-stimulation.34 To determine whether exposure to beryllium can induce TNF-α secretion in THP-1 macrophages, the TNF-α levels of the culture supernatants were measured by ELISA for THP-1 macrophages that were unstimulated or treated with 10 μM BeSO4, 100 μM BeSO4, 10 μM BeO or 100 μM BeO for 24 h or 48 h. For both 24 h and 48 h treatments, the TNF-α levels were all around 1000 pg/mL (Table 1). The TNF-α level of THP-1 monocytes was also measured but was undetectable.| Treatment of THP-1 macrophages | 24 h Treatment (pg/mL) | 48 h Treatment (pg/mL) |
|---|---|---|
| Unstimulated | 1152.6 ± 40.5 | 1103.0 ± 16.8 |
| 10 μM BeSO4 | 1061.4 ± 149.2 | 1081.6 ± 71.7 |
| 100 μM BeSO4 | 1088.2 ± 45.3 | 935.0 ± 37.6 |
| 10 μM BeO | 1026.5 ± 56.1 | 1132.9 ± 38.8 |
| 100 μM BeO | 1046.4 ± 82.5 | 1145.7 ± 55.1 |
| THP-1 monocytes | Non-detectable | Non-detectable |
Discussion
Investigation of cellular metal uptake using cells of human origin is important in understanding metal-related toxicity and diseases.35 In the present work, human monocytecell line THP-1 was induced with PMA to differentiate into a macrophage. This cell with characteristics of human alveolar macrophages was employed to study cellular beryllium uptake and related biological effects. Morphological changes, phagocytosis of fluorescent latex beads, and cell surface CD14 expression were used to verify the successful differentiation of THP-1 monocytes into macrophages by PMA induction. An improved mass spectrometry method for quantitative analysis of intracellular beryllium as opposed to the traditional radioisotopic approach was developed using ICP-MS, and satisfactory recovery rate (92–108%) was obtained. Sulfosalicylic acid, a beryllium ligand, was used in the cell washing procedure to ensure efficient rinsing of the beryllium nonspecifically adsorbed onto the cell surface.Evidence was obtained in our experiment that THP-1 macrophages took up a considerable amount of beryllium, with values varying from several to tens of nmol Be/106 cells depending on the compound solubility, exposure duration, and compound concentration administrated. These values are comparable to the rates of beryllium uptake for cultured canine pulmonary alveolar macrophages when beryllium sulfate or beryllium oxide was added12 and for the rat liver parenchymal cell line and Kupffer cells in culture when particulate beryllium phosphate, soluble beryllium sulfate, or a beryllium sulfosalicylate complex was administrated.15,16 Our data support the hypothesis that phagocytosis is the major mechanism by which the alveolar macrophage incorporates beryllium.11,12 It is possible that soluble forms of beryllium could gain entrance into the macrophageviaphagocytosis by first forming insoluble forms of beryllium in the culture medium. The work of Cheng, and Reeves and Vorwald suggested that, once in blood, soluble beryllium salts form and are transported as colloidal particles of beryllium phosphate and hydroxide.36,37 Due to the presence of phosphate in RPMI 1640 culture medium and its neutral pH, soluble beryllium sulfate could form both beryllium phosphate and hydroxide in the culture medium. On the other hand, the addition of serum globulins to the culture medium can improve the solubility of beryllium. For beryllium chloride at a concentration of 55 μM, the percentage of insoluble beryllium decreased from 80% in basal minimal Eagle’s medium to 10.3% with the presence of fetal calf serum at a final concentration of 10%.11 So, the uptake of soluble beryllium sulfate measured in our experiment might be explained by the balanced effects of the formation of insoluble complex of beryllium phosphate and hydroxide in the RPMI-1640 culture medium and the enhanced beryllium desolvation in the presence of fetal calf serum.
Subsequent apoptosis studies on chromosomal DNA fragmentation and PARP cleavage revealed that incorporated beryllium has negligible cytotoxicity to THP-1 macrophages. Though adherent CBD/BAL and BeS/BAL macrophages and DEOHS-1 human CBD macrophagecell line have been reported to undergo beryllium-stimulated apoptosisin vitro,38,39 a beryllium-induced apoptosis study has not been conducted on macrophages from the BAL of normal, healthy, non-Be-exposed individuals. TNF-α is a potent trigger of cellular apoptosis and TNF-α elevation has been reported in beryllium stimulated CBD/BAL cells.34,40,41 In the present work, ELISA measurement of TNF-α secretion indicated that for THP-1 macrophages, TNF-α expression was not upregulated upon beryllium stimulation in comparison to the untreated control cells. Both Be-stimulated and unstimulated THP-1 macrophages produced a constitutive level of TNF-α around 1000 pg/mL. This enhanced expression, in comparison to the non-detectable value for THP-1 monocytes reported in literature and our experiments34,42 could be attributed to the PMA treatment.43 It is worthy of note that the regulation in TNF-α production caused by beryllium treatment could have been masked by this high background level. Anyway, our results are in agreement with the literature finding that upon in vitro beryllium stimulation, no upregulation of TNF-α was identified in most mouse and human macrophage cell lines, even including BeS/BAL cells.34,38–40 As the human CBD/BAL cell population is a mixed cell population, consisting of macrophages, lymphocytes and other chronic inflammatory cells, and those individuals who progressed from BeS to CBD had a significantly higher percentage of lymphocytes in their BAL fluid on baseline clinical evaluation,2 the measured TNF-α elevation in the CBD/BAL cells could be mainly due to production by the T lymphocytes. Investigations on cell responses of human BAL macrophages of normal volunteers to air pollution particulates indicated that cytokine production, inhibition of host defense mechanisms, and apoptosis were specifically caused by insoluble components of ambient particulate matter in the size range of 2.5–10 μm diameter as compared to insoluble particles of 0.1–2.5 μm diameter and soluble particles.44 Since BeO and colloidal particles of beryllium phosphate and hydroxide formed in the culture medium are much smaller than micron size,12alveolar macrophages could have some level of tolerance to beryllium. This may explain why most Be-exposed individuals remain healthy throughout life.
Conclusion
In this study, a new analytical approach for measuring cellular beryllium content was developed using ICP-MS in place of conventional radioactive isotope labeling. The influence of the solubility of beryllium compound, exposure duration, and beryllium concentration on the incorporation of beryllium was studied. Our experimental results indicated that THP-1 differentiated macrophages can take up a substantial amount of beryllium and the major cellular uptake pathway is through phagocytosis of the particulate form of the beryllium compound. Subsequent DAPI nuclear staining and Western blot analysis of PARP cleavage revealed that intracellular beryllium uptake had a negligible effect on the apoptosis of THP-1 macrophages compared to the unstimulated macrophage control. Meanwhile, no substantial variation of TNF-α production was observed for THP-1 macrophages upon beryllium exposure. These data may explain why most Be-exposed individuals remain healthy throughout life. It is speculated that the individual’s genetic susceptibility may be the determining factor in CBD progression and development. Further experiments with alveolar macrophages of normal, healthy, non-Be-exposed individuals need to be conducted to confirm the benign biological effects of cellular beryllium uptake.Acknowledgements
This work was sponsored by the Program for New Century Excellent Talents in University (NCET-07-0729) and by the Scientific Research Foundation for Returned Overseas Chinese Scholars, State Education Ministry (SRF for ROCS, SEM) for which we are most grateful. The authors gratefully acknowledge Dr Jinzhang Zeng of Xiamen University for technical help in apoptosis measurement.References
- L. S. Newman, Science, 1993, 262, 197–198 CrossRef CAS.
- L. S. Newman, M. M. Mroz, R. Balkissoon and L. A. Maier, Am. J. Respir. Crit. Care Med., 2005, 171, 54–60 Search PubMed.
- P. K. Henneberger, D. Cumro, D. D. Deubner, M. S. Kent, M. McCawley and K. Kreiss, Int. Arch. Occup. Environ. Health, 2001, 74, 167–176 CrossRef CAS.
- K. Kreiss, M. M. Mroz, B. Zhen, J. W. Martyny and L. S. Newman, Am. Rev. Respir. Dis., 1993, 148, 985–991 Search PubMed.
- K. Kreiss, M. M. Mroz, B. Zhen, H. Wiedemann and B. Barna, Occup. Environ. Med., 1997, 54, 605–612 CrossRef CAS.
- L. Richeldi, R. Sorrentino and C. Saltini, Science, 1993, 262, 242–244 CAS.
- A. P. Fontenot, M. Torres, W. H. Marshall, L. S. Newman and B. L. Kotzin, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 12717–12722 CrossRef CAS.
- M. Amicosante, N. Sanarico, F. Berretta, J. Arroyo, G. Lombardi, R. Lechler, V. Colizzi and C. Saltini, Hum. Immunol., 2001, 62, 686–693 CrossRef CAS.
- Z. Wang, G. M. Farris, L. S. Newman, Y. Shou, L. A. Maier, H. N. Smith and B. L. Marrone, Toxicology, 2001, 165, 27–38 CrossRef CAS.
- B. L. Scott, T. M. McCleskey, A. Chaudhary, E. Hong-Geller and S. Gnanakaran, Chem. Commun., 2008, 2837–2847 RSC.
- B. A. Hart and D. G. Pittman, J. Reticuloendothel. Soc., 1980, 27, 49–58 Search PubMed.
- A. F. Eidson, A. Taya, G. L. Finch, M. D. Hoover and C. Cook, J. Toxicol. Environ. Health, 1991, 34, 433–448 CAS.
- H. P. Witschi and W. N. Aldridge, Biochem. J., 1968, 106, 811–820 CAS.
- D. N. Skilleter and R. J. Price, Chem.-Biol. Interact., 1978, 20, 383–396 CrossRef CAS.
- D. N. Skilleter and A. J. Paine, Chem.-Biol. Interact., 1979, 24, 19–33 CrossRef CAS.
- D. N. Skilleter and R. J. Price, Toxicol. Appl. Pharmacol., 1981, 59, 279–286 CrossRef CAS.
- H. Schwende, E. Fitzke, P. Ambs and P. Dieter, J. Leukoc. Biol., 1996, 59, 555–561 CAS.
- R. W. Stokes and D. Doxsee, Cell. Immunol., 1999, 197, 1–9 CrossRef CAS.
- B. Janic, T. M. Umstead, D. S. Phelps and J. Floros, J. Immunol. Methods, 2003, 272, 125–134 CrossRef CAS.
- S. Tsuchiya, M. Yamabe, Y. Yamaguchi, Y. Kobayashi, T. Konno and K. Tada, Int. J. Cancer, 1980, 26, 171–176 CrossRef CAS.
- M. R. Hjort, A. J. Brenyo, J. N. Finkelstein, M. W. Frampton, M. B. LoMonaco, J. C. Stewart, C. J. Johnston and C. T. D'Angio, Inflammation, 2003, 27, 137–145 CrossRef CAS.
- P. R. Kramer and S. Wray, J. Steroid Biochem. Mol. Biol., 2002, 81, 203–216 CrossRef CAS.
- K. Ueki, K. Tabeta, H. Yoshie and K. Yamazaki, Clin. Exp. Immunol., 2002, 127, 72–77 CrossRef CAS.
- H. Sakamoto, M. Aikawa, C. C. Hill, D. Weiss, W. R. Taylor, P. Libby and R. T. Lee, Circulation, 2001, 104, 109–114 CrossRef CAS.
- K. Shigeta, K. Sato and N. Furuta, J. Anal. At. Spectrom., 2007, 22, 911–916 RSC.
- V. F. Taylor, B. P. Jackson and C. Y. Chen, Anal. Bioanal. Chem., 2008, 392, 1283–1290 CrossRef CAS.
- S. Trumpler, W. Lohmann, B. Meermann, W. Buscher, M. Sperling and U. Karst, Metallomics, 2009, 1, 87–91 RSC.
- A. P. Fontenot, L. S. Newman and B. L. Kotzin, Clin. Immunol., 2001, 100, 4–14 CrossRef CAS.
- G. Lombardi, C. Germain, J. Uren, M. T. Fiorillo, R. M. du Bois, W. Jones-Williams, C. Saltini, R. Sorrentino and R. Lechler, J. Immunol., 2001, 166, 3549–3555 CAS.
- W. Zou, Z. Liu, A. Jiang and C. Li, Chinese J. Pathophysiol., 2007, 23, 1195–1198 Search PubMed (in Chinese).
- M. A. Dobrovolskaia and S. N. Vogel, Microbes Infect., 2002, 4, 903–914 CrossRef CAS.
- A. E. Egger, C. Rappel, M. A. Jakupec, C. G. Hartinger, P. Heffeter and B. K. Keppler, J. Anal. At. Spectrom., 2009, 24, 51–61 RSC.
- N. Gambi, F. Tramontano and P. Quesada, Biochem. Pharmacol., 2008, 75, 2356–2363 CrossRef CAS.
- R. T. Sawyer, L. A. Kittle, H. Hamada, L. S. Newman and P. A. Campbell, Toxicology, 2000, 143, 235–247 CrossRef CAS.
- E. P. Chan, A. Mhawi, P. Clode, M. Saunders and L. Filgueira, Metallomics, 2009, 1, 166–174 RSC.
- K. K. Cheng, J. Pathol. Bacteriol., 1956, 71, 265–276 Search PubMed.
- A. L. Reeves and A. J. Vorwald, J. Occup. Med., 1961, 3, 567–574 CAS.
- R. T. Sawyer, V. A. Fadok, L. A. Kittle, L. A. Maier and L. S. Newman, Toxicology, 2000, 149, 129–142 CrossRef CAS.
- L. A. Kittle, R. T. Sawyer, V. A. Fadok, L. A. Maier and L. S. Newman, Sarcoidosis Vasc. Diffuse. Lung Dis., 2002, 19, 101–113 Search PubMed.
- S. S. Tinkle and L. S. Newman, Am. J. Respir. Crit. Care Med., 1997, 156, 1884–1891 Search PubMed.
- R. T. Sawyer, C. E. Parsons, A. P. Fontenot, L. A. Maier, M. M. Gillespie, E. B. Gottschall, L. Silveira and L. S. Newman, Am. J. Respir. Cell Mol. Biol., 2004, 31, 122–130 Search PubMed.
- G. M. Galbraith, J. P. Pandey, M. G. Schmidt, P. Arnaud and J. M. Goust, Arch. Environ. Health, 1996, 51, 29–33 CAS.
- C. Hwang, M. Gatanaga, G. A. Granger and T. Gatanaga, J. Immunol., 1993, 151, 5631–5638 CAS.
- J. M. Soukup and S. Becker, Toxicol. Appl. Pharmacol., 2001, 171, 20–26 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |