The metallomics approach: use of Fe(II) and Cu(II) footprinting to examine metal binding sites on serum albumins
Michael R.
Duff, Jr.
and
Challa V.
Kumar
*
University of Connecticut, Department of Chemistry, 55 North Eagleville Road, U-3060, Storrs, Connecticut 06269, USA. E-mail: Challa.Kumar@uconn.edu; Web: http://jasmin.chem.uconn.edu
First published on 2nd September 2009
Abstract
Metal binding to serum albumins is examined by oxidative protein-cleavage chemistry, and relative affinities of multiple metal ions to particular sites on these proteins were identified using a fast and reliable chemical footprinting approach. Fe(II) and Cu(II), for example, mediate protein cleavage at their respective binding sites on serum albumins , in the presence of hydrogen peroxide and ascorbate. This metal-mediated protein-cleavge reaction is used to evaluate the binding of metal ions, Na+, Mg2+, Ca2+, Al3+, Cr3+, Mn2+, Co2+, Ni2+, Zn2+, Cd2+, Hg2+, Pb2+, and Ce3+ to albumins , and the relative affinities (selectivities) of the metal ions are rapidly evaluated by examining the extent of inhibition of protein cleavage. Four distinct systems Fe(II)/BSA, Cu(II)/BSA, Fe(II)/HSA and Cu(II)/HSA are examined using the above strategy. This metallomics approach is novel, even though the cleavage of serum albumins by Fe(II)/Cu(II) has been reported previously by this laboratory and many others. The protein cleavage products were analyzed by SDS PAGE, and the intensities of the product bands quantified to evaluate the extent of inhibition of the cleavage and thereby evaluate the relative binding affinities of specific metal ions to particular sites on albumins . The data show that Co(II) and Cr(III) showed the highest degree of inhibition, across the table, followed by Mn(II) and Ce(III). Alakali metal ions and alkaline earth metal ions showed very poor affinity for these metal sites on albumins . Thus, metal binding profiles for particular sites on proteins can be obtained quickly and accurately, using the metallomics approach.
Introduction
Identifying metal binding sites on proteins is critical for understanding the role of metal ions in biological processes and in the elucidation of their role in the bioinorganic chemistry of metallo-proteins. Even though X-ray crystallography and NMR spectroscopy have contributed immensely to the identification of metal sites on proteins, rapid location of metal binding sites on numerous proteins, especially the binding of metal ions which are not present in the native form of the protein, is still challenging. Recent developments in proteomics contributed to a very rapid increase in the number of metallo-proteins whose metal binding sites are yet to be determined. This situation urgently demands the design of novel methods for the rapid identification of a variety metal binding sites on proteins. Another important but related challenge in this field is determining the selectivity of binding of a specific metal ion to particular sites on the protein. This information is critical in assessing the bioinorganic chemistry and the toxicological profiles of groups of metal ions.To address these challenges, we describe a novel metallomics approach to rapidly identify if a given metal ion binds to a particular site on a target protein, and also quantify the relative affinities of a small set of metal ions to these sites. Our strategy is two-fold. The first part is to use a well-known, metal-based reagent to cleave the peptide backbone, and then identify the binding site by amino acid sequencing of the daughter fragments. For example, metal-ions,1,2 metal-oxo species3 and metal complexes,4,5 are activated to cleave the peptide backbone at the metal binding site, thereby providing a footprint of the metal site on the protein. We chose the well-known cleavage reactions of serum albumins 2,5 by Fe(II) or Cu(II)-mediated Fenton chemistry6 or Haber–Weiss chemistry.7 In these reactions, the proteins are cleaved into two or more peptide fragments near the metal binding site which facilitates the identification of the metal binding site on the protein. In addition to using the cleavage chemistry mediated by the metal ions, one could use metal mediated oxidation of amino acid residues at the metal binding site. For example, the residues surrounding the Cu(II) binding site in Cu/Zn superoxide dismutase were identified through the analysis of the Cu(II)-mediated oxidation.8 Thus, metal-mediated chemical footprinting methods could be exploited to identify their binding sites on proteins. However, one limitation of this approach has been that it is limited to a few metal ions which can either cleave the protein backbone or modify the surrounding residues.
The second part of our strategy, which is novel, is to use specific metal ions to evaluate the extent of inhibition of the above described metal-mediated protein cleavage via competitive inhibition, and assess the relative binding affinities of specific metal ions to particular sites. By combining the cleavage chemistry with competitive inhibition, the binding sites of metal ions which do not induce protein cleavage or modify amino acid residues can be footprinted by this approach. The data show that the binding of a number of transition metal ions as well as alkaline earth metal ions to specific sites on serum albumins can be examined by this approach.
Therefore, metal-footprinting with Fenton chemistry is a versatile tool to search for metal binding sites (known or unknown sites) on proteins by an indirect approach, where the metal ion of interest may not produce a spectroscopic or chemical signature to identify its interaction with the target protein. Even in cases where specific signatures of metal binding can be monitored, identifying the binding site, and its relationship to other known metal binding sites can be still challenging. Using a competitive cleavage-inhibition approach, the relative affinities of metal ions to particular sites on proteins can be quantified, rapidly and accurately. The approach is rapid (<24 hours from start of the reaction to data analysis), does not require the metal to be spectroscopically or chemically active on binding to the protein, and the method is amenable to evaluating the relative affinities of a set of ions to particular sites on proteins.
Serum albumins (SA) are chosen for the current studies because they are the most abundant proteins in the serum, they bind and transport a multitude of metal ions,9 and have a well characterized metal binding site at their N-terminus (N-Site). SA bind Cu(II) and Ni(II) at the N-site and a second binding site (Site II) has not been firmly established.10 Site II binds Cu(II), Ni(II), Cd(II) and Zn(II) and it is suggested to consist of His105, His147, and His246 in bovine serum albumin (BSA)11 or His67, Asn99, His247, Asp249 in human serum albumin (HSA).12 Recently, a third site has been suggested for Ni(II) and Zn(II) but not identified, and relative affinities of multiple metal ions to specific sites on albumins have not been established.13 Because of the multitude of metals known to bind to serum albumins , at multiple sites,14 this protein is a good candidate to quantify the relative affinities of multiple ions. The metallomics approach outlined above can be effective in rapidly assessing the relative affinities of a small set of metal ions to particular sites on serum albumins . Such data could be useful to evaluate the intrinsic metallo-biochemistry of metal ions and in assessing the toxicities of these ions via binding to the serum proteins such as SA.
Results and discussion
We first examined metal binding to SA by Isothermal Titration Calorimetry and then evaluated the location of metal binding sites by protein cleavage, followed by amino acid sequencing of the daughter fragments. Subsequently, inhibition of protein cleavage by a small set of metal ions is examined to evaluate their binding to particular sites on the protein and their relative affinities. Our observations are enumerated below.Isothermal titration calorimetry (ITC)
The thermodynamics of ligand binding to proteins can be quantitatively determined by measuring the heat absorbed or released during this interaction by ITC. Thus, ITC serves as a powerful tool to evaluate ΔH, ΔS, ΔG, the number of binding sites (n) and the binding constant (Kb), in one experiment. A solution of FeSO4 (1.5 mM, 10 mM MES, pH 6.4, 298 K) was titrated (42 injections of 7 μL each) into a solution of HSA (25 μM, 10 mM MES, pH 6.4, 298 K) and the heat produced during each injection has been recorded. After subtracting the corresponding heats of dilution, protein dilution and metal ion dilution into the buffer, the titration indicated a net release of heat (Fig. 1A). The heat produced during each injection was integrated and total heat produced as a function of progress of the titration plotted (Fig. 1B).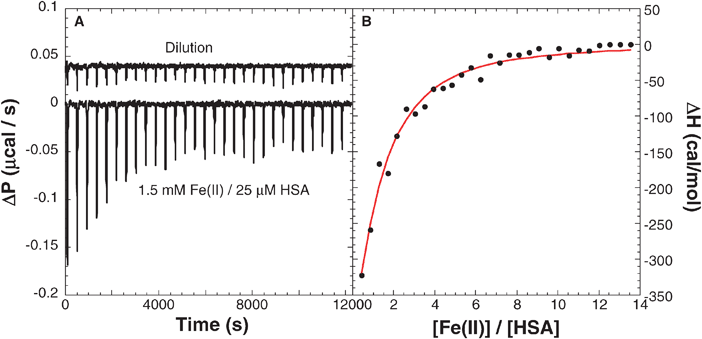 | ||
| Fig. 1 (A) Binding of Fe(II) (1.5 mM) to HSA (25 μM) by ITC (10 mM NaMES pH 6.4). (B) Integrated heat release was analyzed with a number of models and indicated that the single-set of indistinguishable binding sites model gave best fits with a binding constant of 2.0 ± 0.3 × 104 M−1, enthalpy change of −2.9 ± 1.0 kcal/mol and entropy change of 9.9 ± 3.7 cal/mol K. The titration was repeated three times and the data averaged. Note that no precipitate was noted at the end of the titration. | ||
The enthalpy data were analyzed using different binding models by non-linear least squares method, and the best fit to the data indicated the validity of a single-set of indistinguishable binding sites model. The binding parameters extracted from the best fit are, Kb = 2.0 ± 0.3 × 104 M−1, ΔH = −2.9 ± 1.0 kcal/mol, stoichiometry (n) = 1.1 ± 0.1, and ΔS = 9.9 ± 3.7 cal/mol/K. Note that the binding is weakly exothermic, but it is accompanied by an increase in the entropy of the system which is likely due to protein unfolding and/or solvent release from the binding surfaces. Thus, binding of Fe(II) to HSA is both enthalpy and entropy driven.
In a similar manner, titration of FeSO4 into a solution of BSA (50 μM, 10 mM PIPES pH 6.1) indicated a binding constant of 2.4 ± 0.6 × 105 M−1, ΔH of 0.8 ± 0.3 kcal/mol, stoichiometry of 3.3 ± 0.3, and ΔS of 26.8 ± 1.6 cal/mol/K. In contrast to the exothermic binding of Fe(II) to HSA, its binding to BSA is endothermic and entirely entropy driven. Protein structural changes and desolvation play a major role in controlling Fe(II) binding to BSA. Note that binding of Fe(II) to BSA is nearly an order of magnitude stronger than to HSA, under similar conditions of pH/ionic strength, and this observation is interesting.
If Fe(II) binds primarily to the N-terminal site on serum albumin , then binding to the bovine form with residues Asp-Thr-His-Lys at this site is better than binding to Asp-Ala-His-Lys of the corresponding human protein. Thus, the binding sites for Fe(II) on BSA and HSA are different enough to alter the affinity for Fe(II). However, the binding of Fe(II) to SA is not well documented, thermodynamic data are sketchy, but the binding of Fe(III) to BSA has been reported to be stronger than Fe(II).15,16 In contrast, Zn(II) has essentially the same affinity for BSA and HSA, but it binds to Sites A/B on albumin but not at the N-Site.17 On the other hand, UO22+ binds to an internal site on BSA but does not bind to HSA where this site is somehow altered.13 Thus, multiple metal binding sites exist on albumins , and their binding to metal ions can be quite complex and varied.
Note that ITC studies of Cu(II) binding to BSA (100 mM TrisBorate at pH 9) suggested that BSA has only one detectable Cu(II)-binding site with an estimated affinity of 1013 M−1.18 We used these metal binding data in conjunction with the footprinting data shown below to assess the relative affinities of metal ions to particular sites on SA.
Fe(II) or Cu(II)-mediated cleavage of SAs
We re-examined the cleavage of SA by Fe(II)–ascorbate–H2O2 under our experimental conditions (FeSO4 (15 μM), HSA or BSA (15 μM), ascorbate (500 μM), and H2O2 (500 μM) in 10 mM TrisHCl pH 7 or 10 mM MES pH 6).The reaction was quenched after 1.5 hours by the addition of 2-mercaptoethanol (1 mM) and EDTA (1 mM) and the products analyzed by SDS PAGE (Fig. 2). The appearance of two product bands, one at ∼39 kDa (20%) and another at ∼27 kDa (20%), is immediately clear (lane 3). A significant portion of the starting material is left unreacted and low conversions are not unsual for these reactions.1 For example, partial cleavage of pigeon liver malic enzyme by FeSO42 into two fragments and the Fe(II) cleavage of poly(A)-specific ribonuclease into two fragments and a majority of uncleaved starting material has been noted previously.5 As demonstrated in Fig. 2, the cleavage reaction required all three reagents (Fe(II), ascorbate, and H2O2, lane 3) and the reaction did not occur when any one of these were omitted (lanes 4–9).
 | ||
| Fig. 2 Fe(II)-induced cleavage of HSA with FeSO4–H2O2–ascorbate. Lane 1 contained molecular weight markers listed in kDa, and each lane contained multiple reagents. For example, lanes 2 through 9 contained 15 μM HSA; lanes 3, 4, 7, and 8 contained 15 μM FeSO4, lanes 3, 5, 7, and 9 contained 500 μM H2O2; and lanes 3, 6, 8, and 9 contained 500 μM ascorbate. Thus, lane 3 contained the protein, metal ion, hydrogen peroxide and ascorbate, reacted for 1.5 h. | ||
In contrast to the low reactivity of Fe(II)-mediated cleavage of HSA, Cu(II)-mediated cleavage of HSA, under similar conditions, produced at least five distinct product bands (Fig. 3), and these bands are similar to those reported earlier, from this laboratory.19 Lanes 4–9 served as controls where one of the reagents is absent. Significant activity was noted in lane 8, which contained HSA, Cu(II) and sodium ascorbate but not hydrogen peroxide. This reactivity in the absence of hydrogen peroxide is mostly likely due to O2 dissolved in the air saturated solutions where it could serve as the oxidant to promote the oxidative cleavage reactions with Cu(II). Note that such reactivity is not noticed with Fe(II). Thus, there are some distinctions between the reactivities of Fe(II) and Cu(II) as well as the bovine versus human forms of serum albumins . Taken together, these four systems served as excellent reagents to footprint metal binding sites, and they provided opportunities to quantify relative affinities of multiple metal ions to particular sites.
 | ||
| Fig. 3 Cu(II)-induced cleavage of HSA with CuSO4–H2O2–ascorbate. Lane 1 contained molecular weight markers listed in kDa, and each lane contained multiple reagents. For example, lanes 2 through 9 contained 15 μM HSA; lanes 3, 4, 7, and 8 contained 15 μM CuSO4, lanes 3, 5, 7, and 9 contained 500 μM H2O2; and lanes 3, 6, 8, and 9 contained 500 μM ascorbate. Thus, lane 3 contained the protein, metal ion, hydrogen peroxide and ascorbate, reacted for 1.5 h. | ||
To test the hypothesis that the protein cleavage in Fig. 2–3 is due to the metal ions bound to the protein and not due to ions that are free in the solution, quenching experiments were conducted. Attempts were made to quench the reactive oxygen species in these reactions by the addition of radical scavengers such as ethanol, 2-propanol and D-mannitol.20 There was only minimal quenching (<15%) by any of the quenchers (up to 3 M), and thus any reactive oxygen species generated in the solution or on the exterior of the protein is unlikely to be responsible for the observed protein cleavage. Given the fact that these metal ions bind to SA, and that the quenchers are not able to inhibit the chemistry to a measurable extent, data suggest that protein cleavage occurs essentially at metal binding sites on proteins which are not readily accessible to solution-borne quenchers .
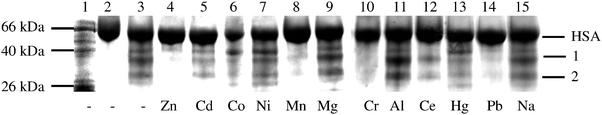 | ||
| Fig. 4 Inhibition of Fe(II) cleavage of HSA by specific metal ions. Lane 1 contained molecular weight markers. Lane 2 is 15 μM HSA, lanes 3–9 contained 15 μM HSA, 15 μM (NH4)2Fe(SO4)2, 500 μM H2O2, and 500 μM ascorbate, in 10 mM TrisHNO3 pH 7.2. Lanes 4–14 contained 1 mM Zn(II), Cd(II), Co(II), Ni(II), Mn(II), Mg(II), Cr(III), Al(III), Ce(III), Hg(II) and Pb(II), respectively. Lane 15 contained 10 mM NaCl and Na(I) did not inhibit the reaction. | ||
The cleavage site on HSA was identified by isolating, purifying and N-terminal sequencing of the two product bands from the Fe(II)–HSA reaction. The analysis showed that the 39 kDa band had the sequence DAHKS (60% of total product) which is the known N-terminal sequence of HSA. In addition, this product band had an additional peptide with the N-terminal sequence, XXKDL (40% of total product), and XXKDL is an interior sequence of HSA which uniquely occurs at residue 10. Thus, two distinct fragments are responsible for the 39 kDa band. N-terminal sequencing of the second product (band 2) indicated one peptide with the sequence DAHKS (100%), the known N-terminal sequence of HSA, and there were no other products found in the sequencing studies. Thus, Fe(II) cleaves HSA near the N-terminal metal binding site (residues 9–10) and two other interior sites. Modeling with Rasmol (v. 2.7.2 by Roger Sayle, freeware) using the known crystal structure of HSA indicates that metal binding to the N-terminal site could account for all the three products identified in the sequencing studies, but the involvement of additional metal binding sites in the protein cleavage cannot be ruled out.
Competitive inhibition studies
The bands in Fig. 4 were quantified and percentage inhibition estimated for each lane, and these have been collected in Table 1. The data clearly show that the order of inhibition for the Fe(II)–HSA reaction is: Cr(III) ∼ Pb(II) > Ce(II) > Co(II) ∼ Ni(II) > Zn(II) ∼ Hg(II) ∼ Mn(II) > Cd(II) ⋙ Na(I), Mg(II), Ca(II) and Al(III). Specific metal ions inhibited the cleavage reaction to a much greater extent than others, but the formation of both product bands was inhibited to similar extents, thereby supporting the hypothesis that metal binding to one site could be responsible for both the product bands.
| Redox system | Buffer | Percent inhibitionabc | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Calculated with NIH Image v. 1.62. b Calculated as a percentage of the difference of the redox system with and without metal over just the redox system. c Estimated error is ±10%. | ||||||||||||||
| Na+ | Mg2+ | Ca2+ | Al3+ | Cr3+ | Mn2+ | Co2+ | Ni2+ | Zn2+ | Cd2+ | Hg2+ | Pb2+ | Ce3+ | ||
| HSA–Fe(II) | TrisNO3 | <10 | <10 | <10 | <10 | 100 | 50 | 60 | 60 | 50 | 40 | 50 | 100 | 70 |
| BSA–Fe(II) | TrisNO3 | <10 | <10 | <10 | 90 | 90 | 60 | 100 | 90 | 50 | <10 | 90 | 40 | 40 |
| HSA–Cu(II) | TrisNO3 | <10 | <10 | <10 | 20 | 40 | <10 | 100 | <10 | 30 | <10 | 100 | 30 | 50 |
| BSA–Cu(II) | TrisNO3 | <10 | <10 | <10 | <10 | 30 | <10 | 40 | 20 | 10 | <10 | <10 | 10 | <10 |
| HSA–Fe(II) | None | <10 | <10 | <10 | <10 | 100 | 80 | 100 | <10 | 100 | 40 | 30 | 90 | 50 |
| BSA–Fe(II) | None | <10 | <10 | <10 | <10 | 100 | 60 | 80 | 70 | 100 | <10 | 80 | 60 | 100 |
| HSA–Cu(II) | None | <10 | <10 | <10 | <10 | 50 | 100 | 20 | 10 | 20 | <10 | <10 | <10 | <10 |
| BSA–Cu(II) | None | <10 | <10 | <10 | 30 | 100 | 100 | 100 | 20 | 20 | <10 | 70 | 10 | 100 |
To test if the cleavage inhibition is due to metal ions or due to metal coordinated buffer ions, we also carried out the cleavage reactions in non-buffered, pH adjusted de-ionized water (Table 1). Overall, there has been greater inhibition in the absence of buffer ions, except for Ni(II), presumably due to higher affinities at lower ionic strengths. These data show that the metal coordinated buffer complexes are unlikely to be responsible for the observed inhibition.
Cleavage inhibition is most likely due to the competitive binding of specific metal ions to particular sites, although allosteric effects can not be ruled out. Therefore, one hypothesis is that the extent of inhibition is proportional to the corresponding affinities of the metal ions to particular sites on the protein. Accordingly, Cr(III) and Pb(II) show the highest affinity for the Fe(II) site on HSA, while Co(II), Ni(II), Mn(II), Zn(II), Hg(II), Cd(II) and Ce(III) show only moderate affinities, and Al(III), Na(I), Mg(II) and Ca(II) show poor or no affinity. Lack of inhibition by 10 mM Na(I) is important because it shows that a mere increase in ionic strength is not responsible for the observed inhibition of the reaction.
Having obtained encouraging results with the Fe(II)–HSA reaction, we then examined the selectivity for the metal binding to Fe(II) binding sites on BSA. SDS PAGE analysis of the reaction mixtures, followed by quantitation of the product band intensities indicated the following trend in the cleavage inhibition for the Fe(II)–BSA reaction: Co(II) > Hg(II) ∼ Cr(III) ∼ Al(III) ∼ Ni(II) > Mn(II) > Zn(II) > Pb(II) ∼ Ce(III) >>> Cd(II), Na(I), Mg(II) and Ca(II). This order is quite different from that of the Fe(II)–HSA reaction. Co(II) competed efficiently with the BSA reaction, but it inhibited only 60% of the HSA reaction. The two proteins differ considerably in their affinities to specific metal ions. Again, the alkaline earth metals and Na(I) did not inhibit the reaction to an appreciable extent. One notable feature is that Al(III) inhibited the Fe(II)–BSA reaction very efficiently (90%), to nearly the same extent as that of Cr(III), Ni(II) or Hg(II). This approach of determining the relative affinities of metal ions to particular binding sites on proteins is further tested by using Cu(II)-mediated protein cleavage.
The data in Table 1 further show that the observed trend in the inhibition of the Cu(II)–BSA reaction is: Co(II) > Cr(III) > Ni(II) > Zn(II) ∼ Pb(II), while the other metals ions examined do not inhibit the reaction appreciably. Indeed, none of the metal ions examined indicated complete inhibition of the Cu(II)–BSA reaction and this particular reaction is least sensitive to the presence of other metal ions. Since the affinity of Cu(II) is generally greater than that of Fe(II) to albumins , it is not surprising that the former reaction is less sensitive to the addition of metal ions but there are more interesting subtle trends.
There are additional, important differences between the human and bovine forms of serum albumins . For example, Al(III) inhibited the Fe(II)–BSA reaction (90%) but not the Fe(II)–HSA reaction (<10%), while Pb(II) inhibited the Fe(II)–HSA reaction (100%) but inhibited only weakly the Fe(II)–BSA reaction (40%). Therefore, the percent inhibition depended on the protein, as well as the type of metal binding site under consideration.
Conclusions
There are subtle differences in the inhibition, and presumably in the binding preferences of specific metal ions, and a discussion of the chemical basis of these trends is beyond the scope of this article. We intend to provide a simple, accurate and rapid method to evaluate the relative binding affinities of a multitude of metal ions to particular sites on proteins.The metallomics method shown here allows for quick (<24 hours) analysis of metal binding to proteins in a parallel manner. Competitive protein-cleavage inhibition could be a useful chemical tool for the rapid determination of the relative affinities of multiple metal ions for a given site on a specific protein. The method developed here complements the direct chemical footprinting of metal binding sites on proteins.13 However, caution about allosteric effects should be born in mind. Even so, this is a powerful method to rapidly screen a number of metal ions for their relative affinities to specific sites on a given protein.
Experimental
Materials
Fatty acid-free BSA and HSA (>95%), sodium ascorbate, AlCl3, CoCl2, Hg(NO3)2, CuSO4, FeSO4, EDTA, PIPES and MES buffers, hydrogen peroxide (30% by weight), 2-mercaptoethanol, sodium dodecylsulfate, and acrylamide were purchased from Sigma-Aldrich (St Louis, MO). All other metal salts were purchased from JT Baker and all metal salts used were of analytical grade. Purities of the protein samples were estimated by SDS PAGE, circular dichroism, and protein samples were used without further purification.Isothermal titration calorimetry
Isothermal titration calorimetry was performed on a VP-ITC 100 from MicroCal (Amherst, MA, USA) operated by the software provided by the manufacturer, as reported previously.13 For BSA, 1.5 mM Fe(II) was dissolved in 10 mM NaPIPES, pH 6.1 and the pH readjusted with 0.1 M NaOH to within ±0.1 pH units. The Fe(II) was titrated into BSA (50 μM, 10 mM NaPipes, pH 6.1) and the heat evolved or absorbed measured. Control experiments were performed where Fe(II) was titrated into 10 mM NaPIPES (pH 6.1) buffer to account for the heat released from dilution. The data, corrected for the dilution, was analyzed using the Origin v. 5.0 software supplied by MicroCal, part of GE Healthcare. The binding constant, binding site size, enthalpy of binding (ΔH), entropy of binding (ΔS), and the free energy of binding (ΔG) were estimated in triplicate measurements. The heat released or absorbed (Q) during the titration is related to ΔH, volume of the sample cell (Vo), the bulk concentration of the ligand (Xt), protein concentration (Mt), the intrinsic binding constant (Kb), and the number of binding sites (n) are related by eqn (1).22 The experiment was repeated multiple times and the values averaged.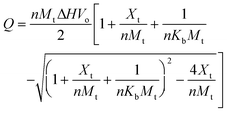 | (1) |
Protein cleavage
To a 15 μM solution of BSA or HSA, in either DI adjusted to pH 7 with dilute NaOH or in 10 mM TrisHNO3 pH 7, the desired amount of inhibiting metal (i.e. 1 mM Zn(II)) was added and binding allowed to reach equilibrium for 10 minutes. Then either 15 μM of Cu(II) or Fe(II) was added and the binding equilibrium was established for 10 minutes. Finally, addition of 500 μM H2O2 and 500 μM of NaAscorbate were added to initiate the reaction. After 1.5 hours, 1 mM 2-mercaptoethanol and 1 mM EDTA were used to inhibit the reaction process. The samples were dried down in a speedvac (Savant Integrated System ISS10).SDS PAGE
Analysis of the protein cleavage studies was performed using SDS PAGE according to a previously published protocol.19 Briefly, the samples were loaded in a reducing, denaturing buffer and loaded onto 9% SDS PAGEgels . The gels were run for 0.5 hours at 60 V, and then at 110 V for 2.5–3 hours. The gels were stained overnight with a 0.02% Coomassie stain in 10% acetic acid and the background destained with 10% acetic acid. High quality images of the proteingels were obtained by scanning the dye-stained gels with a Umax Astra 1220U scanner. Band intensities were quantified using NIH image software (v. 1.63 from the NIH Image FTP site, freeware) and product yields estimated (Yield of band I = intensity of band I/(intensity of band I plus that of the unreacted protein)). The distance of migration of the protein band depended linearly on the logarithm of the protein molecular mass.24 The molecular masses of the photofragments were assessed from a calibration graph constructed using proteins of known molecular masses. For sequencing, the gels were Western blotted on a PVC membrane,18 and sent to MidWest Analytical (St Louis, MO, USA) for amino acid sequencing analysis. At least five residues were sequenced from the N-termini of the product bands.Acknowledgements
Financial support from the Donors of the Petroleum Research Fund (AC4-33821), and the NSF (DMR-0604815) is gratefully acknowledged.References
- J. J. Hlavaty, J. S. Benner, L. J. Hornstra and I. Schildkraut, Biochemistry, 2000, 39, 3097–3105 CrossRef CAS; W. Y. Chou, W. P. Tsai, C. C. Lin and G. G. Chang, J. Biol. Chem., 1995, 270, 25935–25941 CrossRef CAS; S. Luo, H. Ishida, A. Makino and T. Mae, J. Biol. Chem., 2002, 277, 12382–12387 CrossRef CAS.
- C. W. Wei, W. Y. Chou, S. M. Huang, C. C. Lin and G. G. Chang, Biochemistry, 1994, 33, 7931–7936 CrossRef CAS.
- I. R. Gibbons and G. Mocz, Methods Enzymol., 1991, 196, 428–442 CAS.
- C. V. Kumar and J. Thota, Inorg. Chem., 2005, 44, 825–827 CrossRef CAS; C. V. Kumar, A. Buranaprapuk, A. Cho and A. Chaudhari, Chem. Commun., 2000, 597–598 RSC; L. Zhu, R. Bakhtiar and N. M. Kostic, JBIC, J. Biol. Inorg. Chem., 1998, 3, 383–391 CrossRef CAS; L.-M. Dutca, K.-S. Ko, N. L. Pohl and N. M. Kostic, Inorg. Chem., 2005, 44, 5141–5146 CrossRef CAS; G. N. Godson, J. Schoenich, W. Sun and A. A. Mustaev, Biochemistry, 2000, 39, 332–339 CrossRef CAS; E. Zaychikov, E. Martin, L. Denissova, M. Kozlov, V. Markovtosv, M. Kashlev, H. Heumann, V. Nikiforov, A. Goldfarb and A. Mustaev, Science, 1996, 273, 107–109 CrossRef CAS.
- Y. G. Ren, J. Martinez and A. Virtanen, J. Biol. Chem., 2002, 277, 5982–5987 CrossRef CAS.
- H. J. H. Fenton, J. Chem. Soc., Trans., 1894, 65, 899–910 RSC; E. Cadenas, Annu. Rev. Biochem., 1989, 58, 79–110 CrossRef CAS.
- F. Haber and J. Weiss, Proc. R. Soc. London, Ser. A, 1934, 147, 332 Search PubMed.
- J. D. Bridgewater, J. Lim and R. W. Vachet, J. Am. Soc. Mass Spectrom., 2006, 17, 1552–1559 CrossRef CAS.
- F. Friedberg, FEBS Lett., 1975, 59, 140–141 CrossRef CAS; C. Harford and B. Sarkar, in Handbook of Metal–Ligand Interactions in Biological Fluids. Bioinorganic Chemistry, ed. G. Berthod, Marcel Dekker, New York, 1995, pp. 405–410 Search PubMed; R. G. Reed, in Transport by Proteins, ed. G. Blauer and H. Sund, de Gruyter, New York, 1978, pp. 57–78 Search PubMed.
- J. Masuoka, J. Hegenauer, B. R. van Dyke and P. Saltman, J. Biol. Chem., 1993, 268, 21533–21537 CAS.
- P. J. Sadler and J. H. Viles, Inorg. Chem., 1996, 35, 4490–4496 CrossRef CAS.
- A. J. Stewart, C. A. Blindauer, S. Berezenko, C. Sleep and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3701–3706 CrossRef CAS.
- M. R. Duff, Jr. and C. V. Kumar, Angew. Chem., Int. Ed., 2006, 45, 137–139 CrossRef.
- G. Ming, K. Liang, M. Xiqin, L. Xin and Z. Hafna, Sci. China, Ser. B: Chem., 2002, 45, 151–157 CrossRef.
- R. C. Hider, Eur. J. Clin. Invest., 2002, 32, 50–54 CrossRef CAS.
- X. Xu, L. Zhang, D. Shen, H. Wu and Q. Liu, J. Fluoresc., 2008, 18, 193–201 CrossRef CAS.
- J. Masuoka and P. Saltman, J. Biol. Chem., 1994, 269, 25557–25561 CAS.
- Y. Zhang and D. E. Wilcox, JBIC, J. Biol. Inorg. Chem., 2002, 7, 327–337 CrossRef CAS.
- A. Buranaprapuk, S. P. Leach, C. V. Kumar and J. R. Bocarsly, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1998, 1387, 309–316 Search PubMed.
- N. Ettner, J. W. Metzger, T. Lederer, J. D. Hulmes, C. Kisker, W. Hinrichs, G. A. Ellestad and W. Hillen, Biochemistry, 1995, 34, 22–31 CrossRef CAS.
- D. E. Wilcox, Inorg. Chim. Acta, 2007, 361, 857–867.
- J. M. Sturtevant, Annu. Rev. Phys. Chem., 1987, 38, 463–88 CrossRef CAS.
- I. Jelesarov and H. R. Bosshard, J. Mol. Recognit., 1999, 12, 3–18 CrossRef CAS.
- H. Schagger and G. Von Jagow, Anal. Biochem., 1987, 166, 368 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |